
Abstract
High levels of unconjugated bilirubin (UCB) can be neurotoxic. Nevertheless, the mechanism of UCB interaction with neural cells is still unknown. This study investigates whether cultured rat neurons and astrocytes respond differently to UCB exposure. UCB toxicity was evaluated by lactate dehydrogenase release, induction of apoptosis, cytoskeleton degeneration, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction, and glutamate uptake. Primary cultures of rat brain astrocytes and neurons were incubated at 37°C with 85.5 μM UCB plus 28.5 μM albumin for 4 h. In assays of glutamate uptake, cells were exposed to 80–120 μM UCB plus 100 μM albumin for 15 min. The results showed that after incubation with 85.5 μM UCB, lactate dehydrogenase release was greater in neurons than in astrocytes (38%versus 14%, p < 0.05). Also, levels of apoptosis were markedly enhanced in neurons (29%versus 19%, p < 0.01). In accordance, neuronal cytoskeleton disassembly was evident during incubation with 85.5 μM UCB, whereas equivalent effects on astrocytes required as much as 171 μM. Conversely, inhibition of MTT metabolism and glutamate uptake by UCB was more pronounced in astrocytes than in neurons (74%versus 60%, p < 0.05 and 41% to 56%versus 25% to 33%, p < 0.05, respectively). In conclusion, the study demonstrates that astrocytes are more susceptible to inhibition of glutamate uptake and MTT reduction by UCB, whereas neurons are more sensitive to cell death by necrosis or apoptosis. These results suggest that UCB is toxic to both astrocytes and neurons, although through distinct pathways.
Similar content being viewed by others

Main
Neonatal jaundice is a very common predicament, usually requiring therapeutic intervention in early life. The significance of neonatal hyperbilirubinemia can vary from a minor nontoxic phenomenon to death by kernicterus (1–3). In contrast to kernicterus, where permanent lesions or death are likely to occur (4–6), bilirubin encephalopathy can result in transient or sometimes definitive effects of UCB toxicity, such as auditory and visual impairment (7–9). However, the mechanisms of bilirubin encephalopathy and kernicterus remain unclear.
Nerve cells are the main target for UCB cytotoxicity. In fact, several effects of UCB toxicity to neurons have been reported either in brain sections (10–12), cultured cell lines (13, 14), or isolated nerve terminals (15–18). Although neurons have been suggested as more susceptible than astrocytes (13), more recent reports describe compromised astrocyte function following UCB interaction (19–22). Evidence is also arising on the influence of astrocytes in neuronal function, metabolism, and survival (23–26). The role of astrocytes on UCB toxicity is further reinforced by electron microscopic and autoradiographic studies on experimental kernicterus, indicating that these cells represent the main transporters of UCB from blood to neurons and are also involved in the release of waste products from UCB damaged neurons (27, 28).
We have previously shown that UCB significantly reduces glutamate uptake in astrocytes (22) and induces apoptosis both in neurons and astrocytes (29). Nevertheless, comparative data on cell susceptibility to UCB toxicity are still scarce. In this study, we evaluate several toxic effects of UCB on cultured rat neurons and astrocytes to differentiate cell response and, therefore, better understand the underlying mechanism leading to UCB encephalopathy and/or kernicterus. The results show that neurons are more sensitive than astrocytes to necrotic or apoptotic cell death, and to cytoskeleton disruption by UCB. Conversely, astrocytes demonstrate a greater inhibition of both MTT metabolism and glutamate uptake.
METHODS
Materials.
Dulbecco's medium modified by Eagle (DMEM), Neurobasal medium, B-27 Supplement 50×, Hanks' balanced salt solution (HBSS-1), Hanks' balanced salt solution without Ca2+ and Mg2+ (HBSS-2), gentamicin (50 mg/mL), and FCS, were purchased from Invitrogen (Carlsbad, CA, U.S.A.). Bilirubin, antibiotic antimycotic solution (100×), HSA fraction V, fatty acid free, MTT, rabbit antibodies against GFAP, tubulin and neurofilaments 200 kD, and goat antibody anti-rabbit labeled with FITC were from Sigma Chemical (St. Louis, MO, U.S.A.). 3H-glutamate (specific activity 49 Ci/mmol) was obtained from Amersham Life Science (Little Chalfont, Buckinghamshire, U.K.). Cell culture clusters (12-well) were from Corning Costar (Cambridge, MA, U.S.A.). All other chemicals were of analytical grade and were purchased from Merck (Darmstadt, Germany). Commercial bilirubin was further purified according to the method of McDonagh and Assisi (30) before use.
Cell cultures.
Astrocytes were isolated from 2-d-old Wistar rats as described previously (20), with minor modifications (22). Briefly, the rat brain was collected after decapitation in DMEM containing 11 mM sodium bicarbonate, 71 mM glucose, and 1% antibiotic and antimycotic solution. Meninges, blood vessels, and white matter were removed. The cortex was homogenized by mechanical fragmentation, and the cell suspension passed sequentially through steel screens of 230-, 104-, and 73.3-μm pore size. Cells were collected by centrifugation at 700 ×g for 10 min and resuspended in DMEM supplemented with 10% FCS. Finally, 2.0 × 105 cells/cm2 were plated on 12-well tissue culture plates and maintained at 37°C in a humidified atmosphere of 5% CO2. Culture medium was replaced at d 7 and 10, and the confluent cultures used at d 11. Neurons were isolated from fetuses of 17–18 d pregnant Wistar rats as previously described (31) with minor modifications. In short, pregnant rats were ether anesthetized and decapitated. The fetuses were collected in HBSS-1 and rapidly decapitated. After removal of meninges and white matter, the brain cortex was collected in HBSS-2 and mechanically fragmented. The cortex fragments were transferred to a 0.025% trypsin in HBSS-2 solution and incubated for 15 min at 37°C. Following trypsinization, cells were washed twice in HBSS-2 containing 10% FCS, and resuspended in Neurobasal medium supplemented with 0.5 mM l-glutamine, 25 μM l-glutamic acid, 2% B-27 Supplement, and 0.12 mg/mL gentamicin. Aliquots of 1 × 105 cells/cm2 were plated on 12-well tissue culture plates and maintained at 37°C in a humidified atmosphere of 5% CO2. Every 3 d, 0.5 mL of old medium was removed by aspiration and replaced by the same volume of fresh medium without glutamic acid. Cells were used after 8 d in culture. Our institutional animal care and use committee approved all animal procedures.
The use of fetal brains at 2–3 d before delivery increases the efficiency of neuronal cultures, because embryonic tissue has fewer cell connections than older tissue. The use of older brains makes the isolation of neurons more difficult because irreversible damage to cell contacts and to shared axons and dendrites is more likely to occur. Furthermore, contamination by glial cells, already observed in cultures from brain cortex of 1-d postnatal rats, is avoided by the use of fetal brains. Finally, preparation of astrocyte cultures from brain cortex of 2-d-old rats minimizes the presence of cortical neurons.
UCB treatment of astrocytes and neurons.
UCB was dissolved in 0.1 N NaOH to prepare an 8.6 mM stock solution. The stock solution was added to either DMEM (astrocytes) or Neurobasal medium (neurons), containing HSA, to obtain a final UCB concentration of 85.5 μM, at a UCB/HSA molar ratio of 3:1. Cultured cells were then exposed to UCB for 4 h, at 37°C, in the dark. In selected experiments, astrocytes were exposed to 171 μM UCB and 57 μM HSA. In addition, cells were also incubated for 15 min, at 37°C, with 80, 100, and 120 μM UCB, in the presence of 100 μM HSA, for the assessment of glutamate uptake. In all UCB incubations, the pH was restored to 7.4 by adding 0.1 M HCl. Although rarely found in jaundiced newborns, UCB to HSA molar ratio of 3 was used to achieve, in short incubation periods, significant interaction of UCB with the cell, mimicking the toxic conditions of a prolonged hyperbilirubinemia (6, 32). Also, we decided to use lower molar ratios to evaluate alterations in the glutamate uptake by neural cells to reproduce initial steps of UCB cytotoxicity and because previous studies in astrocytes evidenced a reduced uptake in these milder conditions (22).
Cell viability.
The assay used to evaluate cell viability was based on the release of LDH by nonviable cells. LDH was evaluated in the incubation medium of cells exposed to UCB using the Cytotoxicity Detection kit, LDH (Roche Molecular Biochemicals, Mannheim, Germany). All readings were corrected for the possible interference of UCB absorption, and in control experiments UCB was excluded. Results were expressed as percent of total LDH release, obtained by treating nonincubated cells with 0.5% Triton X-100 for 5 min.
Assessment of apoptosis.
Cells exposed to UCB were washed twice with PBS, pH 7.4, and fixed for 15 min, at room temperature, with freshly prepared 4% formaldehyde in PBS. Apoptosis was assessed by DNA fragmentation using the TUNEL assay (Roche Molecular Biochemicals). The specific technique uses terminal deoxynucleotidyl transferase to attach fluorescein-labeled dUTP to free 3′-OH DNA ends. Astrocyte permeabilization was achieved using 0.1% Triton X-100 in 0.1% sodium citrate, for 5 min at 4°C, while 5% Triton X-100 was needed to permeabilize neurons. The number of TUNEL-positive cells (i.e. red nuclei) were counted in at least five random microscopic fields (×400) for each sample and the mean values expressed as the percentage of apoptotic nuclei.
Immunocytochemistry.
Cells for immunostaining were fixed with 4% formaldehyde as described above. After three washings in PBS containing 15% sucrose and 0.01% sodium azide, cells pretreated with 0.1% Triton X-100 in PBS for 30 min at room temperature were placed in contact with rabbit diluted primary antiserum against either GFAP (astrocytes), neurofilaments 200 kD (neurons), or tubulin, and incubated overnight at 4°C. Cells were then washed twice in PBS and incubated with a FITC-labeled secondary goat anti-rabbit antibody for 1 h at room temperature. Fluorescence was examined using a Axioskop fluorescence microscope (Carl Zeiss GmbH, Jena, Germany).
MTT reduction.
A stock solution of MTT at 5 mg/mL in DMEM was prepared weekly, filter-sterilized, and stored at −20°C. Cellular reduction of MTT was measured in cells exposed to 85.5 μM, as described before (33), with modifications. After removing UCB, cells were incubated for 1 h at 37°C with 500 μL of MTT at 0.5 mg/mL, prepared by dilution of the stock solution with DMEM. After incubation, medium was discarded and MTT formazan crystals dissolved by addition of 1 mL isopropanol/HCl 0.04 M and gentle shaking in an orbital shaker for 15 min at room temperature. After centrifugation, absorbance values at 570 nm were determined in an Unicam UV2 spectrophotometer (Unicam Limited, UV2, Cambridge, UK). Results were expressed as percentage of control, which was considered as 100%.
Glutamate uptake.
Glutamate uptake was estimated in cells exposed for 15 min to 80, 100, and 120 μM UCB in the presence of 100 μM HSA by an adaptation of the method of Swanson et al.(34) as previously described (22). 3H-glutamate (25 nCi) and unlabeled glutamate, to a final concentration of 5 μM (neurons) or 50 μM (astrocytes), were added to each culture well and incubated for 7 min. At the end of the incubation period, medium was removed and glutamate uptake stopped by three washes with ice-cold PBS followed by immediate addition of 1 mL NaOH 1 M to promote cell lysis. Aliquots of each cell lysate were taken for protein estimation using the method of Lowry et al.(35), as well as for glutamate radioassay. Scintillation counts were performed in a Beckman LS 6000LL liquid scintillation spectrometer with internal quench correction (Beckman Instruments, Inc., Fullerton, CA, U.S.A.). Quench correction was further validated by using an aliquot of a cell lysate from a parallel incubation with UCB in the absence of labeled glutamate. Controls were performed without UCB. Results of glutamate uptake in experiments with UCB were compared with absolute values obtained in the absence of the pigment (controls) and expressed as relative uptakes, i.e. as percentage of controls. The use of lower unlabeled glutamate concentrations in assays using neuronal cells is intended to avoid cell death by an excitotoxic mechanism (36).
Statistical analysis.
Differences between control and UCB-treated cells were compared using the two-tailed t test for a two-sample population and were considered statistically significant when p < 0.05. Results are expressed as mean ± SEM.
RESULTS
Loss of cell viability is greater in neurons than in astrocytes after exposure to UCB.
LDH release increased from 4.5 ± 0.5% in control astrocytes to 13.9 ± 2.1% (p < 0.01) in treated cells, after 4 h exposure to 85.5 μM UCB. In neurons, similar incubation conditions resulted in an increase in LDH release from 7.2 ± 1.5% in controls to 37.0 ± 5.7% (p < 0.01) in exposed cells (Fig. 1). No significant difference was, however, found between control values for the different cell types (p > 0.1). Therefore, exposure of neural cells to UCB produces loss of cell survival, as demonstrated by the release of intracellular LDH to the incubation medium, both in astrocytes and neurons. However, neurons exhibited a three-fold increased sensitivity to UCB compared with astrocytes (p < 0.01).

Loss of cell viability is greater in neurons than in astrocytes after incubation with UCB. Astrocytes or neurons in culture were incubated with 85.5 μM UCB plus 28.5 μM HSA for 4 h, at 37°C. In control experiments, UCB was omitted. Viability was assessed by the release of LDH by nonviable cells, as described in “Materials and Methods.” After incubation with UCB, viability is approximately 3-fold lower in neurons than in astrocytes. Results are expressed as percentage of total LDH release. Each bar represents the mean ± SEM of six separate experiments. *p < 0.01 from control; ‡p < 0.01 from astrocytes.
Apoptosis is more marked in neurons than in astrocytes after exposure to UCB.
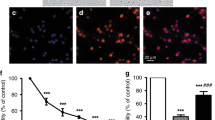
To evaluate other differences on cell response to UCB-induced toxicity, we determined levels of apoptosis in cells after incubation with UCB. Apoptotic cell death was significantly elevated in both cultured rat astrocytes and neurons after incubation with UCB, as assessed by evaluation of DNA fragmentation using the TUNEL assay (Fig. 2). In fact, treatment with 85.5 μM UCB for 4 h caused a four- and five-fold increase in glial and neuronal apoptosis, respectively (p < 0.01). Control values of apoptosis in astrocytes and neurons showed no statistical difference (p > 0.6). As for cell viability, comparison of apoptotic levels between the two cell types revealed that neuronal cells were 1.6-fold more susceptible to UCB toxicity than glial cells (p < 0.01).

Apoptosis is more marked in neurons than in astrocytes after incubation with UCB. Cells were incubated with 85.5 μM UCB plus 28.5 μM HSA, for 4 h, at 37°C. Control was performed in the absence of UCB. DNA fragmentation was assessed by the TUNEL assay, as described in “Materials and Methods.” UCB-induced apoptotic cell death is 1.6-fold higher in neurons than in astrocytes. The results are means ± SEM for at least six different experiments. *p < 0.01 from control; ‡p < 0.01 from astrocytes.
Cytoskeletal alterations are more pronounced in neurons than in astrocytes following exposure to UCB.
To evaluate the effect of UCB on glial and neuronal cytoskeleton, we used indirect immunocytochemical analysis of specific intermediate filaments present in astrocytes (GFAP) and neurons (neurofilaments 200 kD), as well as microtubule tubulin. As expected, in the absence of UCB, cultured astrocytes displayed a strong immunoreactivity for GFAP, with the filaments distributed as a dense network in the cell cytoplasm, surrounding the nucleus and reaching the cell surface (Fig. 3A). In contrast, labeling was considerably reduced in the presence of 171 μM UCB, whereas the filamentous distribution was almost completely converted into dispersed particles throughout the cytoplasm (Fig. 3B). In addition, the polygonal morphology of cell has changed to an elongated shape, and yellow deposits of UCB became evident. In the same way, tubulin filamentous distribution was affected by UCB treatment, revealing loss of immunoreactivity, granular distribution (Fig. 3, C and D) and intense cellular yellow discoloration.

Cytoskeletal alterations are more pronounced in neurons than in astrocytes after incubation with UCB. Cells were incubated with 171 and 85.5 μM UCB, at a UCB to HSA molar ratio of 3, for 4 h, at 37°C. In control experiments, UCB was omitted. Astrocytes were immunostained for GFAP (A and B) and tubulin (C and D), whereas neurons were immunostained for neurofilaments 200 kD (E and F) and tubulin (G and H), as described in “Materials and Methods.” Cells in control experiments show strong immunofluorescent reactivity either for GFAP (A) or neurofilaments (E), and also for tubulin (C and G). Alterations on astrocyte cytoskeleton (B and D) require 171 μM UCB, whereas modifications of neuronal cytoskeleton filaments (F and H) occur at 85.5 μM UCB.
Neurons in culture for 8 d displayed a marked immunoreactivity for neurofilaments (Fig. 3E) and tubulin (Fig. 3G), and a characteristic morphology of small, bipolar or multipolar cells, with a distinct neuritic network. Similar to that observed in astrocytes, neuronal cytoskeleton was disrupted by UCB, despite the lower concentration used (85.5 μM), showing loss of immunostaining and filament reticular distribution, as well as destruction of the neuritic network (Fig. 3, F and H). Yellow discoloration was less evident in UCB-treated neurons compared with astrocytes, probably as a consequence of the different concentrations used in both experiments. Equivalent effects were produced when cells were exposed to UCB at different pH values, such as those of 7.0 and 8.0, although higher UCB precipitation was apparent at the former pH (data not shown).
MTT metabolism is more affected in astrocytes than in neurons after exposure to UCB.
Having shown that neurons are more sensitive than astrocytes to both cell death and cytoskeletal alterations induced by UCB, we next investigated whether this toxic agent influences cell function in a similar manner. Thus, the conversion of MTT to a blue formazan product was measured in astrocytes and neurons exposed to 85.5 μM UCB for 4 h. Incubation with UCB markedly reduced the ability of astrocytes to convert MTT to 26.0 ± 1.2% of control values (p < 0.01). Neurons also demonstrated a 40.0 ± 5.8% decrease in the conversion of MTT compared with nontreated cells (p < 0.01) (Fig. 4). However, it is meaningful that astrocytes were at this point significantly more disturbed than neurons (p < 0.05), in contrast with previous data regarding cell death and cytoskeleton disassembly.

MTT metabolism is more affected in astrocytes than in neurons after incubation with UCB. Cells were exposed to 85.5 μM UCB plus 28.5 μM HSA, for 4 h, at 37°C. Control was performed in the absence of UCB. MTT conversion to a blue formazan product was assessed as described in “Materials and Methods.” MTT reduction is 1.5-fold lower in UCB exposed astrocytes than in neurons. Results, expressed as percentage from controls, are mean ± SEM of six separate experiments. *p < 0.01 from control; †p < 0.05 from astrocytes.
Inhibition of glutamate uptake is higher in astrocytes than in neurons following exposure to UCB.
To gain further comprehension on different cell response to toxicity of UCB, we examined the effect of 80, 100, and 120 μM UCB, at a constant 100 μM HSA, on the rate of glutamate uptake in astrocytes and neurons (Fig. 5). Both astrocytes and neurons revealed a reduced uptake of glutamate over a 7-min incubation period with the 3H-labeled amino acid (p < 0.05), irrespective of the UCB concentration used. Nevertheless, although impairment varied between 42% and 56% in astrocytes, no more than 25% to 33% were attained in neurons. Moreover, increased susceptibility of astrocytes toward neurons proportionally expanded by the concentration of UCB (80 μM, p < 0.05; 100 μM, p < 0.01; 120 μM, p < 0.001).

Inhibition of glutamate uptake is higher in astrocytes than in neurons after incubation with UCB. Cells were incubated with 80, 100, and 120 μM UCB, plus 100 μM HSA, for 4 h, at 37°C. Glutamate uptake assessed as described in “Materials and Methods.” In control experiments, UCB was omitted. UCB has a concentration-dependent inhibitory effect on glutamate uptake in both astrocytes and neurons. However, inhibition is approximately 1.6-fold greater in glial than in neuronal cells. Results, expressed as percentage from controls, are mean ± SEM of 10 separate experiments. ‡p < 0.01 and †p < 0.05 from neurons;p < 0.05 all data points from control.
DISCUSSION
In this study, we demonstrate that astrocytes and neurons exhibit a different cell response to UCB-induced toxicity. Using similar UCB concentrations, we found that neural cells present a higher rate of cell death by necrosis or apoptosis, whereas astrocytes rather revealed an impairment on glutamate uptake and MTT metabolism. Toxicity of UCB to neural cells has been demonstrated, either in vivo(27, 28, 37–39) or in vitro(12, 14, 15, 18, 19, 22), and a higher sensitivity of neurons to UCB has previously been suggested (13). However, as far as cell function is concerned, this is one of the first studies demonstrating that astrocytes can be more susceptible than neurons to UCB.
The use of the terms kernicterus and bilirubin encephalopathy to interchangeably refer to UCB toxicity in the neonatal period has recently been questioned (2, 3). In fact, it appears that kernicterus and bilirubin encephalopathy are two different manifestations of UCB toxicity; the first corresponding to the classic deposition of the pigment in defined brain areas with neuronal degeneration as a result of high levels of serum UCB, and the second comprehending diffuse staining of the brain and edema as a result of lower serum levels (3, 40). Whereas irreversible neurologic damage and death may result from kernicterus, a more subtle expression, including reversible or transitory effects such as lethargy and alterations in evoked potentials, was associated with the bilirubin encephalopathy syndrome (2). Once again, the question as to whether both manifestations share a common graded mechanism or reflect different entities remains unanswered. In addition, it is important to clarify the molecular basis for the interaction between neural cells and UCB.
Involvement of both neurons and glia in UCB toxicity was demonstrated in autopsy findings of subjects with kernicterus, showing decreased neuronal elements and reactive glial proliferation in several brain areas (3). Moreover, in an in vivo model of experimental kernicterus, astrocytes seem to play an important role both in the delivery of UCB to neurons, and elimination of toxic waste products from neurons to the blood stream (27, 28).
In the present study, several techniques directed at assessing both cell death and function were used to investigate whether astrocytes and neurons respond differently to UCB toxicity. Our results showed that the release of LDH by neurons was increased three-fold when compared with astrocytes, indicating an increased sensitivity of the former cells to UCB-induced necrotic death. Accordingly, apoptosis, a distinct form of cell death, was also significantly higher in neurons than in astrocytes after exposure to UCB. It is then conceivable that binding of UCB to neuronal cells is favored over that to astrocytes, facilitating the access of this molecule to intracellular targets, thus rendering those cells more vulnerable to irreversible cell damage. This assumption is corroborated by alterations on cytoskeletal filaments, an intracellular target of both astrocytes and neurons, where disassembly of neuronal intermediate filaments and tubulin required much lower concentrations of UCB. Such type of cytotoxic phenomena is apparently due to the acid species of UCB (6) involving UCB aggregation and deposition into cell membranes or intracellularly (14, 41, 42). Indeed, if the anionic species of UCB were responsible for the cytoskeleton disassembly, then increased alterations would be expected as the pH rose to 8.0, which did not occur.
In contrast with data on cell death, our studies on MTT metabolism indicate that astrocytes are more disturbed by UCB than neurons, as evidenced by the lower ability of the former cells to promote MTT reduction. The use of the MTT conversion as an indicator of mitochondrial function has been recently questioned based on the assumption that alterations on MTT metabolism rather result from deficient endocytic or exocytic pathways. In fact, the intracellular reduction of MTT, which is preceded by MTT endocytosis, originates a formazan yellow product that is taken out by exocytosis (43–47). We have shown that UCB is also able to inhibit the endocytosis of ferritin by astrocytes (48), suggesting that a similar effect may have accounted for the lower MTT reduction observed in the present study. Even considering that mitochondria are not involved, conversion of MTT is comprehensively used as an indicator of cell function. Our findings regarding the enhanced susceptibility of astrocytes to UCB toxicity may be of great relevance because glial cells seem to play a key role in the maintenance of brain function (23). In addition, interdependence of astrocytes and neurons has been demonstrated in studies reporting that astrocytes deliver glutamine and glucose to neurons (24, 49), and protect neuronal cells from toxic agents (25, 50, 51).
Although we cannot exclude the involvement of UCB aggregates on the impairment of MTT metabolism, the results are equally consistent with an interaction at a superficial level of the membrane leaflet according to the multistep concept of UCB cytotoxicity (14, 18, 52). This latter hypothesis was tested by evaluating the uptake of the neurotransmitter glutamate, as a different approach to estimate cell function. Thus, we used UCB/HSA molar ratios of 0.8, 1.0, and 1.2 which are closer to the physiologic conditions and less favorable to UCB aggregation. Moreover, the use of lower molar ratios and short incubation periods (15 min) ensured the maintenance of cell viability during the experiments (cell death was not >1%, even when a molar ratio of 3 was used). The greater impairment of glutamate transport by UCB in astrocytes compared with neurons may therefore result from an alternative mechanism to that inducing cell death. Indeed, there is evidence supporting that the reduced uptake of glutamate induced by UCB in astrocytes is due to monoanion species, and thus partially reverted by albumin (22).
It is conceivable that astrocytes are more resilient to UCB damage, through a higher expression of active efflux pumps (53), or a greater capacity to handle UCB detoxification (54). However, other cellular functions are more affected in astrocytes than in neurons. This raises the possibility that differences in cell vulnerability are related to distinct cell responses to UCB toxicity, depending on the pathway(s) involved. Further studies are needed, however, to definitively clarify this issue.
In conclusion, the results suggest that UCB toxicity in neural cells is promoted at least by two distinct mechanisms: one more severe, inducing cell death and most likely responsible for irreversible lesions, to which neurons are particularly susceptible; and the other milder, compromising some aspects of cell function and probably related with transitory or reversible damage, to which astrocytes showed increased vulnerability. Thus, although less sensitive to cell death than neurons, astrocytes are still greatly affected by UCB and most likely participate as well in the pathogenesis of bilirubin encephalopathy.
Abbreviations
- FCS:
-
fetal calf serum
- FITC:
-
fluorescein-isothyocianate
- GFAP:
-
glial fibrillary acidic protein
- HSA:
-
human serum albumin
- LDH:
-
lactate dehydrogenase
- TUNEL:
-
terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end-labeling
- UCB:
-
unconjugated bilirubin
References
Rubaltelli FF 1993 Bilirubin metabolism in the newborn. Biol Neonate 63: 133–138
Hansen TWR 1994 Bilirubin in the brain. Distribution and effects on neurophysiological and neurochemical processes. Clin Pediatr 33: 452–459
Gourley GR 1997 Bilirubin metabolism and kernicterus. Adv Pediatr 44: 173–229
Odell GB 1980 Neonatal Hyperbilirubinemia. Grune & Straton, New York,
Blanckaert N, Fevery J 1990 Physiology and pathophysiology of bilirubin metabolism. In: Zakim D, Boyer TD (eds) Hepatology. A Textbook of Liver Disease. WB Saunders, London, 254–303
Brodersen R, Stern L 1990 Deposition of bilirubin acid in the central nervous system—a hypothesis for the development of kernicterus. Acta Paediatr Scand 79: 12–19
Fenwick JD 1975 Neonatal jaundice as a cause of deafness. J Laryngol Otol 89: 925–932
Perlman M, Fainmesser P, Sohmer H, Tamari H, Wax Y, Pevsmer B 1983 Auditory nerve-brainstem evoked responses in hyperbilirubinemic neonates. Pediatrics 72: 658–664
Chen Y, Kang W 1995 Effects of bilirubin on visual evoked potentials in term infants. Eur J Pediatr 154: 662–666
Mayor F Jr, Pagés M, Diez-Guerra J, Valdivieso F, Mayor F 1985 Effect of postnatal anoxia on bilirubin levels in rat brain. Pediatr Res 19: 231–236
Hansen TWR, Paulsen O, Gjerstad L, Bratlid D 1988 Short-term exposure to bilirubin reduces synaptic activation in rat transverse hippocampal slices. Pediatr Res 23: 453–456
Danbolt C, Hansen TWR, Oyasaeter S, Storm-Mathisen J, Bratlid D 1993 In vitro binding of [3H] bilirubin to neurons in rat brain sections. Biol Neonate 63: 35–39
Notter MF, Kendig JW 1986 Differential sensitivity of neural cells to bilirubin toxicity. Exp Neurol 94: 670–682
Amit Y, Fedunec S, Thomas PD, Poznansky MJ, Shiff D 1990 Bilirubin-neural cell interaction: characterization of initial cell surface binding leading to toxicity in the neuroblastoma cell line N-115. Biochim Biophys Acta 1055: 36–42
Amit Y, Cashore W, Schiff D 1992 Studies of bilirubin toxicity at the synaptosome and cellular levels. Semin Perinatol 16: 186–190
Hansen TWR, Bratlid D, Walaas SI 1988 Bilirubin decreases phosphorylation of synapsin I, a synaptic vesicle-associated neuronal phosphoprotein, in intact synaptosomes from rat cerebral cortex. Pediatr Res 23: 219–223
Mayor F Jr, Diez-Guerra J, Valdivieso F, Mayor F 1986 Effect of bilirubin on the membrane potential of rat brain synaptosomes. J Neurochem 47: 363–369
Vazquez J, Garcia-Calvo M, Valdivieso F, Mayor F, Mayor F Jr 1988 Interaction of bilirubin with the synaptosomal plasma membrane. J Biol Chem 263: 1255–1265
Amit Y, Brenner T 1993 Age-dependent sensitivity of cultured rat glial cells to bilirubin toxicity. Exp Neurol 121: 248–255
Blondeau J-P, Beslin A, Chantoux F, Francon J 1993 Triiodothyronine is a high-affinity inhibitor of amino acid transport system L1 in cultured astrocytes. J Neurochem 60: 1407–1413
Chantoux F, Chuniaud L, Dessante M, Trivin F, Blondeau J-P, Francon J 1993 Competitive inhibition of thyroid hormone uptake into cultured rat brain astrocytes by bilirubin and bilirubin conjugates. Mol Cell Endocrinol 97: 145–151
Silva R, Mata LR, Gulbenkian S, Brito MA, Tiribelli C, Brites D 1999 Inhibition of glutamate uptake by unconjugated bilirubin in cultured cortical rat astrocytes: role of concentration and pH. Biochem Biophys Res Commun 265: 67–72
Montgomery DL 1994 Astrocytes: form, functions, and roles in disease. Vet Pathol 31: 145–167
Forsyth R, Fray A, Boutelle M, Fillenz M, Middleditch C, Burchell A 1996 A role for astrocytes in glucose delivery to neurons?. Dev Neurosci 18: 360–370
Mena MA, Casarejos MJ, Carazo A, Paino CL, Yébenes JG 1996 Glia conditioned medium protects fetal rat midbrain neurones in culture froml-DOPA toxicity. Neuroreport 7: 441–445
Fróes MM, Correia AHP, Garcia-Abreu J, Spray DC, Carvalho ACC, Neto VM 1999 Gap-junctional coupling between neurons and astrocytes in primary central nervous system cultures. Proc Natl Acad Sci U S A 96: 7541–7546
Chen H, Tsai D, Wang C, Chen Y 1969 An electron microscopic and radioautographic study on experimental kernicterus. I. Bilirubin transport via astroglia. Am J Pathol 56: 31–58
Chen H, Wang C, Tsan K-W, Chen Y 1971 An electron microscopic and radioautographic study on experimental kernicterus. II. Bilirubin movement within neurons and release of waste products via astroglia. Am J Pathol 64: 45–66
Silva RFM, Rodrigues CMP, Brites D 2001 Bilirubin-induced apoptosis in cultured rat neural cells is aggravated by chenodeoxycholic acid but prevented by ursodeoxycholic acid. J Hepatol 34: 402–408
McDonagh AF, Assisi F 1972 The ready isomerization of bilirubin IX-α in aqueous solution. Biochem J 129: 797–800
Brewer GJ, Torricelli JR, Evege EK, Price PJ 1993 Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free combination. J Neurosci Res 35: 567–576
Perlman JM, Rogers BB 1997 Kernicteric findings at autopsy in two sick near term infants. Pediatrics 99: 612–615
Mosmann T 1983 Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65: 55–63
Swanson RA, Farrel K, Simon RP 1995 Acidosis causes failure of astrocyte glutamate uptake during hypoxia. J Cereb Blood Flow Metab 15: 417–424
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ 1951 Protein measurements with the Folin phenol reagent. J Biol Chem 193: 265–275
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF 2000 Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686
Diamond I, Schmid R 1966 Experimental bilirubin encephalopathy. The mode of entry of bilirubin-14C into the central nervous system. J Clin Invest 45: 678–689
Hoffman DJ, Zanelli SA, Kubin J, Mishra OP, Delivoria-Papadopoulos M 1996 The in vivo effect of bilirubin on the N-methyl-d-aspartate receptor/ion channel complex in the brains of newborn piglets. Pediatr Res 40: 804–808
McDonald JW, Shapiro SM, Silverstein FS, Johnston MV 1998 Role of glutamate receptor-mediated excitotoxicity in bilirubin-induced brain injury in the Gunn rat model. Exp Neurol 150: 21–29
Turkel SB, Miller CA, Guttenberg ME, Moynes DR, Hodgman JE 1982 A clinical pathologic reappraisal of kernicterus. Pediatrics 69: 267–272
Brites D, Silva R, Brito A 1997 Effect of bilirubin on shape and haemolysis, under hypotonic, aggregating or non-aggregating conditions, and correlation with cell age. Scand J Clin Lab Invest 57: 337–350
Brito MA, Silva R, Tiribelli C, Brites D 2000 Assessment of bilirubin toxicity to erythrocytes. Implication in neonatal jaundice management. Eur J Clin Invest 30: 239–247
Berridge MV, Tan AS 1993 Characterization of the cellular reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT): subcellular localization, substrate dependence, and involvement of mitochondrial electron transport in MTT reduction. Arch Biochem Biophys 303: 474–482
Liu Y, Schubert D 1997 Cytotoxic amyloid peptides inhibit cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction by enhancing MTT formazan exocytosis. J Neurochem 69: 2285–2293
Liu Y, Peterson DA, Kimura H, Schubert D 1997 Mechanism of cellular 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction. J Neurochem 69: 581–593
Liu Y, Schubert D 1998 Steroid hormones block amyloid fibril-induced 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) formazan exocytosis: relationship to neurotoxicity. J Neurochem 71: 2322–2329
Shearman MS, Hawtin SR, Tailor VJ 1995 The intracellular component of cellular 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction is specifically inhibited by β-amyloid peptides. J Neurochem 65: 218–227
Silva RFM, Mata LM, Gulbenkian S, Brites D 2001 Endocytosis in rat cultured astrocytes is inhibited by unconjugated bilirubin. Neurochem Res 26: 793–800
Westergaard N, Sonnewald U, Schousboe A 1995 Metabolic trafficking between neurons and astrocytes: the glutamate/glutamine cycle revisited. Dev Neurosci 17: 203–211
Koops A, Kappler J, Junghans U, Kuhn G, Kresse H, Müller HW 1996 Cultured astrocytes express biglycan, a chondroitin/dermatan sulfate proteoglycan supporting the survival of neocortical neurons. Mol Brain Res 41: 65–73
Brown DR 1999 Dependence of neurones on astrocytes in a coculture system renders neurones sensitive to transforming growth factor β1-induced toxicity. J Neurochem 72: 943–953
Eriksen EF, Danielsen H, Brodersen R 1981 Bilirubin-liposome interaction. Binding of bilirubin dianion, protonization, and aggregation of bilirubin acid. J Biol Chem 256: 4269–4274
Decleves X, Regina A, Laplanche JL, Roux F, Boval B, Launay JM, Scherrmann JM 2000 Functional expression of P-glycoprotein and multidrug resistance-associated protein (Mrp1) in primary cultures of rat astrocytes. J Neurosci Res 60: 594–601
Hansen TWR, Allen JW 1997 Oxidation of bilirubin by brain mitochondrial membranes—dependence on cell type and postnatal age. Biochem Mol Med 60: 155–160
Acknowledgements
The authors thank Cláudia Simões for her contribution during part of the experiments.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported by grant PRAXIS/PSAU/C/SAU/127/96 from Fundação para a Ciência e a Tecnologia (FCT), Lisbon, Portugal.
Rights and permissions
About this article
Cite this article
Silva, R., Rodrigues, C. & Brites, D. Rat Cultured Neuronal and Glial Cells Respond Differently to Toxicity of Unconjugated Bilirubin. Pediatr Res 51, 535–541 (2002). https://doi.org/10.1203/00006450-200204000-00022
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200204000-00022
This article is cited by
-
Bilirubin potentiates etomidate-induced sedation by enhancing GABA-induced currents after bile duct ligation
BMC Pharmacology and Toxicology (2023)
-
The effect of obstructive jaundice on the sensitivity of intravenous anesthetic of remimazolam: study protocol for a controlled multicenter trial
Trials (2022)
-
Bilirubin Encephalopathy
Current Neurology and Neuroscience Reports (2022)
-
High unbound bilirubin for age: a neurotoxin with major effects on the developing brain
Pediatric Research (2019)
-
Histone acetylation as a new mechanism for bilirubin-induced encephalopathy in the Gunn rat
Scientific Reports (2018)
