
Abstract
In seven patients with mucopolysaccharidoses (1 Hurler, 1 Hurler-Scheie, 4 Hunter, 1 Sly), cranial 1H-magnetic resonance spectroscopy was performed to evaluate the accumulation of mucopolysaccharides and biochemical changes in the CNS in vivo before and after bone marrow transplantation (BMT). In two of seven patients, 1H-magnetic resonance spectroscopy was performed before and after BMT. Nuclear magnetic resonance spectra of dermatan sulfate and chondroitin sulfate-C and magnetic resonance spectroscopy of chondroitin sulfate-C and urine from patients with mucopolysaccharidoses showed resonance higher than the chemical shift of myoinositol in the brain (3.7 ppm). The resonance was considered to contain signals from mucopolysaccharide molecules. The resonance was measured as presumptive mucopolysaccharides (pMPS). In white matter lesions detected by magnetic resonance imaging, pMPS/creatine ratios and choline/creatine ratios were consistently higher than control ratios. In white matter without lesions, choline/creatine ratios were higher than control ratios. Patients with higher developmental quotient or intelligence quotient tended to show higher N-acetylaspartate/creatine ratios and lower pMPS/creatine ratios in basal ganglia. After BMT, the pMPS/creatine ratio in white matter lesions of patient 3, with Hunter syndrome, was slightly decreased, but in none of the patients was the ratio ever below the control ratios, even 7 y after BMT. In white matter without lesions, the pMPS/creatine ratio in patient 3 was decreased to the control ratios after BMT, but although the choline/creatine ratios were gradually decreased, they remained higher than the control ratio, 2 y after BMT. These results suggest that evaluation of pMPS, choline, and N-acetylaspartate by 1H-magnetic resonance spectroscopy is an important technique that may provide useful biochemical information in vivo on the neurologic process and the efficacy of BMT in patients with mucopolysaccharidoses.
Similar content being viewed by others

Main
Mucopolysaccharidosis is a lysosomal storage disease involving the visceral organs, bones, CNS, and other tissues (1). Patients exhibit serious clinical manifestations that cause various disturbances in daily life, including mental deterioration in some subtypes of mucopolysaccharidoses (1). For a long time, there was no effective treatment, until Hobbs, in 1981 (2), performed successful BMT in a patient with Hurler syndrome. Since then, many patients have been treated by BMT (3–6), which in the early stage of the disease can reduce the accumulation of MPS in the visceral organs. Recently, ERT for mucopolysaccharidoses is undergoing clinical trial (7–9). However, the clinical and biochemical effects of BMT and ERT on the CNS are not clear, because the in vivo analysis of accumulated MPS in the CNS has not been possible.
Localized proton MRS of the brain has become an additional diagnostic tool for use in patients with neurodegenerative or metabolic disorders (10–17). Cranial MRS can detect the metabolites, myoinositol, choline, creatine, NAA, lactate, and others, and suggest the accumulation of these compounds and the neuronal damage they can cause. Although cranial MRS has been performed in patients with mucopolysaccharidoses, a previous report touched only on changes of choline or NAA (18). A study on the analysis of chondroitin sulfate by NMR spectroscopy revealed resonance from MPS molecules (19). Using the noninvasive technique of MRS, we tried to establish the measurement of MPS in the CNS in vivo to evaluate the effect of treatment on mucopolysaccharidoses.
METHODS
Patients.
We studied seven patients with mucopolysaccharidoses (Table 1). The diagnosis was made by the urinary secretion pattern of MPS and lysosomal enzyme assay (20–22). The informed consents for the examinations were obtained from patients or their parents. The extent and morphology of brain lesions on MRI were shown in Table 1. White matter lesions in patients with Hurler syndrome, Hurler-Scheie compound, or Hunter syndrome were small and round, with T2 high intensity. Basal ganglia lesions in patient 6 were honeycomb-shaped, with T2 high intensity. DQ and IQ were measured by Kinder Infancy Developmental Scale, WISC-R, or WAIS-R.
Proton NMR spectroscopy.
Proton NMR spectroscopy of the standard MPS, DS (10 mg/mL in 95% H2O/5%2H2O) and CS-C (100 mg/mL in 95% H2O/5%2H2O) was performed with VARIAN INOVA 500 (Varian, Inc, Palo Alto, CA, U.S.A.) at room temperature. Presaturation of water resonance was performed before acquisition.
MRS of standard MPS and urine.
MRS of standard CS-C and urine from the patients was performed at 1.5 T with a conventional whole-body MRI system (Signa Horizon 1.5T, General Electric, Milwaukee, WI, U.S.A.). The standard solution and urine in 50-mL tubes were placed in the conventional whole-body MRI system and measured.
Cerebral MRS.
Localized proton MRS of the brain in patients with mucopolysaccharidosis was performed at 1.5 T with a conventional whole-body MRI system. Voxels of 15 × 15 × 15 mm were positioned over the white matter lesions, the homotopic contra lateral area without lesions, and the basal ganglia. Water resonance was suppressed by means of the point-resolved spectroscopy (PRESS) technique, and the water-suppressed MRS examination was followed by an unsuppressed MRS measurement, performed at a transmission time of 2000 ms and an echo time of 30 ms. Control subjects were patients with localization-related epilepsy, and did not manifest any neurologic signs at interictal period. The mean age was 8.2 ± 3.4 y (n = 5). MRS was performed over brain regions without atrophy or an epileptogenic focus.
Quantification of brain metabolites.
An image scanner (Sharp JX330M, SHARP, Osaka) and computer software (Photoshop, Adobe, San Jose, CA, U.S.A.) were used to transfer the MRS images to a personal computer (Macintosh), and computer software (National Institutes of Health Image 1.55) was used to analyze the area of each peak on the MRS images. Peaks of NAA, choline, creatine, and myoinositol spectra were identified by their chemical shifts. Areal ratios of each peak to creatine were measured to evaluate the quantity of metabolites in the brain (Fig. 1A).

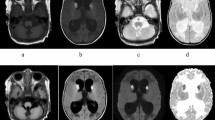
Quantification of the metabolites pMPS, choline, creatine, and NAA by MRS. A, Resonances of pMPS are composed of peaks with chemical shifts higher than those of myoinositol. Choline, creatine, and NAA have peak signals at 3.20, 3.041, and 2.024, respectively. Raw MRS data from a white matter lesion (B), white matter without lesion (C), and left basal ganglia (D), at the age of 5 y 2 mo in patient 3. The highest peak of pMPS is higher than the peak of myoinositol in white matter lesion (B), and is lower than that in white matter without lesion (C). Positions of voxels are shown on the MRI insets.
Informed consent was obtained from all participants, and the Institutional Review Board of the Gifu University Hospital approved the study.
RESULTS
In patient 1, with Hurler syndrome, the DQ increased from 77 to 105, and lesions in the white matter were slightly reduced, although dilatation of ventricles was aggravated transiently after BMT. Patient 2, with Hurler-Scheie compound, had few white matter lesions at 7 y after BMT. In patient 3, with severe Hunter syndrome, the DQ and ventricular dilatation worsened after BMT. In patients 4, 5, and 6, with Hunter syndrome, DQ or IQ was not involved, but all had white matter or basal ganglia lesions. Patient 7, with Sly disease, became able to ride a bicycle and diffuse T2 high-intensity lesions in white matter disappeared after BMT.
NMR spectra of DS and CS-C showed a common resonance at approximately 2.0 ppm that was characteristic of the N-acetyl group protons of GalNAc, and variable resonance around chemical shifts of 3.7 ppm (Fig. 2). Although the resonance at approximately 3.7 ppm is not assigned to a specific residue of MPS because of its molecular complexity, it contains characteristic signals derived from MPS molecules.

NMR spectra of CS-C and DS, measured by VARIAN INOVA 500 spectrometer. NAC, N-acetyl group protons of Ga1NAc.
MRS of CS-C had broad resonance, with a peak signal at approximately 3.7 ppm (Fig. 3A). The spectrum of urine from control subjects, containing CS-C as a main MPS component, showed four major signals, one of which peaked at approximately 3.7 ppm, and the signal has morphology similar to that of CS-C (Fig. 3B). Considering the characteristic resonance of MPS at approximately 3.7 ppm in NMR spectra, it seems likely that resonance at approximately 3.7 ppm in MRS also contains signals from MPS.

MRS of CS-C (A), control urine (B), urine from patients (C–H), and white matter lesion in patient 1 (I), measured by Signa Horizon 1.5 T (General Electric).
MRS of urine from patients (mucopolysaccharidoses I–IV, VI, and VII) was measured (Fig. 3C–H). The spectrum of urine from every patient had a peak at approximately 3.7 ppm, and the morphology of the peak varied according to the subtypes of mucopolysaccharidoses, probably owing to the variable composition of MPS in the urine. These results suggest that MRS can detect resonance from MPS in urine at around 3.7 ppm.
Cranial MRS in patient 1, with Hurler syndrome, showed resonance higher than the chemical shift of myoinositol in the brain (3.626 ppm) (Fig. 3I). The resonance was similar to that in the urine (Fig. 3C). Cranial MRS in patient 3, with Hunter syndrome, also showed resonance higher than the chemical shift of myoinositol in the brain (Fig. 1B–D). These results suggest that MRS can detect resonance from MPS in the brain at the chemical shift at approximately 3.7 ppm.
Because resonance higher than chemical shifts of myoinositol was considered to contain the resonance from MPS molecules in the brain, we measured the area of resonance as pMPS (Fig. 1A). The ratios of metabolites to creatine are shown in Table 2, and the raw data of MRS in patient 3, with Hunter syndrome, are shown in Figure 1B–D.
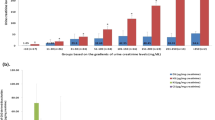
In white matter lesions, pMPS/creatine ratios were consistently higher than those of control ratios (Fig. 4A, p = 0.019). In white matter without lesions before BMT, two of three pMPS/creatine ratios exceeded the control ratio (patients 3 and 5), but one (patient 4, mild form of Hunter syndrome) was within the range of control ratios (Table 2 and Fig. 4B). After BMT, three of five pMPS/creatine ratios were within the control ratio range, and two were slightly higher than the control ratio. Although the mean NAA/creatine ratio in white matter lesions was not significantly different from that in white matter without lesions, the mean pMPS/creatine ratio and the mean pMPS/NAA ratio in white matter lesions were significantly higher than that in white matter without lesions (p = 0.010 and p = 0.041, Mann-Whitney U test).

Relationships between DQ or IQ and pMPS/creatine ratios in white matter lesions (A) or white matter without lesions on MRI (B).
In white matter lesions and white matter without lesions, all choline/creatine ratios were higher than the control ratios, and the mean choline/creatine ratio (1.17 ± 0.08) in white matter lesions was not significantly different from that in white matter without lesions (0.99 ± 0.05;p = 0.12, Mann-Whitney U test;Table 2).
In basal ganglia, the relationships between DQ or IQ and the ratios of NAA/creatine, pMPS/creatine, and pMPS/NAA are shown in Figure 5. There tended to be an association between higher DQ or IQ and higher NAA/creatine ratios and lower pMPS/creatine and pMPS/NAA ratios, but the correlation was not significant (r = 0.39, −0.04, −0.26, respectively; Spearman's correlation coefficient by rank test).

Relationships between DQ or IQ and NAA/creatine ratios, pMPS/creatine ratios, and pMPS/NAA ratios in basal ganglia of patients with mucopolysaccharidoses. Lines show least squares regression lines.
The time course of the pMPS/creatine ratio before and after BMT is shown in Figure 6. After BMT, in white matter lesions, the ratio was decreased in patient 3 (Hunter syndrome), but in none of the patients was the ratio ever below control ratio, even 7 y after BMT. In white matter without lesions, pMPS/creatine ratio was decreased to the control ratios after BMT in patient 3, and in the remainder of the patients the ratio after BMT was within or slightly higher than the control ratios. The time course of the choline/creatine ratio in white matter of patients 1 and 3 showed that the ratio gradually decreased after BMT, but remained higher than the control ratio, 2 y after BMT (Table 2).

Time course of pMPS/creatine ratio before and after BMT in white matter lesions and white matter without lesions on MRI. Shaded region shows range of pMPS/creatine ratios in control subjects. The data were obtained before and after BMT in patient 3 and after BMT in patients 1, 2, and 7.
DISCUSSION
We used NMR data from MRS to evaluate the accumulation of MPS in the CNS in vivo. The results suggest that resonance higher than the chemical shift of myoinositol in MRS contains resonance from MPS molecules, and that the resonance can be detected by cranial MRS with a conventional MRI system. Because the resonance also contained signals derived from molecules other than MPS, we named the resonance pMPS.
We could not quantify the accumulated MPS itself, but by measuring pMPS/creatine ratios we were able to evaluate the accumulation of MPS in the CNS in vivo. pMPS/creatine ratios and choline/creatine ratios in white matter lesions were elevated before BMT and slightly decreased thereafter. Increased choline/creatine ratios usually suggest that the examined region contains myelin damage, increased cell membrane synthesis, or glial proliferation (23–27). Histologic reports of mucopolysaccharidosis also describe gliosis and demyelination around the lesions in white matter (28–30). Therefore, our findings of increased pMPS/creatine ratios and choline/creatine ratios in white matter lesions are compatible with those of previous pathologic or neuroimaging studies.
Some pMPS/creatine ratios in white matter without lesions were higher than control ratios and others were not. The mean pMPS/creatine ratio in white matter was significantly lower than that in white matter lesions. Choline/creatine ratios were elevated in both white matter and white matter lesions. These results suggest that MPS accumulates and related histologic changes (gliosis and/or demyelination) occur in white matter even before the development of lesions on MRI. Then, as the accumulation of MPS increases to some critical level, white matter lesions develop.
The time course of pMPS/creatine ratios before and after BMT suggests that accumulated MPS in white matter lesions cannot be normalized and that accumulated MPS in white matter without lesions can be normalized. Choline/creatine ratios in white matter were not normalized after BMT. Therefore, early treatment with BMT or ERT, before the development of lesions in the CNS, might contribute to a better neuropsychological outcome in mucopolysaccharidoses type I or II. In patient 1, with Hurler syndrome, early BMT, at age 2 y 2 mo, when dilatation of the ventricles was slight, brought a better outcome; the DQ increased from 77 to 105. In patient 3, with severe Hunter syndrome, late BMT, at age 4 y 8 mo, when dilatation of the ventricles had progressed, resulted in a poor neuropsychological outcome; DQ and ventricular dilatation worsened.
NAA is distributed in the brain primarily in neurons and neuronal processes (31). In basal ganglia, patients with higher DQ or IQ tended to have higher NAA/creatine ratios and lower pMPS/creatine ratios. This might suggest that the accumulation of MPS in neurons contributes to neuronal damage, resulting in decreased NAA/creatine ratios and DQ or IQ. In patient 1, who had a better outcome after BMT, the NAA/creatine ratio was 1.20 before BMT, but in patient 3, who had a poor outcome, the ratio was 0.93 before BMT. This might suggest that the NAA/creatine ratio in basal ganglia is important in estimating the effect of BMT or ERT. We must await further investigation before reaching conclusions.
Although our findings on MPS, NAA, and choline in the CNS have not been proved by biochemical analyses of brain tissue, their evaluation by 1H-MRS is an important technique that may provide useful biochemical information on the neurologic process and an indication of treatment in patients with mucopolysaccharidoses.
Abbreviations
- BMT:
-
bone marrow transplantation
- MPS:
-
mucopolysaccharide
- ERT:
-
enzyme replacement therapy
- MRS:
-
magnetic resonance spectroscopy
- NAA:
-
N-acetylaspartate
- DS:
-
dermatan sulfate
- CS-C:
-
chondroitin sulfate-C
- DQ:
-
developmental quotient
- IQ:
-
intelligence quotient
- pMPS:
-
presumptive MPS
- NMR:
-
nuclear magnetic resonance
- MRI:
-
magnetic resonance imaging
References
Neufeld EF, Muenzer J 1995 The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Bases of Inherited Disease, 7th Ed. McGraw-Hill, New York, pp 2465–2494
Hobbs JR 1981 Bone marrow transplantation for inborn errors. Lancet 2: 735–739
Hobbs JR 1990 Displacement bone marrow transplantation for some inborn errors. J Inherit Metab Dis 13: 572–596
Cowan MJ 1991 Bone marrow transplantation for the treatment of genetic diseases. Clin Biochem 24: 375–381
Whitley CB, Belani KG, Chang P, Summers CG, Blazar BR, Tsai MY, Latchaw RE, Ramsay NKC, Kersey JH 1993 Long-term outcome of Hurler syndrome following bone marrow transplantation. Am J Med Genet 46: 209–218
Yamada Y, Kato K, Sukegawa K, Tomatsu S, Fukuda S, Emura S, Kojima S, Matsuyama T, Sly WS, Kondo N, Orii T 1998 Treatment of MPS VII (Sly disease) by allogeneic BMT in a female with homozygous A619V mutation. Bone Marrow Transplant 21: 629–634
Shull RM, Kakkis ED, McEntee MF, Kania SA, Jonas AJ, Neufeld EF 1994 Enzyme replacement in a canine model of Hurler syndrome. Proc Natl Acad Sci USA 91: 12937–12941
Kakkis ED, McEntee MF, Schmidtchen A, Neufeld EF, Ward DA, Gompf RE, Kania S, Bedolla C, Chien SL, Shull MR 1996 Long-term and high-dose trials of enzyme replacement therapy in the canine model of mucopolysaccharidosis I. Biochem Mol Med 58: 156–167
Kakkis ED, Muenzer J, Tiller J, Waber L, Belmont J, Passage M, Izykowski B, Phillips J, Walot J, Doroshow R, Hoft R, Yu KT, Okazaki S, Lewis D, Lachman R, Neufeld EF 1999 Recombinant α-l-iduronidase replacement therapy in mucopolysaccharidosis the canine model of mucopolysaccharidosis. I: Results of a human clinical trial. Abstracts of the 5th International Symposium on Mucopolysaccharide and Related Diseases. Vienna, March 18–21, p 104(abstr)
Grodd W, Krägeloh-Mann I, Klose U, Sauter R 1991 Metabolic and destructive brain disorders in children: findings with localized proton MR spectroscopy. Radiology 181: 173–181
Bruhn H, Kruse B, Korenke GC, Hanefeld F, Hänicke W, Merboldt KD, Frahm J 1992 Proton NMR spectroscopy of cerebral metabolic alterations in infantile peroxisomal disorders. J Comput Assist Tomogr 16: 335–344
Aria Tzika A, Ball WS, Vigneron DB, Dunn RS, Kirks DR 1993 Clinical proton MR spectroscopy of neurodegenerative disease in childhood. AJNR Am J Neuroradiol 14: 1267–1281
Bergman AJI, Van Der Knaap MS, Smeitink JAM, Duran M, Dorland L, Valk J, Poll-The BT 1996 Magnetic resonance imaging and spectroscopy of the brain in propionic acidemia: clinical and biochemical considerations. Pediatr Res 40: 404–409
Ashwal S, Holshouser BA, Tomashi LG, Shu S, Perkin RM, Nystrom GA, Hinshaw DB 1997 1H-magnetic resonance spectroscopy-determined cerebral lactate and poor neurological outcomes in children with central nervous system disease. Ann Neurol 41: 470–481
Seitz D, Grodd W, Schwab A, Seeger U, Klose U, Nägele T 1998 MR imaging and localized proton MR spectroscopy in late infantile neuronal ceroid lipofuscinosis. AJNR Am J Neuroradiol 19: 1373–1377
Pavlakis SG, Kingsley PB, Kaplan GP, Stacpoole PW, O'Shea M, Lustbader D 1998 Magnetic resonance spectroscopy: use in monitoring MELAS treatment. Arch Neurol 55: 849–852
Novotony E, Ashwal S, Shevell M 1998 Proton magnetic resonance spectroscopy: an emerging technology in pediatric neurology research. Pediatr Res 44: 1–10
Seto T, Tanaka A, Furukawa K, Hamazaki K, Baba K, Hayashi K, Hattori E, Matsuoka O, Mototsuka K, Inoue Y, Ishiki G 1998 Cranial MRS in congenital mucopolysaccharidoses. Nihon Shounikagakkaizasshi 102: 295 (in Japanese)
Nadanaka S, Sugahara K 1997 The unusual tetrasaccharide sequence GlcAβ1–3GalNac(4-sulfate) β1–4GlcA(2-sulfate)β1–3GalNac(6-sulfate) found in the hexasaccharides prepared by testicular hyaluronidase digestion of shark cartilage chondroitin sulfate D. Glycobiology 7: 253–263
Huang K-C, Sukegawa K, Orii T 1985 Glycosaminoglycan excretion in random samples of urine. Clin Chim Acta 151: 141–146
Huang K-C, Sukegawa K, Orii T 1985 Screening test for urinary glycosaminoglycans and differentiation of various mucopolysaccharidoses. Clin Chim Acta 151: 147–156
Sukegawa K, Song XQ, Masuno M, Fukao T, Shimozawa N, Fukuda S, Isogai K, Nishio H, Matsuo M, Tomatsu S, Kondo N, Orii T 1997 Hunter disease in a girl caused by R468Q mutation in the iduronate-2-sulfatase gene and skewed inactivation of the X chromosome carrying the normal allele. Hum Mutat 10: 361–367
Davie CA, Hawkins CP, Barker GJ, Brennan A, Tofts PS, Miller DH, McDonald WI 1994 Serial proton magnetic resonance spectroscopy in acute multiple sclerosis lesions. Brain 117: 49–58
Kugel H, Heindel W, Ernestus R, Bunke J, du Mesnil R, Friedmann G 1992 Human brain tumors: spectral patterns detected with localized H-1 MR spectroscopy. Radiology 183: 701–709
Negendank W 1992 Studies of human tumors by MRS: a review. NMR Biomed 5: 303–324
Connelly A, Jackson GD, Duncan JS, King MD, Gadian DG 1994 Magnetic resonance spectroscopy in temporal lobe epilepsy. Neurology 44: 1411–1417
Cross JH, Gordon I, Connelly A, Jackson CL, Neville BG, Gadian DG 1997 Interictal 99mTc HMPAO SPECT and 1H MRS in children with temporal lobe epilepsy. Epilepsia 38: 338–345
Lee C, Dineen TE, Brack M, Kirsch JE, Runge VM 1993 The mucopolysaccharidoses: characterization by cranial MR imaging. AJNR Am J Neuroradiol 14: 1285–1292
Murata R, Nakajima S, Tanaka A, Miyagi N, Matsuoka S, Inoue Y 1989 MR imaging of the brain in patients with mucopolysaccharidosis. AJNR Am J Neuroradiol 10: 1165–1170
Shimamura K, Hakozaki H, Takahahsi K, Kimura A, Fujino J 1976 Sanfilippo B syndrome: a case report. Acta Pathol Jpn 26: 739–764
Birken DL, Oldendorf WH 1989 N-acetyl-aspartic acid: a literature review of a compound prominent in 1H-NMR spectroscopic studies of brain. Neurosci Behav Rev 13: 23–31
Author information
Authors and Affiliations
Additional information
Supported in part by a Grant-in-Aid for Scientific Research (C) No. 10670722 and Research Grants (10A-1 and 12B-2) for Nervous and Mental Disorders from the Ministry of Health and Welfare.
Yukitoshi Takahashi, M.D., Department of Pediatrics, Gifu Prefectural Gifu Hospital, 4-6-1, Noishiki Gifu, 500-8717, Japan.
Rights and permissions
About this article
Cite this article
Takahashi, Y., Sukegawa, K., Aoki, M. et al. Evaluation of Accumulated Mucopolysaccharides in the Brain of Patients with Mucopolysaccharidoses by 1H-Magnetic Resonance Spectroscopy before and after Bone Marrow Transplantation. Pediatr Res 49, 349–355 (2001). https://doi.org/10.1203/00006450-200103000-00008
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200103000-00008
This article is cited by
-
Clinical 1H MRS in childhood neurometabolic diseases — part 2: MRS signatures
Neuroradiology (2022)
-
Mucopolysaccharidoses: overview of neuroimaging manifestations
Pediatric Radiology (2018)
-
Imaging findings of mucopolysaccharidoses: a pictorial review
Insights into Imaging (2013)
-
Proton nuclear magnetic resonance spectroscopic detection of oligomannosidic n glycans in alpha‐mannosidosis: a method of monitoring treatment
Journal of Inherited Metabolic Disease (2011)
-
Neurological findings in Hunter disease: Pathology and possible therapeutic effects reviewed
Journal of Inherited Metabolic Disease (2008)
