
Abstract
The disease gene for Pendred syndrome has been recently characterized and named PDS. It codes for a transmembrane protein called pendrin, which is highly expressed at the apical surface of the thyroid cell and functions as a transporter of chloride and iodide. Pendrin is also expressed at the inner ear level, where it appears to be involved in the maintenance of the endolymph homeostasis in the membranous labyrinth, and in the kidney, where it mediates chloride-formate exchange and bicarbonate secretion. Mutations in the PDS gene and the consequent impaired function of pendrin leads to the classic phenotype of Pendred syndrome, i.e. dyshormonogenic goiter and congenital sensorineural hearing loss. In the present study, we performed a detailed clinical, radiologic, and molecular analysis of six families presenting with clinical diagnosis of Pendred syndrome. In two families a homozygous pattern for PDS mutations was found, whereas the affected members of the other four families were compound heterozygotes. One family did not harbor PDS mutations. Among the four novel mutations described, one is a transversion in exon 2 (84C>A), leading to the substitution S28R. Two other novel mutations lie in exon 4 (398T>A) and in exon 16 (1790T>C), leading to the substitutions S133T and L597S, respectively. The fourth novel mutation (1614+1G>A) is located in the first base pair of intron 14, probably affecting the splicing of the PDS gene. Clinically, all patients had goiter with positive perchlorate test, hypothyroidism, and severe or profound sensorineural hearing loss. In all the individuals harboring PDS mutations, but not in the family without PDS mutations, inner ear malformations, such as enlargement of the vestibular aqueduct and of the endolymphatic duct and sac, were documented. The pseudo-Pendred phenotype exhibited by the family without PDS mutations is likely caused by an autoimmune thyroid disease associated with a sensorineural hearing loss of different origin.
Similar content being viewed by others
Main
PS is an autosomal recessive disease first described in 1896 as the combination of congenital deafness and goiter (1). It is considered the most common cause of congenital deafness, but the actual incidence of the syndrome is difficult to establish because of the variable phenotype of affected subjects, even within the same family (2–4). In the last 3 y PS has been associated, with a homozygous or compound heterozygous pattern, to mutations in the PDS gene (5–14). PDS maps on chromosome band 7q22–31.1 and codes for pendrin (7), a transmembrane protein, which was first found to be expressed in the thyroid and in particular at the apical surface of the thyroid cell, where it likely functions as an iodide-chloride transporter (15–17). The effect of impaired pendrin function is thus a deficient transport of iodide into the colloid space and its accumulation into the cytoplasm. The consequent altered iodide gradient between thyroid and serum is revealed by the perchlorate test (18), which returns a positive result in the majority of affected patients. Indeed, the thyroid clearance of iodide, measured before and after the administration of perchlorate, is notably higher in PS patients than in normal subjects (131I discharge, 15–80%versus <10%, respectively). However, the defect is only partial, as in most patients the thyroid hormone status is normal or a subclinical hypothyroidism is shown (4). Furthermore, goiter is not a constant feature of the syndrome and can vary from a slight enlargement to a large multinodular goiter, with a time of onset ranging from the first months of life until puberty (3, 4, 19). Both endogenous and environmental factors could explain such variability in the expression of thyroid phenotype.
More recently, pendrin has been immunolocalized in the inner ear of the mouse and in particular in the ED and ES (20), both regulating endolymph secretion and resorption (21). Additional studies are needed to identify which anion(s) is transported by pendrin within the inner ear, although defects in chloride transport might produce both toxic and osmotic damages of the neuroepithelium (20), accounting for the SNHL of PS patients. In particular, in PS the deafness is generally profound (>60 dB) with a prelingual onset (19), and sometimes a fluctuating but worsening course (5, 22, 23), consistent with a progressive lesion of the sensory organ. Moreover, the increased osmotic pressure and volume of the endolymph leads to enlargement of the membranous labyrinth, i.e. the ED and ES, and of the surrounding bony structures such as the VA and the cochlea. Cochlear abnormalities (Mondini cochlea) are present in a minority of patients (24, 25), whereas the enlargement of the ED and ES and of the VA have been recently documented by high-resolution CT and MRI in 80–100% of patients affected with PS or LVAS, in which the SNHL is not associated with thyroid abnormalities (2, 6, 26–34).
Finally, pendrin has been recently immunolocalized in the proximal tubule of the kidney, where it seems to function as a chloride-formate exchanger (35), and in the intercalated cells of cortical collecting ducts, where it has an essential role in bicarbonate secretion (36). Probably because of the redundancy of compensatory mechanisms all along the nephron, no phenotypic alterations have been reported at the renal level in patients with PDS mutations.
In the present study, we report the clinical and radiologic investigations and the genetic analyses in six Italian families referred to our institution with a clinical diagnosis of PS.
METHODS
Patients.
We studied nine patients from six unrelated Italian families and from nonconsanguineous parents referred to us with a clinical diagnosis of PS (Table 1). The affected members showed childhood deafness related to a sensorineural defect and goiter, with positive perchlorate discharge test. Three of the affected patients are males (family 1 and 5). The age range was 14–41 y (mean, 26.7 y) at the time of the present study. The two sisters of family 6 are monozygotic twins. Thyroid function analyses showed, in patients from families 1–5, a subclinical hypothyroidism with normal values of free thyroid hormones and TSH concentrations ranging between 5 and 6.5 mU/L (normal values, 0.25–4.5 mU/L). The two sisters of family 6 were subclinical hypothyroid at the diagnosis (9 y of age) with TSH levels of 5.3 and 6 mU/L, although a worsening of hypothyroid state was seen during levothyroxine withdrawal at 16 y of age (TSH, 40 and 36 mU/L, respectively). Antithyroglobulin and antithyroid peroxidase antibodies were positive (350 IU/mL and 649 IU/mL, respectively; normal values, <100 IU/mL) only in family 6. In all patients a multinodular goiter of different sizes was documented at ultrasound examination (from 18 to 60 mL). A hypoechoic pattern was shown in patients from family 6. It is worth noting that all patients developed goiter after the first months or years of life. The perchlorate test, performed by measuring radiolabeled iodide thyroid uptake before and after the oral administration of 1 g of KClO4, was positive in all patients with discharge rates of 40–60% of basal radioiodine uptake.
In all patients a bilateral SNHL was diagnosed during childhood. The degree of deafness, assessed by pure-tone audiometry in each ear, was bilaterally severe (60–80 dB) in the affected members of families 1, 3, 5, and 6 and severe or profound (>80 dB) in families 2 and 4. In the patient of family 4, the SNHL showed a characteristic fluctuating and progressive pattern. In addition, she suffered from a vestibular syndrome. Mild language disturbances, related to the SNHL, were observed in families 1, 2, 3, and 5. In the twins of family 6 a diagnosis of SNHL with severe language disturbances and mental retardation was reached at the age of 5.5 y. Subclinical hypothyroidism with goiter was incidentally discovered at 9 y of age, therefore leading to the diagnosis of PS. In monitoring control subjects (age, 15.5 y), neurologic examination revealed a global cognitive retardation (IQ, 68 on WISC-R test) and mental age of 10.5 y in both twins. It is likely that such mental retardation derives from a not readily cured deafness, consistent with the poor level of instruction and social background of the family. Nevertheless, clinical history revealed that their father was deaf-mute but without evidence of thyroid disease, suggesting a possible genetic origin of the hearing defect.
The six families come from different Italian regions with moderate iodine deficiency.
Radiologic studies.
In all patients, the presence of alterations of the cochlea and the VA was tested by high-resolution CT of temporal bones in coronal and axial planes (1-mm contiguous sections). The VA was considered enlarged when its diameter at the midpoint between the common crus and the external aperture was ≥1.5 mm or more on thin CT sections (32).
High-resolution FSE T2-weighted MRI was performed (in axial and coronal planes) to study the membranous labyrinth and in particular the ED and ES. The size of the normal ED at its midpoint ranges from not visible to 1.4 mm (29). ES is rarely seen in normal subjects, despite high-quality FSE MR images, and it is considered enlarged when >2.5 mm (29, 31). All the images were photographed individually and as a three-dimensional composite.
Molecular studies.
DNA was extracted by standard methods from whole blood of all patients and their available parents. The experimental studies were approved by the ethics committees of each institution. An informed consent was obtained for all screened subjects.
In each affected member all 20 coding PDS exons (2–21) were amplified using primers flanking each exon (7). Samples were subjected to 5 min of denaturation at 98°C, followed by 35 three-step cycles (55°C for 1 min, 72°C for 2 min, 94°C for 1 min), and 72°C for 10 min in a TouchDown Thermal Cycler (Hybaid, Middlesex, U.K.). PCR products were directly sequenced after removal of unincorporated deoxynucleotides and primers by GFX PCR DNA purification kit (Amersham Pharmacia Biotech, Uppsala, Sweden). An aliquot of 3–10 ng/100 bp of purified DNA and 3.2 pmol of either the forward or reverse primer were used in standard cycle-sequencing reactions with ABI PRISM Big Dye terminators and run on an ABI PRISM 310 Genetic Analyzer (Applied Biosystems, Foster City, CA, U.S.A.). The cycle-sequencing conditions consisted of 25 cycles of 96°C for 30 s, 50°C for 15 s, and 60°C for 4 min. One sequence read from each direction across the entire coding region and including intron-exon boundaries was obtained for each patient.
RESULTS
Radiologic examination.
In all patients from families 1–5, malformations of the inner ear were found. At CT scan, the VA was enlarged bilaterally, with a diameter ranging from 2 to 4 mm. No cochlear alterations were found. The ED and ES were enlarged in all patients at MRI examination, with an ES diameter ranging from 4 to 11 mm (Table 1). The higher dimensions belong to the patient from family 4, who exhibited in the left ear a huge ES (11 mm), leading to the complete disruption of the corresponding VA, in which the endolymphatic system is contained (Fig. 1). In the two sisters of family 6, no abnormalities of the inner ear structures were detected by either CT or FSE MRI.

Radiologic studies of the inner ear in a normal subject and in a patient of family 4. N1, high-resolution FSSE T2-weighted MRI scan of the petrous temporal bones. In the normal subject the endolymphatic sac cannot be identified. A1, in the proband the ES is enlarged in both ears, with a diameter of 5 mm in the right and 11 mm in the left ear (arrows). A2, a particular of the left ES in which axial, contiguous, 1-mm images have been summated into a single composite three-dimensional image. N2, high-resolution CT scan of the petrous temporal bones (axial view). Normal VA. B, in the proband the VA of the right ear (arrow) is enlarged (2.4 mm). The left VA is disrupted by the very enlarged endolymphatic system (arrow).
Mutation analysis.
PDS sequence analyses of the entire 2–20 exons and of the coding portion of exon 21 showed gene mutations in all patients but those of family 6. All mutations have been classified according to the nomenclature recommendations, with the A of the ATG of the initiator Met codon denoted as +1 (37). In the proband of family 1, a homozygous missense mutation in exon 2 (84C>A) was found (Fig. 2). This is a novel mutation leading to the substitution of a serine for an arginine at codon 28 (S28R). No mutations were found in all the other PDS coding exons. The parents of the proband showed the same mutation in heterozygosity. In the two sisters of family 2, a compound heterozygous pattern was found: a known mutation in intron 8 (1001+1G>A) and a novel missense mutation in exon 16 (1790T>C;Fig. 2). The first mutation affects a splicing site and the second leads to the substitution of a leucine for a serine at codon 597 (L597S). The proband of family 3 was compound heterozygote for a known single base pair deletion in exon 10 (1197delT) and for a novel intronic mutation in intron 14 (1614+1G>A) (Fig. 2). In particular the exon 10 deletion leads to a frameshift at codon 400 (FS400) with the creation of a stop codon at 431, whereas the intronic mutation affects a splicing site.

Sequence analysis of the four novel mutations described in patients of familes 1, 2 (2a, 2b), 3, and 5 (5a, 5b). The homozygous 84C>A mutation and the other three heterozygous mutations are indicated by arrows.
The proband of family 4 resulted homozygous for an already described missense mutation in exon 10 (1226G>A). This mutation leads to the substitution of an arginine for a histidine at codon 409 (R409H) and was found in heterozygosity in the mother of the proband, although the father was not available for investigation.
In the two brothers of family 5 a compound heterozygous pattern has been found: a known missense mutation in exon 10 (1229C>T), leading to the substitution of a threonine for a methionine at codon 410 (T410M), and a novel mutation in exon 4 (398T>A), leading to the substitution of a serine for a threonine at codon 133 (S133T;Fig. 2). The parents showed a heterozygous pattern, T410M/wild-type for the mother, and S133T/wild-type for the father.
Besides the amino terminal mutation (S28R) and the splice site substitutions, the other mutations are located in the II transmembrane domain (S133T), in the IX transmembrane domain (FS400), in the V extracellular domain (R409H and T410M), and in the C- terminus (L597S), according to the model proposed by Everett and collaborators (7).
DISCUSSION
After the characterization of the PDS gene in 1997, more than 40 different mutations have been described scattered all along the 20 coding exons, in families with PS or LVAS (5–13, 34, 38, 39). The majority of the mutations (approximately 60%) are missense substitutions and are localized in the second half of the protein. However, neither hot-spot regions nor phenotype-genotype correlation have been clearly identified. The present study, reporting the characterization of four novel PDS mutations and the precise description of the associated clinical picture, aims to give more insights on mutations clustering and genotype-phenotype relationship.
The three novel substitutions described in the present work have not been observed in the normal control population and affect conserved amino acids, as results from the alignments of the PDS sequence with two highly homologous human genes, such as DRA (down-regulated in adenoma) and DTD (diastrophic dysplasia) (7). Thus, it can be expected that these mutations result in impairment of pendrin function. In particular, S133T is located in the II transmembrane domain and likely impairs the stability of the protein at the cell membrane level, as previously reported for mutations affecting other transmembrane domains (8). The L597S lies in the C-terminus, where the majority of PDS mutations have been localized (6), therefore suggesting an important role of this domain in pendrin function. Furthermore, after submission of this paper, Campbell et al. (39) reported the same mutation in three families with enlarged VA. The substitution leading to the amino-terminal mutation (S28R) was heterozygous in the unaffected parents, and the proband inherited both mutated alleles. The change of an uncharged amino acid (serine) with a bulkier, highly basic, and positively charged amino acid (arginine) is predicted to affect the protein function. Moreover, an amino acid change in the adjacent residue (E29Q) has been very recently reported in a patient affected with enlarged VA (39). The fourth novel mutation (1614+1G>A, family 3) is located in the first base pair of intron 14, and most probably affects splicing of the PDS gene. As no tissues expressing the PDS gene were available from this patient, we were unable to analyze the effect of the mutation at the mRNA or protein level. However, there are several examples of mutations occurring at AG/GT invariant intronic sequences of splice sites in other genes, which have been found to impair protein expression or function and therefore cause genetic diseases [reviewed in Krawczak et al. (40)]. Intronic mutations affecting splice sites seem to be frequent also for PDS gene, and involve intron 4 (12), intron 7 (10), intron 8 [(8, 13), present study], and intron 14 (present study). Moreover, the splice site mutation in intron 8 (1001+1G>A) belongs to the group of the most common mutations reported to date. Indeed, the revision of the literature shows that seven PDS mutations are recurrent: L236P [n = 21 families;(8, 9, 38, 39)], T416P [n = 18;(8, 9, 38, 39)], 1001+1G>A [n = 18;(8, 13, 39)], E384G [n = 7;(5, 8)], L445W [n = 6;(5, 9, 10, 14, 38)], FS400 [n = 5;(6, 7, 9)], and T410M [n = 4;(8, 38)]. Consistent with these findings, our families harbored, besides the novel, also the mutations 1001+1G>A, FS400, and T410M. Only one mutation (T410M) is a transition in CpG dinucleotides that are frequent sites of point mutation in several other genes (41), and no mutations in G-C-rich regions (defined as four or more consecutive Gs or Cs) have been recorded. Although a cluster of mutations appears to localize in exon 10 (Fig. 3), a founder effect for some of these mutations cannot be ruled out, as most of the families studied originate from Northern Europe.

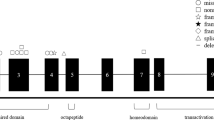
Schematic representation of the PDS gene (all but exon 1 and 21 are drawn to scale). The seven more common mutations reported in the literature are shown in bold characters, and their distribution on the PDS gene is indicated by arrows. The number of families harboring each mutation is also reported (references in parentheses). The mutation located in intron 8 (1001+1G>A) is predicted to lead to an altered mRNA splicing. All the other mutations are within the coding sequence and are located in exon 6 (L236P), in exon 10 (E384G, FS400, T410M, and T416P), and in exon 11 (L445W). All mutations are uniformly classified according to the recent nomenclature recommendations (37).
No phenotypic differences have been found among individuals with different mutations, even considering the patient showing a very premature truncated protein (family 1) and those harboring mutations in the C terminus (family 2). However, although all the patients reported in the present study showed a congenital, severe or profound hearing loss, only one patient (family 4) shared a fluctuating and progressive course associated with a vestibular syndrome. Moreover, in this case the enlargement of the ED and ES of the left ear was so huge to determine the complete disruption of the VA. Further studies are needed to establish whether the amino acid substitution found in this patient (R409H) is associated with an extremely severe ear phenotype accompanied by vestibular symptoms.
As far as the two sisters without PDS mutations (family 6) is concerned, the radiologic examinations and the genetic analysis allowed us to exclude the initial clinical diagnosis of PS. They are likely to be affected with Hashimoto's thyroiditis, as suggested by the presence of antithyroid antibodies and a typical pattern at the ultrasound. We have recently reported a similar case (6) in which, despite a clinical diagnosis of PS, no PDS mutations were found, although biochemical and ultrasonography signs of thyroid autoimmunity were present. In both cases an SNHL was shown without inner ear malformations detectable at CT and MRI. Thus, among a total of nine families presenting with a clinical diagnosis of PS [(6) and present study], two exhibit a pseudo-Pendred condition, in which the association of thyroid and hearing impairment is not caused by PDS mutations. Because congenital deafness is extremely common, affecting 1 of 2000 newborns (4), and >50 different genes have been found to be involved in it (42), the origin of hearing loss in these cases may be owing to mutations in other deafness genes. However, considering the high frequency of pseudo-Pendred in our series, the hypothesis of a correlation between the thyroid and the ear disorders should be considered. Indeed, it has been postulated that the hearing loss is associated with autoimmunity (43). Positive anti-Tg antibodies and thyroid-stimulating antibodies were found in PS patients by Friis et al. (44), and Mayot et al. (45) reported anti-thyroid as well as anti-cochlear antibodies in a group of 40 patients with progressive SNHL, suggesting a link between autoimmune inner ear disease and autoimmune thyroid disease. More studies are needed to establish the importance of these observations, but we postulate that in the two families presenting with pseudo-Pendred, the hearing loss may be linked to the thyroid disorder on an autoimmune basis. Clinically, inasmuch as the perchlorate test is not necessarily conclusive of PS as positive tests (20–80%) are found in Hashimoto's thyroiditis (46), the differential diagnosis between PS and pseudo-PS should be based on the study of inner ear malformations that are extremely specific signs and can be considered the only clinical feature always corresponding to the presence of PDS mutations.
In conclusion, our data confirm that molecular genetic analysis of the PDS gene is an essential requisite to the identification of PS, mostly because of the extreme high variability of the phenotype making the clinical diagnosis difficult to secure. However, as confirmed by our data, PDS mutations are widely distributed along the gene, highlighting how time- and money-consuming genetic analysis can be. Hence, efforts should be made to recognize the full range of presentation of the syndrome and to make a correct diagnosis before performing the molecular studies, taking into account the more specific clinical criteria.
Abbreviations
- PS:
-
Pendred syndrome
- ED:
-
endolymphatic duct
- ES:
-
endolymphatic sac
- SNHL:
-
sensorineural hearing loss
- CT:
-
computed tomography
- MRI:
-
magnetic resonance imaging
- VA:
-
vestibular aqueduct
- LVAS:
-
large vestibular aqueduct syndrome
- FSE MRI:
-
fast spin-echo MRI
References
Pendred V 1896 Deaf-mutism and goiter. Lancet 2: 532
Reardon W, Coffey R, Phelps PD, Luxon LM, Stephens D, Kendall-Taylor P, Britton KE, Grossman A, Trembath R 1997 Pendred syndrome: 100 years of underascertainment?. Q J Med 90: 443 443–447
Reardon W, Coffey R, Pembrey ME, Rajput K, Phelps PD, Luxon LM, Britton K, Grossman A, Kendall-Taylor P, Trembath R 1997 Pitfalls in practice—diagnosis and misdiagnosis in Pendred syndrome. J Audiol Med 6: 1–9
Reardon W, Coffey R, Chowdhury T, Grossman A, Jan H, Britton K, Kendall-Taylor P, Trembath R 1999 Prevalence, age of onset, and natural history of thyroid disease in Pendred syndrome. J Med Genet 36: 595–598
Cremers CWRJ, Bolder C, Admiraal RJC, Everett LA, Joosten FB, Van Hauwe P, Green ED, Otten BJ 1998 Progressive sensorineural hearing loss and a widened vestibular aqueduct in Pendred syndrome. Arch Otolaryngol Head Neck Surg 124: 501–505
Fugazzola L, Mannavola D, Cerutti N, Maghnie M, Pagella F, Bianchi P, Weber G, Persani L, Beck-Peccoz P 2000 Molecular analysis of the Pendred's syndrome gene and magnetic resonance imaging studies of the inner ear are essential for the diagnosis of true Pendred's syndrome. J Clin Endocrinol Metab 85: 2469–2475
Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED 1997 Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 17: 411–422
Coyle B, Reardon W, Herbrick JA, Tsui LC, Gausden E, Lee J, Coffey R, Grueters A, Grossman A, Phelps PD, Luxon L, Kendal-Taylor P, Scherer SW, Trembath RC 1998 Molecular analysis of the PDS gene in Pendred syndrome (sensorineural hearing loss and goitre). Hum Mol Genet 7: 1105–1112
Van Hauwe P, Everett LA, Coucke P, Scott DA, Kraft ML, Ris-Stalpers C, Bolder C, Otten B, de Vijlder JJM, Dietrich NL, Ramesh A, Srisailapathy SCR, Parving A, Cremers CWRJ, Willems PJ, Smith JH, Green ED, Van Camp G 1998 Two frequent missense mutations in Pendred syndrome. Hum Mol Genet 7: 1099–1104
Coucke PJ, Van Hauwe P, Everett LA, Demirhan O, Kabakkaya Y, Dietrich NL, Smith RJH, Coyle E, Reardon W, Trembath R, Willems Green ED, Van Camp G 1999 Identification of two different mutations in the PDS gene in an inbred family with Pendred syndrome. J Med Genet 36: 475–477
Kopp P, Karamanoglu Arseven O, Sabacan L, Kotlar T, Dupuis J, Cavaliere H, Santos CLS, Jameson JL, Medeiros-Neto G 1999 Phenocopies for deafness and goiter development in a large inbred Brazilian kindred with Pendred's syndrome associated with a novel mutation in the PDS gene. J Clin Endocrinol Metab 84: 336–341
Lopez-Bigas N, Rabionet R, de Cid R, Govea N, Gasparini P, Zelante L, Arbones ML, Estivill X 1999 Splice-site mutation in the PDS gene may result in intrafamilial variability for deafness in Pendred syndrome. Hum Mutat 14: 520–526
Bogazzi F, Raggi F, Ultimieri F, Campomori A, Cosci C, Berrettini S, Neri E, La Rocca R, Ronca G, Martino E, Bartalena L 2000 A novel mutation in the pendrin gene associated with Pendred's syndrome. Clin Endocrinol 52: 279–285
Masmoudi S, Charfedine I, Hmani M, Grati M, Ghorbel AM, Elgaied-Boulila A, Drira M, Hardelin JP, Ayadi H 2000 Pendred syndrome: phenotypic variability in two families carrying the same PDS missense mutation. Am J Med Genet 90: 38–44
Scott DA, Wang R, Kreman TM, Sheffield VC, Karnishki LP 1999 The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 4: 440–443
Royaux IE, Suzuki K, Mori A, Kato R, Everett LA, Kohn LD, Green ED 2000 Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology 141: 839–845
Bidart JM, Mian C, Lazar V, Russo D, Filetti S, Caillou B, Schlumberger M 2000 Expression of pendrin and the Pendred syndrome (PDS) gene in human thyroid tissue. J Clin Endocrinol Metab 85: 2028–2033
Morgans ME, Trotter WR 1958 Association of congenital deafness with goitre: the nature of the thyroid defect. Lancet 1: 607–609
Fraser GR, Morgans ME, Trotter WR 1960 The syndrome of sporadic goitre and congenital deafness. Q J Med 29: 279–295
Everett LA, Morsli H, Wu DK, Green ED 1999 Expression pattern of the mouse ortholog of the Pendred's syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc Natl Acad Sci USA 96: 9727–9732
Ferrary E, Sterkers O 1998 Mechanisms of endolymph secretion. Kidney Int 53 ( suppl): S98–S103
Fraser GR 1965 Association of congenital deafness with goitre (Pendred's syndrome). Hum Genet 28: 201–249
Johnsen T, Larsen C, Friis J, Hougaard-Jensen F 1987 Pendred's syndrome: acoustic, vestibular and radiological findings in 17 unrelated patients. J Laryngol Otol 101: 1187–1192
Phelps PD, Coffey RA, Trembath RC, Luxon LM, Grossman AB, Britton KE, Kendall-Taylor P, Graham JM, Cadge BC, Stephens SGD, Pembrey ME, Reardon W 1998 Radiological malformations of the ear in Pendred syndrome. Clin Radiol 53: 268–273
Cremers CWRJ, Admiraal RJC, Huygen PLM, Older C, Everett LA, Joosten FBM, Green ED, Van Camp G, Otten BJ 1998 Progressive hearing loss, hypoplasia of the cochlea and widened vestibular aqueducts are very common features in Pendred's syndrome. Int J Pediatr Otorhinolaryngol 45: 113–123
Mafee MF, Charletta D, Kumar A, Belmont H 1992 Large vestibular aqueduct and congenital sensorineural hearing loss. Am J Neuroradiol 13: 805–819
Johnsen T, Jorgensen MB, Johnsen S 1986 Mondini cochlea in Pendred's syndrome. Acta Otolaryngol 102: 239–247
Mondini C 1791 In: Opuscula Caroli Mundini: Anatomica Surdi Nati Sectio. De Bononiensi Scientarium et Artium Instituto atque Academia Commentarii. Bononiae. 7: 419–428
Harnsberger HR, Dahlen RT, Shelton C, Gray SD, Parkin JL 1995 Advanced techniques in magnetic resonance imaging in the evaluation of the large endolymphatic duct and sac syndrome. Laryngoscope 105: 1037–1042
Griffith AJ, Arts A, Downs C, Innis JW, Shepard NT, Sheldon S, Gebarski SS 1996 Familial large vestibular aqueduct syndrome. Laryngoscope 106: 960–965
Okamoto K, Ito J, Furusawa T, Sakai K, Horikawa S, Tokiguchi S 1998 MRI of enlarged endolymphatic sacs in the large vestibular aqueduct syndrome. Neuroradiology 40: 167–172
Valvassori GE, Clemis JD 1978 The large vestibular aqueduct syndrome. Laryngoscope 88: 723–748
Walsh RM, Ayshford CA, Chavda SV, Proops DW 1999 Large vestibular aqueduct syndrome. ORL J Otorhinolaryngol Relat Spec 61: 41–44
Li XC, Everett LA, Lalwani AK, Desmukh D, Friedman TB, Green ED, Wilcox ER 1998 A mutation in PDS causes non-syndromic deafness. Nat Genet 18: 215–217
Scott DA, Karnisky LP 2000 Human pendrin expressed in Xenopus laevis oocytes mediates chloride/formate exchange. Am J Physiol 278: C207–C211
Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED 2001 Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 98: 4221–4226
Antonarakis SE, Nomenclature Working Group 1998 Recommendations for a nomenclature system for human gene mutations. Hum Mutat 11: 1–3
Reardon W, O'Mahoney CF, Trembath R, Jan H, Phelps PD 2000 Enlarged vestibular aqueduct: a radiological marker of Pendred syndrome, and mutation of the PDS gene. Q J Med 93: 99–104
Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB, Galer CE, Karniski LP, Sheffield VC, Smith RJ 2001 Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat 17: 403–11
Krawczak M, Reiss J, Cooper DN 1992 The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 90: 41–54
Cooper DN, Yousofian H 1998 The CpG dinucleotide and human genetic disease. Hum Genet 78: 151–155
Steel KP, Bussoli TJ 1999 Deafness genes: expression of surprise. Trends Genet 15: 207–211
Hariri MA 1993 Autoimmune inner-ear disease. J Audiol Med 2: 41–52
Friis J, Johnsen T, Feldt-Rasmussen U, Bech K, Friis T 1988 Thyroid function in patients with Pendred's syndrome. J Endocrinol Invest 11: 97–101
Mayot D, Bene MC, Dron K, Parrin C, Faure GC 1993 Immunologic alterations in patients with sensorineural hearing disorders. Clin Immunol Immunopathol 68: 41–45
O'Mahoney CF, Luxon LM, Chew SL, Wass JAH 1996 When the triad of congenital hearing loss, goitre and perchlorate positive is not Pendred syndrome. J Audiol Med 5: 157–165
Acknowledgements
The authors thank Dr. Luca Persani for critical comments and advice.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fugazzola, L., Cerutti, N., Mannavola, D. et al. Differential Diagnosis between Pendred and Pseudo-Pendred Syndromes: Clinical, Radiologic, and Molecular Studies. Pediatr Res 51, 479–484 (2002). https://doi.org/10.1203/00006450-200204000-00013
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200204000-00013
This article is cited by
-
Mutation analysis of SLC26A4 (Pendrin) gene in a Brazilian sample of hearing-impaired subjects
BMC Medical Genetics (2018)
-
The R130S mutation significantly affects the function of prestin, the outer hair cell motor protein
Journal of Molecular Medicine (2016)
-
Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents
Human Genetics (2016)
-
Genetic mutations in nonsyndromic deafness patients of Chinese minority and han ethnicities in Yunnan, China
Journal of Translational Medicine (2013)
-
Extremely discrepant mutation spectrum of SLC26A4 between Chinese patients with isolated Mondini deformity and enlarged vestibular aqueduct
Journal of Translational Medicine (2011)
