
Abstract
The neuroprotective effect of hypothermia instituted after resuscitation from asphyxic cardiac arrest has not been studied in immature brain, particularly in a large animal model with recovery periods greater than 4 d. Moreover, protection from severe hypoxia seen with 3 h of hypothermia was reported to be lost when hypothermic duration was extended to 24 h in unsedated piglets, in contrast to the neuroprotection reported by 72 h of intrauterine head cooling in fetal sheep. Piglets (5–7 postnatal days) were subjected to asphyxic cardiac arrest followed by 24 h of either hypothermia (34°C) or normothermia (38.5–39°C). Comparisons were made with normothermic and hypothermic surgical sham animals without asphyxia. All of these groups were sedated, paralyzed, and mechanically ventilated for the first 24 h to prevent shivering and possible depletion of glucose stores. Hypothermia per se did not cause remarkable structural abnormalities. Ischemic damage was evaluated in putamen at 1 d of recovery without rewarming and at 11 d (10 d ± SD after rewarming). Ischemic cytopathology affected 60 ± 12% of neurons in putamen of normothermic animals compared with 9 ± 6% in hypothermic animals at 1 d of recovery without rewarming. At 11 d of recovery from hypoxia-ischemia, the density of viable neurons (neuron profiles/mm2) in putamen was markedly reduced in normothermic animals (81 ± 40) compared with hypothermic animals (287 ± 22), which was the same as in sham normothermic (271 ± 21), sham hypothermic (288 ± 46) and naïve animals (307 ± 51). These data demonstrate that 24 h of hypothermia at 34°C with sedation and muscle relaxation after asphyxic cardiac arrest prevents necrotic striatal neuronal cell death in immature brain before rewarming, and that the effect is sustained at 11 d after injury without deleterious side effects.
Similar content being viewed by others


Main
Transient cerebral hypoxia-ischemia (HI) in newborns often results from difficulties during delivery or cardiorespiratory arrest. Perinatal HI in its most severe form may cause permanent neurodevelopmental impairment, which typically includes spastic quadriparesis, severe cognitive deficiency and epilepsy (1). Advances in obstetric and neonatal care have not significantly altered the outcome of HI encephalopathy (2). Treatment for newborns with HI remains supportive despite the efforts of a variety of investigators to evaluate neuroprotective interventions (3). In adults resuscitated from cardiac arrest, mild hypothermia lasting 12–24 h is one of the few interventions found to benefit neurologic outcome (4, 5). In term newborns, selective head cooling is the subject of an on-going multicenter trial, where results to date have been encouraging (6). Preliminary data support the safety of mild hypothermia in sick newborns (7).
Mild-to-moderate hypothermia after HI provides neuroprotection in several experimental models of HI injury in immature brain (8, 9) including fetal sheep (10) and piglets (11, 12). Although short-term neuroprotection was seen when hypothermia was induced for 1–3 h after HI in postnatal day 7 rats, long-term protection was lost (13). In contrast, when the duration of hypothermia was extended to 6 h after HI, protection was sustained over a 6-wk recovery period (14). Protection also was seen with 72 h of hypothermia in fetal sheep (10). Thus prolonged periods of hypothermia are presumably required to provide long-term neuroprotection. However, neuroprotection seen with 3 h of mild hypothermia after HI in piglets (15) was lost with 24 h of hypothermia, possibly because of the prolonged cold stress in unsedated piglets (16). Thus, larger postnatal animals may require sedation and muscle relaxation during hypothermia to reduce the potential adverse effects of cold stress. Moreover, in large animal models of perinatal HI, such as HI in fetal sheep and newborn piglet, evaluation of efficacy has been limited to within 72 h of rewarming, and the degree of neuroprotection before and after rewarming has not been compared.
In the current study, asphyxic cardiac arrest in piglets was chosen as a model to simulate neonatal HI. This model of systemic hypoxia followed by asphyxic cardiac arrest is well characterized physiologically and pathologically (17, 18), and the pattern of brain injury closely resembles human newborn HI encephalopathy (19, 20). This study focused on striatum because this is the most vulnerable region in the piglet and because cell death matures most rapidly in this region, thereby requiring early intervention. The overall goal was to determine whether hypothermia (34°C) after resuscitation from systemic hypoxia and asphyxia provides short-term and long-term striatal neuroprotection in piglets that are kept sedated during hypothermia. The first experiment was designed to determine whether whole body hypothermia after HI ameliorates neuronal cell death before rewarming in the putamen and caudate nucleus at 24 h, at which time striatal injury is near maximal (18, 21). To determine whether the benefit from hypothermia is transient or sustained, striatal damage was evaluated at 11 d after HI in the second experiment. The hypotheses tested were that sedated piglets subjected to 24 h of whole body hypothermia (34°C) after asphyxic cardiac arrest demonstrate 1) decreased acute neuronal necrosis in striatum at 24 h in the absence of rewarming, 2) sustained neuroprotection at 11 d of recovery, and 3) improved functional recovery as measured by EEG and neurobehavioral scores.
METHODS
Animal preparation.
All procedures on piglets were approved by the Animal Care and Use Committee at the Johns Hopkins University. Animal preparation was performed as previously described (22). Five to seven-day-old male piglets, weighing 3.0–4.5 kg, were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneal), intubated and mechanically ventilated to maintain normoxia and normocarbia. Normal rectal temperature during the preparation was maintained at 38.5 ± 0.5°C using heating blankets and overhead lamps. Femoral arterial and venous catheters were placed via sterile technique into the descending aorta and inferior vena cava through the right groin, tunneled s.c. and secured to a right flank exit incision. All animals received i.v. fentanyl (10 μg/kg) and pancuronium (0.3 mg/kg) after catheterization. i.v. fluids of 0.45% saline with 5% dextrose were given continuously at 10 mL/h, adjusting the rate to maintain euvolemia and normoglycemia (arterial glucose 3–5 mM). Brain temperature was measured in a subset of 24-h survivors (n = 4) using a sterile thermistor placed via a burr hole into parietal cortex to a depth of 1 cm. Mean arterial blood pressure (MABP), heart rate, rectal temperature and ECG were continuously monitored. Arterial blood samples were obtained for measurements of Po2 (Pao2) and Pco2 (Paco2), pH, arterial O2 saturation, Hb concentration, and glucose concentration.
Asphyxia and recovery protocol.
Piglets were subjected to hypoxia for 30 min by decreasing inspired O2 to ∼10%, adjusted as needed to maintain Pao2 at 2.7–3.3 kPa (20–25 mm Hg) (22). This was followed by 5 min of room air ventilation, and then by 7 min of airway occlusion, resulting in circulatory arrest by 5–7 min. It was found necessary to briefly reoxygenate before asphyxia to achieve a high rate of successful cardiac resuscitation. Cardiopulmonary resuscitation (CPR) consisted of mechanical ventilation with 100% O2, manual chest compressions, and i.v. injections of epinephrine (0.1 mg/kg) and sodium bicarbonate (1 mmol/kg), which were repeated if needed until return of spontaneous circulation (ROSC), defined as MABP ≥ 8 kPa (60 mm Hg). Defibrillation (2–5 J/kg) was performed if ventricular fibrillation occurred. After ROSC, inspired O2 concentration was reduced to ∼30% to maintain arterial O2 saturation. Two hours after resuscitation, animals were placed on continuous i.v. infusions of fentanyl (15–40 μg/kg/h) and pancuronium (0.3–0.6 mg/kg/h) for the duration of mechanical ventilation. The dose of fentanyl was increased as needed for a rise in heart rate or MABP. During the first day of recovery, animals received a continuous i.v. infusion of 5% dextrose in 0.45% saline at a rate of 10–15 mL/h. During intubation, routine care included repositioning every 2 h, endotracheal suctioning every 4 h, ocular lubrication, and a suprapubic bladder tap if needed.
In hypothermic groups, cooling commenced at 5 min after ROSC by applying cooling blankets (10°C) underneath the supine piglet, and applying ice packs around the head and torso. When rectal temperature reached 36°C, the ice packs were removed from the head. When rectal temperature reached 35°C, the ice was removed from the torso. Target rectal temperature of 34°C was maintained by adjusting the temperature of the circulating water blanket. Twenty-four-hour survivors did not undergo rewarming or extubation. Eleven-day survivors were rewarmed beginning at 20 h after ROSC at a rate of 1°C/h over 4 h by adjusting the temperature of the cooling blanket until the rectal temperature reached normothermia (38.5–39°C). Fentanyl was decreased by 5–10 μg/kg/h and pancuronium was decreased by 0.1 mg/kg/h (∼25% decrease per hour). In normothermic animals, rectal temperature was maintained near a target temperature range of 38.5–39°C with a warming blanket and overhead heating lamps during the first 24 h of recovery. Normothermic piglets were treated in the same manner as the hypothermic piglets with regard to sedation, neuromuscular paralysis, ventilation and postoperative care.
Eleven-day survivors were extubated after recovery of ventilatory effort and were moved to a padded cage where they were observed, cleaned, fed and mobilized at 2- to 4-h intervals during the 2nd day and evening of recovery. Monitoring continued through the night if animals were unable to feed. They drank reconstituted swine milk ad libitum, or were syringe fed milk, supplemented with i.v. fluids until taking feedings well by mouth (at least 30 mL every 3 h). After adequate recovery, animals ambulated and fed independently (∼1100 mL of milk per 24 h), and were observed frequently, remaining in a pigpen during the day, and co-crated with another pig at night. On postoperative day 4, weaning pellets of dry food were introduced and supplemented with piglet formula. Cephalothin (100 mg i.v.) was given as a prophylactic antibiotic once daily for 5 d.
Experimental design.
In the first experiment, neuropathologic damage was evaluated in two groups of 24-h survivors. Animals were randomly assigned at the start of surgery to normothermic (n = 5) or hypothermic (n = 6) groups. In the second experiment, recovery of EEG, neurobehavioral deficits and neuropathologic damage were evaluated in four groups of 11-d survivors: HI-normothermic (n = 5), HI-hypothermic (n = 5), sham-normothermic (n = 3), sham-hypothermic (n = 3). Surgical sham animals underwent the same anesthesia, surgery, and 24 h of sedation, muscle paralysis and temperature manipulations as asphyxiated animals. An additional group of naïve-uninjured animals (n = 4) did not undergo surgery or anesthesia, and served as a control group for developmental changes while being cared for in the laboratory for 11 d.
Electroencephalogram.
Three bipolar channels of EEG and one channel of ECG were recorded at a gain of 7 μV/mm with low- and high-pass filters set to 0.3 and 35 Hz, respectively, using gold cup electrodes and a Grass model 10–20 EEG machine, converted and stored in digital form (Grass Polyview). EEG was recorded continuously during HI and the first 4 h of recovery. Subsequently 30-min epochs were recorded every 4 h until the time of extubation. After extubation, 30-min epochs were recorded at 36 h and 48 h of ROSC. Recordings were analyzed visually by an investigator (RNI) blinded to treatment group.
Neurobehavioral assessment.
Neurobehavioral assessment was performed by an investigator (RNI) blinded to treatment group, using a scoring system which assigns points for deficits in consciousness, brain stem reflexes, motor and sensory function and common behaviors (Table 1). The scoring system has been used previously in piglets (22) and was adapted from that used by others in dogs (23), cats (24), and piglets (25) with some modifications of the weighting of the scores to emphasize more complex behavior and functioning of higher brain centers. Evaluation was performed at baseline before surgery and during recovery at 24-h intervals until d 5. In addition, the “time to feed independently” and weight gain were assessed.
Histologic assessment.
For perfusion, piglets were deeply anesthetized with sodium pentobarbital, 30 mg/kg i.v. in 24-h survivors, or 65 mg/kg intraperitoneally in 11-d survivors, followed by injection of sodium nitrite and heparin. Animals were then exsanguinated by intra-aortic perfusion with ice-cold PBS until venous return was clear (∼10 min). Then the descending aorta was clamped and animals were perfused intra-aortically for 20 min with ice-cold 4% paraformaldehyde prepared in PBS (pH 7.4). Brains were removed and bisected mid-sagittally, and each hemisphere cut into 1 cm coronal blocks. A 3- to 4-mm thick slab was taken from the left forebrain at a mid-striatal level and was processed for paraffin histology. Neuronal damage in this mid-striatal sample was quantified in 10-μm sections stained with hematoxylin and eosin (H&E). Acute neuronal degeneration in 24-h survivors was measured by examination under oil immersion and counting the percent of striatal neurons showing ischemic cytopathology in six random, nonoverlapping, 1000× fields (22). For each animal, an average value of the six fields was obtained on one section where previous examination of serial sections indicated maximum damage. Morphologic criteria for acute ischemic neurodegeneration included cytoplasmic eosinophilia and vacuolation, and nuclear pyknosis. In the 11-d survivors, neuronal damage, defined as a decrease in principal striatal neuron density, was measured by counting viable neuronal cell profiles in H&E sections in 10 nonoverlapping fields in the caudate and the putamen under oil immersion at 1000× magnification with the use of a microscopic grid. An average value from the 10 fields was obtained on one section for each animal. Profile counting was performed in a manner blinded as to treatment group.
Statistical analysis.
All data are presented as mean ± SD. The level of statistical significance was set at p < 0.05. For the 24-h survivors, comparisons of physiologic data and the percent of ischemic neurons were made between the normothermic and hypothermic asphyxic groups using an independent t test. For 11-d survivors, comparisons of physiologic data, neurobehavioral deficit scores, and density of viable neurons were made among all groups using one-way ANOVA (or a t test when comparing only the two HI groups during the asphyxic period). When the F-value of the ANOVA was significant, the Newman-Keuls multiple range test was used to identify differences between groups. The time for EEG recovery was compared between normothermic and hypothermic groups by t test.
RESULTS
Correlation of brain and rectal temperature.
Simultaneous measurements of brain and rectal temperature in four piglets correlated well (r = 0.96) with very little lag time between brain and rectal temperature during the onset of cooling. During steady state cooling, the mean difference between rectal and brain temperature for four piglets averaged 0.2 ± 0.2°C. Remaining piglets were studied without a brain temperature probe to avoid the potential confounding influence of damage from probe insertion.
24-Hour Survival
Physiologic results.
Resuscitation was successful in 85% of animals (11 of 14) in the 24-h recovery experiment. The normothermic and hypothermic groups had similar arterial O2 desaturation, arterial hypertension, and tachycardia during the 30-min period of hypoxia (Table 2) and similar recovery during the 5-min period of room air ventilation, which was interposed to improve cardiac resuscitation. During the 7-min period of asphyxia, both groups exhibited similar degrees of hypoxia, hypercapnia, hypotension, and bradycardia. After resuscitation, target temperature was rapidly achieved in the hypothermic group, reaching a mean of 34.1 ± 0.5°C by 30 min after ROSC. Mean temperature during the first 24 h after ROSC was 39.0 ± 0.1°C in normothermic animals and 33.8 ± 0.1°C in hypothermic animals. During recovery there were no differences between groups in arterial blood gas values, Hb, glucose, or MABP.
Histology.
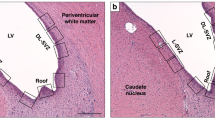
Nonischemic, hypothermic piglets were evaluated to determine whether hypothermia has damaging effects on the newborn piglet striatum (Fig. 1). Striatal histology was normal in nonischemic, hypothermic piglets (Fig. 1B) and was identical to sham normothermic animals (Fig. 1A). The majority of normothermic HI animals (4 of 5) at 24 h had severe ischemic cytopathology in putamen, characterized by extensive ischemic changes in principal neurons with prominent nuclear pyknosis and the presence of many small cell nuclei (Fig. 1C). However, in all hypothermic HI animals (6 of 6), the principal striatal neurons were preserved with minimal evidence of neuronal degeneration and gliosis as revealed by the absence of small cell nuclei or neuronal nuclear pyknosis (Fig. 1D). There were minor vacuolar changes in the neuropil of the putamen in 2 of 6 hypothermic animals characterized by the formation of perineuronal and perivascular spaces. The percentage of neurons in putamen with ischemic cytopathology at 24 h of hypothermic recovery without rewarming was markedly less than at 24 h of normothermic recovery (Fig. 2).

H&E stained sections showing panoramic views of piglet striatum. (A) normothermic piglet 11 d after sham surgery. (B) hypothermic piglet 11 d after sham surgery. (C) normothermic, hypoxic-ischemic (HI) piglet (higher magnification) at 24 h with active neuronal degeneration as indicated by the neuronal shrinkage and pyknotic nuclei (compare with 1D at same magnification). (D) hypothermic-HI piglet (higher magnification) with preserved striatal histology at 24 h. (E) normothermic-HI piglet with profound loss of neurons at 11 d; hypercellularity resulting from inflammatory changes and many small nuclei of nonneuronal cells are present. (F) hypothermic-HI piglet at 11 d with normal striatal histology, not different from A or B. Scale bars = 100 μm (A, same for B, E, and F), 50 μm (C, same for D).

(A) Percentage of neuronal death at 24 h of recovery in putamen after asphyxic cardiac arrest in normothermic (normo) pigs (n = 5) and hypothermic (hypo) pigs (n = 6). (B) Density of viable neurons in putamen at 11 d of recovery after hypoxia-ischemia (HI) in normothermic (n = 5) and hypothermic (n = 6) groups, after sham surgery in normothermic (n = 3) and hypothermic (n = 3) groups, and in a naïve group (n = 4). Values are means ± SD. *p < 0.001 from all other groups.
11-Day Survival
Physiologic results.
Resuscitation was successful in 80% (12 of 15) of animals in the 11-d survivor cohort. Two animals died after resuscitation (one normothermic, one hypothermic) due to cardiopulmonary complications during the first 48 h. Results are reported for five normothermic and five hypothermic piglets that survived 11 d. There were no deaths among sham animals.
There were no differences between normothermic and hypothermic HI groups in PaO2, MABP or heart rate during hypoxia or asphyxia (Fig. 3). Heart rate and MABP returned to baseline values within one hour of ROSC in both groups and remained stable at levels equivalent to those in the corresponding sham groups throughout the intensive care monitoring period and after extubation. Normothermic and hypothermic groups had similar declines in O2 saturation during hypoxia and asphyxia, and similar hypercarbia and acidemia during asphyxia (Table 3). After resuscitation, O2 saturation, pH and PaCO2 recovered to baseline values within 60 min after ROSC in both groups and were similar to those in the sham groups. Hb and glucose concentrations were not different among groups.

Partial pressure of arterial oxygen (Pao2), mean arterial blood pressure (MABP), and heart rate for hypoxic-ischemic (HI) and sham piglets treated with hypothermia or normothermia. Values are means ± SD measured at baseline, at 5, 15, and 27 min of hypoxia, at 5 min of room air (RA) ventilation, during 7 min of asphyxia (Asph), and during the first 26 h of recovery in 11-d survivors. The last measurements were after endotracheal extubation. Error bars in the sham groups were omitted for clarity, but were of similar magnitude as those of the HI groups.
The duration of CPR was 1.5 ± 0.3 min in the normothermic group and 1.4 ± 0.5 min in the hypothermic group. The duration for which MABP was <3.3 kPa (25 mm Hg) at the end of asphyxia and during CPR was not significantly different between normothermic (3.7 ± 1.0 min) and hypothermic (2.3 ± 1.4 min) groups. Normothermic and hypothermic animals received similar doses of epinephrine (0.14 ± 0.09 versus 0.10 ± 0.06 mg/kg, respectively) and sodium bicarbonate (1.40 ± 0.89 versus 1.30 ± 0.87 mmol/kg, respectively). Cardiac defibrillation was required in 2 normothermic and 1 hypothermic animals.
Rectal temperature was rapidly decreased to 34.5 ± 0.5°C by 30 min after ROSC in the hypothermic group and was well maintained during the subsequent 20-h period (Fig. 4). Mean temperature during the first 20 h after ROSC was 38.8 ± 0.2°C in the normothermic-HI group, 38.8 ± 0.1°C in the normothermic sham group, 34.1 ± 0.2°C in the hypothermic-HI group, and 34.2 ± 0.2°C in the hypothermic sham group. During rewarming target temperatures were achieved at the expected rate of approximately 1°C/h (Fig. 4). There were no differences among groups in rectal temperature during the remaining 10 d of recovery. Normothermic-HI and hypothermic-HI animals did not differ from each other with respect to cumulative doses of fentanyl (343 ± 25 μg/kg and 316 ± 26 μg/kg, respectively) or of pancuronium (5.8 ± 3.3 mg/kg, and 5.9 ± 0.9 mg/kg, respectively) between 2 and 24 h after ROSC. Sham normothermic and hypothermic animals received similar cumulative doses of fentanyl (358 ± 90 μg/kg and 294 ± 17 μg/kg, respectively) and pancuronium (6.9 ± 1.5 mg/kg and 5.7 ± 0.6 mg/kg, respectively).

Rectal temperature in hypoxic-ischemic (HI) and sham piglets undergoing whole body mild hypothermia or normothermia for 24 h followed by an additional 10-d recovery. Error bars in sham groups were omitted for clarity.
EEG recovery.
The time of appearance of successive grades of background EEG recovery, as depicted in Fig. 5, was assessed in each HI piglet. This sequence began with a continuation of the isoelectric tracing (Stage 0), which first appeared during asphyxia. The earliest electrographic recovery (Stage 1) was characterized by very brief (< 2 s duration) primitive slow wave complexes separated by long intervals of 4–8 min of an isoelectric or profoundly suppressed (<5 μV) tracing (Fig. 5A). Stage 1 recovery appeared at comparable times after ROSC in both normothermic (19 ± 9 min) and hypothermic (19 ± 10 min) animals. Over the next 8 h the EEG gradually evolved from a discontinuous (Stage 1–3 in Fig. 5) to a continuous pattern (Stage 4–6 in Fig. 5). The time to earliest appearance of a fully continuous tracing, depicted as Stage 4, was not different between normothermic (4.1 ± 2.9 h) and hypothermic (5.2 ± 2.5 h) groups. Spontaneous sleep/wake shifts (Stage 6) appeared after discontinuing the fentanyl infusion in both groups. Electrographic seizures were not observed in any of the recordings of any piglets in the hypothermic or normothermic groups.

Grading scheme for EEG recovery after resuscitation. (A) Stage 1: early discontinuous: isoelectric >50% of the epoch, alternating with low voltage (5–10 μV) slow activity (1–5 Hz). (B) Stage 2: intermediate discontinuous: isoelectric 10% to 50% of the epoch, alternating with low voltage (5–10 μV) slow activity (1–5 Hz). (C) Stage 3: late discontinuous: regular brief intervals (up to 10% of epoch) of isoelectric record alternating with disorganized bursts of medium-to-high voltage (20–70 μV) mostly slow activity. (D) Stage 4: early continuous: mostly low voltage (5–10 μV) slow and medium frequencies (1–8 Hz), no sleep-wake shifts. (E) Stage 5: intermediate continuous: mix of voltages (10–50 μV), greater mix of frequencies (3–16 Hz), no sleep-wake shifts. (F) Stage 6: fully continuous: Similar to 5, with addition of spontaneous sleep-wake shifts, depicted as /*./ (G) Time for EEG to recover to each of 6 stages for hypoxic-ischemic normothermic and hypothermic 11-d survivor groups (means ± SD). There were no differences in recovery times between groups.
Weight gain and neurobehavioral recovery.
Weight gain was similar among groups. Body weight averaged 3.7 ± 0.2 kg at baseline and increased to 5.9 ± 0.3 kg in the normothermic-HI group, 5.7 ± 0.6 kg in the hypothermic-HI group, 6.2 ± 1.0 kg in the normothermic-sham group, 6.3 ± 0.9 kg in the hypothermic-sham group, and 6.4 ± 0.3 kg in the naïve group 11 d later. Piglet neurobehavioral scores (NBS) were assessed at baseline and at 26, 48, 72, and 96 h after ROSC (Fig. 6). Sham animals had moderate deficits as they recovered from anesthesia. Hypothermic HI animals had improved early neurobehavioral recovery, with lower deficit scores than normothermic animals at 26 h (34 ± 14.7 versus 57 ± 14.2) and at 48 h (2 ± 2.7 versus 8 ± 3.3). Early recovery of NBS in the hypothermic HI group was similar to the sham cohorts. By day 3 of ROSC, all animals had similar NBS, without measurable deficits. Compared with hypothermic-HI animals, normothermic-HI animals took significantly longer to independently drink milk by mouth out of a bowl and required additional oral syringe feedings and i.v. fluids. Hypothermic HI animals were not different from the sham groups in their recovery of independent feeding (Fig. 6).

(Top) neurobehavioral score during the first 4 d of recovery in the 11-d survivor piglets undergoing hypoxic-ischemic (HI) with normothermic and hypothermic recoveries and sham surgery with normothermic and hypothermic recoveries from anesthesia. (Bottom) time to independently feed after stopping the infusion of fentanyl and pancuronium in the 11-d survivor groups.
Histology.
Hypothermia produced complete neuroprotection in the vulnerable striatum at 11 d of survival (Fig. 1). Neuronal density in putamen was 287 ± 22 neurons/mm2 in hypothermic-HI animals, which was not different from hypothermic-sham (288 ± 46 neurons/mm2), normothermic-sham (271 ± 21 neurons/mm2), or naïve (307 ± 51 neurons/mm2) groups (Fig. 2). However, severe ischemic neuronal damage in putamen occurred in the normothermic-HI animals (81 ± 40 neurons/mm2). In contrast to putamen, no differences in neuronal densities were detected in caudate nucleus among groups, including the asphyxiated normothermic group (data not shown). Thus, striatal injury in the normothermic group was restricted to putamen. In addition, the cytoarchitecture in sensorimotor cortex appeared normal without consistent evidence of ischemic cytopathology in any group (data not shown).
DISCUSSION
This study established that 24 h of whole body hypothermia at 34°C after asphyxic cardiac arrest prevented striatal neuronal cell death at 24 h, and completely sustained striatal neuronal cell viability at 11 d of recovery after rewarming without deleterious side effects, in a model of neonatal hypoxic-ischemic encephalopathy using piglets.
The use of hypothermia in newborn animals has produced mixed results, related in part to the experimental model and the timing of hypothermic induction and duration (12, 26–28). Trescher et al.(13) studied hypothermia in the 7-d rat pup model of focal HI, and found that 3 h of postischemic hypothermia delayed the maturation of injury but did not improve the final result. In contrast, Bona et al.(14) subjected 7-d old rat pups to 6 h of postischemic hypothermia and demonstrated sustained amelioration of neuropathologic damage and neurologic deficits in 6-wk survivors. Thus, the 3-h duration of hypothermia used by Trescher et al. may have been insufficient. Our study differs from studies of focal HI in postnatal rats not only because we used a global asphyxic insult in piglets, but also because hypothermia was applied for a full 24 h after the injury. Our results are consistent with studies in adult rats indicating that prolonged hypothermia after global cerebral ischemia can provide more long-lasting protection (29, 30) than brief hypothermia (31), possibly by reducing delayed hyperthermia (32).
In other work using systemic hypoxia in piglets, Haaland and colleagues (15) found that 3 h of mild hypothermia reduced neuropathologic damage at 3 d of survival in animals subjected to a modest insult, but not in animals subjected to a more severe insult. Unexpectedly, there was no benefit when the duration of hypothermia was extended to 24 h in the same model in piglets (16). These experiments differed from our protocol in that the piglets were not paralyzed or sedated during hypothermia. The authors suggest that the effects of cold stress may have negated any benefit. The present study avoids some of the potential negative effects of cold stress by maintaining animals paralyzed and sedated through the period of cooling. Furthermore, the period of survival was extended to 11 d, whereas previous studies in piglets subjected to comparable insults have evaluated neuropathologic outcome no later than 4 d of survival. Therefore, the current study is the first to demonstrate that neuropathological improvement can be sustained over an extended period with postischemic hypothermia after systemic hypoxia and asphyxia in newborns of a large animal species.
In the first experiment without rewarming, 24 h of postasphyxic whole body hypothermia was found to provide a 5-fold decrease in the percentage of cells undergoing ischemic neurodegeneration in piglet striatum. In the second experiment with 11 d of recovery, hypothermic animals maintained significantly more viable striatal neurons than normothermic animals and had complete preservation of striatal neuronal cell counts compared with sham and naïve groups. Therefore, hypothermia conferred complete neuroprotection against acute necrosis that normally evolves rapidly over the first 24–48 h of recovery in the vulnerable striatum (18, 21), and the neuroprotection was sustained 10 d after rewarming. However, by preventing necrosis, it is possible that hypothermia permits a delayed apoptosis to emerge at a later time, but this outcome was not the case. Thus rewarming did not appear to induce delayed apoptosis beyond that normally occurring at this stage of development.
The functional significance of our neuropathology findings is, in part, borne out by the beneficial effect of hypothermia on the speed of behavioral recovery. The hypothermic animals had better NBS during the first 48 h after ROSC, in addition to significantly shorter ‘time-to-feed independently' after extubation. Moreover, the complete neuroprotection seen in neuropathologic outcome parallels the degree of protection in behavioral measures. Hypothermic animals were similar to sham animals with regard to rapid recovery of NBS and “time to independent feeding.” However, the significance of the lag in behavioral improvement in the normothermic group is unclear in that both asphyxic groups had normal NBS by 72 h of ROSC. There are several possible explanations for the eventual recovery of NBS to baseline. First, the NBS measure may have limited sensitivity for detecting subtle basal ganglia derived behaviors. Alternatively, neuronal plasticity responses may have compensated, resulting in full recovery of measurable function despite persistence of structural abnormalities seen pathologically. Functional recovery may have been enhanced by the practice of housing injured and uninjured animals together during recovery, which may act to provide social enrichment, to decrease stress, and to permit learning by cues from other piglets.
Early EEG recovery was similar in normothermic and hypothermic animals. Of interest was the fact that early EEG recovery in this protocol was markedly prolonged (Stage 4 at 4.1 ± 2.9 h) compared with previous normothermic cohorts (2.2 ± 1.0 h) subjected to the same HI protocol but recovered without sedation or paralysis (33). In the current protocol, animals received fentanyl and pancuronium starting at 2 h of ROSC, whereas previous cohorts began to awaken from anesthesia, breathe and move spontaneously soon after this time. Moreover, latency to recovery of EEG has correlated with severity of neuropathologic injury in cortex in previous cohorts. Because the prolonged EEG recovery in both the normothermic and hypothermic groups was associated with no cortical injury, that correlation no longer holds in the present study. The only change in the protocol during the first 2–4 h of recovery common to both normothermic and hypothermic animals was the use of sedation and paralysis. During recovery from anesthesia, animals ordinarily experience steadily increasing inputs via all ascending sensory modalities, as motor activity increases and analgesic drug effects wane. Sensory inputs via tactile, pain, and proprioceptive systems would be profoundly depressed in animals maintained on deeply sedating doses of narcotic and pharmacologically paralyzed. A more prolonged EEG recovery in sedated paralyzed animals may indicate that neuronal activity in primary somato-sensory pathways modifies cortical recovery during the immediate postischemic period. In the case of unsedated, unparalyzed animals, delays in early EEG recovery could reflect a greater degree cortical injury. In contrast, in the case of sedated paralyzed animals, delays in early EEG recovery may predominantly reflect the effects of drugs.
Many factors may have influenced the profound neuroprotection observed in our studies. First, hypothermia was initiated immediately after ROSC. Second, the animals remained intubated, sedated and paralyzed during the period of hypothermia to minimize the cold stress. Third, hypothermic animals were slowly rewarmed over 4 h without rebound hyperthermia, which could abate any of the protective effects from hypothermia. Moreover, temperature was rigorously controlled over a narrow range in both groups. Spontaneous hyperthermia was thereby completely prevented during the first 24 h of recovery.
It is also noteworthy that normothermic piglets in the present study had less neuropathologic evidence of injury in cerebral cortex compared with previous cohorts (17) in which cortical cytopathology evolved between 24–48 h. The physiologic parameters during hypoxia and asphyxia were similar to previous cohorts, suggesting that animals in the current experiments experienced a similar HI insult. Perhaps maintaining animals truly normothermic for the 24 h after HI, thereby preventing any degree of hyperthermia, offered some neuroprotection from the evolving cortical damage. It is also possible that sedation and paralysis may directly influence the evolution of neocortical damage through mechanisms that are distinct from their effects on striatal damage. For example, decreased afferent activity may reduce metabolic demand in sensorimotor cortex where cytochrome oxidase activity is selectively enriched at this stage of development (17). Lastly, piglets were previously housed singly in individual cages, whereas they were presently housed in groups of 2–3 after regaining consciousness. The social interaction of animals that are normally together in litters may have acted to reduce the postischemic stress and agitation that could have an impact on delayed cortical injury.
The variability of hypothermic neuroprotection in different injury paradigms and different species of newborns indicates that caution is warranted in extrapolating experimental findings to the clinical setting. Our findings suggest several factors should be born in mind when interpreting the results of hypothermia in human clinical trials. First, sedatives and paralytics are administered at the discretion of the practitioner in clinical trials. It is difficult to assess the level of cold stress in neonates, who rarely shiver in response to cold. Therefore, variability or omissions in the use of sedating and analgesic drugs during trials of hypothermia in newborns may alter the outcome. On the other hand, the physiologic response to cold stress induced by selective head cooling may be less than with whole body hypothermia used in the present study. Second, the degree of injury in the present study was less severe than that of some other studies in piglets (34) or in rat pups with focal infarction. Efficacy of hypothermia is likely to be diminished with more severe insults and with delayed treatment. The clinical trials have strict inclusion criteria for the more severely injured infants in addition to a window to entry of up to 6 h after birth. Restricting enrollment to the most severely injured patients may skew the study population to those least likely to benefit. Striatal injury can rapidly evolve over the first few hours of recovery (21). Neonatal clinical trials which commence cooling as late as 6 h after HI may fail to show a benefit, or may require lower temperatures or more prolonged periods of cooling to show a benefit. The implementation of hypothermia immediately in the delivery room, as part of the resuscitation protocol, to mildly or moderately affected infants, as well as the most severely affected infants, might add significantly to the neuroprotective potential of this intervention. Our data strongly support the view that hypothermia is a promising therapy divorced from major harmful consequences.
Abbreviations
- CPR:
-
cardiopulmonary resuscitation
- H&E:
-
hematoxylin and eosin
- HI:
-
hypoxia-ischemia
- MABP:
-
mean arterial blood pressure
- NBS:
-
neurobehavioral scores
- PaCO2:
-
arterial Pco2
- Pao2:
-
arterial Po2
- ROSC:
-
return of spontaneous circulation
References
Nelson KB, Ellenberg JH 1981 Apgar scores as predictors of chronic neurologic disability. Pediatrics 68: 36–44
Ellenberg JH, Nelson KB 1988 Cluster of perinatal events identifying infants at high risk for death or disability. J Pediatr 113: 546–552
Vannucci RC, Perlman JM 1997 Interventions for perinatal hypoxic-ischemic encephalopathy. Pediatrics 100: 1004–1014
Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K 2002 Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med 346: 557–563
The Hypothermia After Cardiac Arrest Study Group 2002 Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med 346: 549–556
Battin MR, Dezoete JA, Gunn TR, Gluckman PD, Gunn AJ 2001 Neurodevelopmental outcome of infants treated with head cooling and mild hypothermia after perinatal asphyxia. Pediatrics 107: 480–484
Gunn AJ, Gluckman PD, Gunn TR 1998 Selective head cooling in newborn infants after perinatal asphyxia: a safety study. Pediatrics 102: 885–892
Sirimanne ES, Blumberg RM, Bossano D, Gunning M, Edwards AD, Gluckman PD, Williams CE 1996 The effect of prolonged modification of cerebral temperature on outcome after hypoxic-ischemic brain injury in the infant rat. Pediatr Res 39: 591–597
Thoresen M, Bagenholm R, Loberg EM, Apricena F, Kjellmer I 1996 Posthypoxic cooling of neonatal rats provides protection against brain injury. Arch Dis Child Fetal Neonatal Ed 74: F3–F9
Gunn AJ, Gunn TR, de Haan HH, Williams CE, Gluckman PD 1997 Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J Clin Invest 99: 248–256
Edwards AD, Yue X, Squier MV, Thoresen M, Cady EB, Penrice J, Cooper CE, Wyatt JS, Reynolds EO, Mehmet H 1995 Specific inhibition of apoptosis after cerebral hypoxia-ischaemia by moderate post-insult hypothermia. Biochem Biophys Res Commun 217: 1193–1199
Laptook AR, Corbett RJ, Sterett R, Burns DK, Garcia D, Tollefsbol G 1997 Modest hypothermia provides partial neuroprotection when used for immediate resuscitation after brain ischemia. Pediatr Res 42: 17–23
Trescher WH, Ishiwa S, Johnston MV 1997 Brief post-hypoxic-ischemic hypothermia markedly delays neonatal brain injury. Brain Dev 19: 326–338
Bona E, Hagberg H, Loberg EM, Bagenholm R, Thoresen M 1998 Protective effects of moderate hypothermia after neonatal hypoxia- ischemia: short- and long-term outcome. Pediatr Res 43: 738–745
Haaland K, Loberg EM, Steen PA, Thoresen M 1997 Posthypoxic hypothermia in newborn piglets. Pediatr Res 41: 505–512
Thoresen M, Satas S, Loberg EM, Whitelaw A, Acolet D, Lindgren C, Penrice J, Robertson N, Haug E, Steen PA 2001 Twenty-four hours of mild hypothermia in unsedated newborn pigs starting after a severe global hypoxic-ischemic insult is not neuroprotective. Pediatr Res 50: 405–411
Martin LJ, Brambrink A, Koehler RC, Traystman RJ 1997 Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J Comp Neurol 377: 262–285
Martin LJ, Brambrink AM, Lehmann C, Portera-Cailliau C, Koehler RC, Rothstein J, Traystman RJ 1997 Hypoxia-ischemia causes abnormalities in glutamate transporters and death of astroglia and neurons in newborn striatum. Ann Neurol 42: 335–348
Barkovich AJ, Westmark K, Partridge C, Sola A, Ferriero DM 1995 Perinatal asphyxia: MR findings in the first 10 days. Am J Neuroradiol 16: 427–438
Low JA, Robertson DM, Simpson LL 1989 Temporal relationships of neuropathologic conditions caused by perinatal asphyxia. Am J Obstet Gynecol 160: 608–614
Martin LJ, Brambrink AM, Price AC, Kaiser A, Agnew DM, Ichord RN, Traystman RJ 2000 Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol Dis 7: 169–191
Brambrink AM, Martin LJ, Hanley DF, Becker KJ, Koehler RC, Traystman RJ 1999 Effects of the AMPA receptor antagonist NBQX on outcome of newborn pigs after asphyxic cardiac arrest. J Cereb Blood Flow Metab 19: 927–938
Bircher N, Safar P 1985 Cerebral preservation during cardiopulmonary resuscitation. Crit Care Med 13: 185–190
Fleischer JE, Tateishi A, Drummond JC, Scheller MS, Grafe MR, Zornow MH, Shearman GT, Shapiro HM 1989 MK-801, an excitatory amino acid antagonist, does not improve neurologic outcome following cardiac arrest in cats. J Cereb Blood Flow Metab 9: 795–804
LeBlanc MH, Vig V, Smith B, Parker CC, Evans OB, Smith EE 1991 MK-801 does not protect against hypoxic-ischemic brain injury in piglets. Stroke 22: 1270–1275
Gunn AJ, Gunn TR, Gunning MI, Williams CE, Gluckman PD 1998 Neuroprotection with prolonged head cooling started before postischemic seizures in fetal sheep. Pediatrics 102: 1098–1106
Gunn AJ, Bennet L, Gunning MI, Gluckman PD, Gunn TR 1999 Cerebral hypothermia is not neuroprotective when started after postischemic seizures in fetal sheep. Pediatr Res 46: 274–280
Laptook AR, Corbett RJ, Burns DK, Sterett R 1999 A limited interval of delayed modest hypothermia for ischemic brain resuscitation is not beneficial in neonatal swine. Pediatr Res 46: 383–389
Colbourne F, Li H, Buchan AM 1999 Indefatigable CA1 sector neuroprotection with mild hypothermia induced 6 hours after severe forebrain ischemia in rats. J Cereb Blood Flow Metab 19: 742–749
Hicks SD, DeFranco DB, Callaway CW 2000 Hypothermia during reperfusion after asphyxial cardiac arrest improves functional recovery and selectively alters stress-induced protein expression. J Cereb Blood Flow Metab 20: 520–530
Dietrich WD, Busto R, Alonso O, Globus MY, Ginsberg MD 1993 Intraischemic but not postischemic brain hypothermia protects chronically following global forebrain ischemia in rats. J Cereb Blood Flow Metab 13: 541–549
Coimbra C, Drake M, Boris-Moller F, Wieloch T 1996 Long-lasting neuroprotective effect of postischemic hypothermia and treatment with an anti-inflammatory/antipyretic drug. Evidence for chronic encephalopathic processes following ischemia. Stroke 27: 1578–1585
Ichord RN, Agnew DM, Koehler RC, Traystman RJ 2001 Sedation and paralysis after cardiac arrest in piglets delays EEG recovery and is associated with improved short-term neurologic outcome. Abstr Soc Neurosci 27: 3337
Thoresen M, Haaland K, Loberg EM, Whitelaw A, Apricena F, Hanko E, Steen PA 1996 A piglet survival model of posthypoxic encephalopathy. Pediatr Res 40: 738–748
Acknowledgements
The authors are grateful to Michael Giuffre and Erin Simmers for their expert technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported by National Institutes of Health grants NS-20020, F31-07525, and NS-01805
Rights and permissions
About this article
Cite this article
Agnew, D., Koehler, R., Guerguerian, AM. et al. Hypothermia for 24 Hours After Asphyxic Cardiac Arrest in Piglets Provides Striatal Neuroprotection That Is Sustained 10 Days After Rewarming. Pediatr Res 54, 253–262 (2003). https://doi.org/10.1203/01.PDR.0000072783.22373.FF
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/01.PDR.0000072783.22373.FF
This article is cited by
-
Neurologic effects of short-term treatment with a soluble epoxide hydrolase inhibitor after cardiac arrest in pediatric swine
BMC Neuroscience (2020)
-
Rewarming from Therapeutic Hypothermia Induces Cortical Neuron Apoptosis in a Swine Model of Neonatal Hypoxic–Ischemic Encephalopathy
Journal of Cerebral Blood Flow & Metabolism (2015)
-
Adenosine A2A Receptor Contributes to Ischemic Brain Damage in Newborn Piglet
Journal of Cerebral Blood Flow & Metabolism (2013)
-
Comparison of Three Hypothermic Target Temperatures for the Treatment of Hypoxic Ischemia: mRNA Level Responses of Eight Genes in the Piglet Brain
Translational Stroke Research (2013)
-
Dopamine Receptor Modulation of Hypoxic—Ischemic Neuronal Injury in Striatum of Newborn Piglets
Journal of Cerebral Blood Flow & Metabolism (2007)
