
Abstract
The treatment of multiple acyl-CoA-dehydrogenase deficiency (MADD) includes a low-fat, low-protein, high-carbohydrate diet, avoiding long fasting periods. However, there is no useful biochemical marker to determine the response to different diets or fasting periods. The aims of this study are to report a patient with MADD, diagnosed through a newborn screening program using tandem mass spectrometry, to assess her response to different feedings, and to evaluate the usefulness of acylcarnitines and FFA to monitor the response to dietary changes. The patient was diagnosed at 6 d. Family history revealed three dead siblings. Five tests were performed, one with breast milk and the subsequent four after giving the patient a bottle of a low-fat, low-protein formula (F), F with glucose polymers (GP), F+GP plus uncooked corn starch (CS), or F+GP+CS preceded by amylase. The results showed that acylcarnitines, FFA, and total nonesterified fatty acids levels were greatly improved at 2 and 4 h on F+GP compared with breast milk. At 6 mo of age, the test with F+CS was repeated to assess the response to a longer fast. The results were similar at 2 and 4 h, but showed a marked increase of acylcarnitines, FFA, and total nonesterified fatty acids at 6 h. The increase of these metabolites could not be avoided by the use of F+GP+CS, but was prevented when amylase was used simultaneously. The patient is currently 3.9 y old and has normal growth and development. We conclude that diagnosis of MADD through a newborn screening program using tandem mass spectrometry is suitable; acylcarnitines and FFA are useful to monitor the response to treatment; and exogenous amylase allows the use of CS in small children with MADD. This therapeutic approach may be an alternative to the use of continuous overnight feedings used for young children with severe fatty acid oxidation defects. Early diagnosis and treatment may change the natural history of MADD.
Similar content being viewed by others

Main
MADD is a disorder of fatty acid and protein metabolism caused by a defect of the electron transfer flavoprotein or the electron transfer flavoprotein ubiquinone oxidoreductase. Patients with MADD have been classified in three groups: neonatal onset with congenital anomalies, neonatal onset without anomalies, and mild or later onset (1). Patients in the first group are often premature and present soon after birth with hypotonia and hepatomegaly. Some dysmorphic features may be present: high forehead, hypoplastic midface, wide-open anterior fontanel, defects of the anterior abdominal wall, abnormal genitalia, and renal cysts. Most patients die within the first week of life (1, 2). Patients in the second group do not have congenital anomalies, but their clinical presentation is otherwise similar to the first one. Children in this group usually develop severe cardiomyopathy and die during the first few weeks of life (1). The course and presentation of the late-onset patients is extremely variable, ranging from episodes of vomiting with hypoglycemia and hepatomegaly to progressive lipid storage myopathy in adulthood (3–5).
The diagnosis of MADD is suspected on the basis of the clinical and biochemical presentation. Common laboratory abnormalities are hypoglycemia with hypoketosis and mild metabolic acidosis. Analysis of the organic acids or acylglycines in urine shows a characteristic profile. However, these abnormalities may be present only when patients are under stress (6). More recently, the analysis of AC by MS-MS has been used for the biochemical diagnosis of MADD (7, 8). This method is also being used for neonatal diagnosis through newborn screening programs. However, its sensitivity for the neonatal detection of MADD has been questioned, and, to our knowledge, only one patient has been reported to be prospectively diagnosed in a newborn screening program (9).
The recommended long-term treatment of MADD includes riboflavin, carnitine, or glycine, and a low-fat, low-protein, high-carbohydrate diet, avoiding long periods of fasting (1, 2, 10–13). However, assessment of the patients' metabolic status when they are not in crisis is difficult because there is no useful biochemical marker to determine the response to different dietary treatments or periods of fasting. Additionally, there is limited information about the long-term outcome of these patients.
The aims of this study are to report the diagnosis of a patient with MADD through a newborn screening program using MS-MS, to assess her biochemical response to different feeds during short fasting periods, and to evaluate the usefulness of AC and FFA to monitor the response to these dietary changes.
METHODS
Patient
Family history.
There was a history of consanguinity and three dead siblings: the first one (boy) died, unexpectedly, at age 7 d; the second one (girl) died at 1 y of age with a sepsis-like episode. The third one (boy) presented at the age of 9 mo with a Reye-like syndrome. Diagnosis of a severe MADD was established at the time on the basis of the clinical presentation and a typical organic acid profile. Treatment with riboflavin and carnitine was started, but the patient died a few days later.
Initial presentation.
The patient was the female product of a full-term pregnancy. She was born via cesarean section with a birth weight of 3400 g. Physical examination was normal except for a wide-open anterior fontanel and a high forehead. A Guthrie card sample was sent at age 6 d to our laboratory for newborn screening, while patient was asymptomatic. Diagnosis was established on the following day, and treatment with carnitine (Albicar, Casasco, Buenos Aires, Argentina, 100 mg·kg−1·d−1, divided in three doses) was immediately started. Riboflavin (200 mg/d, divided in two doses) was added 4 d later. Pretreatment glucose, liver enzymes, and riboflavin levels, as well as renal ultrasound, were normal.
Diet
From birth until the age of 3 mo, the patient was breast-fed every 3–4 h. From 3 mo on, the patient has been on treatment with a low-fat (15%), low-protein (7%), high-carbohydrate (78%) diet, providing 90–120 calories·kg−1·d−1 (adjusted according to age and weight gain). From 3 to 6 mo of age, the diet was given as F feedings every 4 h.
After 6 mo of age, semisolids were started, and at 9 mo uncooked CS was gradually incorporated. After the results of test number 4 were available (see below), pancreatic enzymes (Prolipase, Janssen-Cilag, Buenos Aires, Argentina), at a dose of 40,000 IU of A, were given before the F.
Protocol
To assess the patient's response to different feeds and fasting periods, five tests were performed during the first year of life. The initial one was done while the patient was receiving BM. The following four tests were performed by giving the patient a bottle, prepared with F (Similac, Abbot, Buenos Aires, Argentina), 1.2 g/kg, a low-fat milk (Molico, Nestlé, Buenos Aires, Argentina), 0.7 g/kg, and different types of carbohydrates: GP (Polimerosa, Kasdorf, Buenos Aires, Argentina), 3 g/kg, or GP +CS, 1.5 g/kg each. This F feed provided 20 calories/kg (approximately one sixth of the total caloric intake) with the recommended caloric distribution for MADD: carbohydrates, 78%; protein, 7%; and fat, 15%. The tests were performed as outlined in Table 1. The protocol was approved by the ethics committee of the Fundación para el Estudio de las Enfermedades Neurometabólicas. An informed consent was obtained from the mother before each test. All the tests were performed while the patient was clinically stable, without any sign of intercurrent illness. Liver enzymes and creatine kinase (obtained in the basal sample of each test) were normal. A heparin lock was maintained throughout each test, and blood was obtained for AC, NEFA, insulin, glucose, 3-hydroxybutyrate, amino acids, and total and free carnitine.
Measurements
All the studies were performed at the Fundación para el Estudio de las Enfermedades Neurometabólicas. For AC analysis, blood samples were spotted in a filter paper (Schleicher & Schuell 903, Keene, NH, U.S.A.) and allowed to dry at room temperature. Sample preparation and analysis were performed as described (14, 15) in a Quattro II, triple quadrupole mass spectrometer (Micromass, U.K.) with electrospray. Acquisition was performed scanning for ions parents of m/z 85 (butyl esters). Free, acetyl- (C2), propionyl- (C3), butyryl- (C4), isovaleryl- (C5), octanoyl- (C8), decanoyl- (C10), dodecanoyl- (C12), tetradecanoyl- (C14), hexadecanoyl- (C16), and octadecanoyl- (C18) carnitines were quantitated with their corresponding isotopes. Hexanoylcarnitine (C6) was quantitated using the C8 isotope. Labeled internal standards were obtained from Dr. Herman J. Ten Brink (Academic Hospital V.U., Amsterdam, The Netherlands). Normal values for the newborn period were obtained from 841 normal neonates screened in our program. Normal values for older age were obtained from 711 children who had normal levels of amino acids, AC, and carnitine in blood and organic acids in urine. None of these controls received a special diet, and blood samples were obtained after their regular fasting period.
Oxidation of [9,10(n)-3H]palmitic and myristic acids in lymphocytes was performed as described (16). The method of Saudubray et al. (17), adapted for lymphocytes, was used for the oxidation of [14C]butyric, [14C]octanoic, and [14C]palmitic. Samples for these assays were obtained while the patient was already receiving riboflavin. When enough sample was available, levels of the individual FFA of carbon length C6, C8, C10, C12, C14, C16, and C18 were measured in plasma by gas chromatography–mass spectrometry, using the method of Costa et al.(18). NEFA, 3-hydroxybutyrate, amino acids, and carnitine were measured as described (19–22). Pearson's correlation was used for statistical analysis.
RESULTS
The newborn screening profile showed low levels of C2 and elevations of short-, medium-, and long-chain AC, except for C3 and C16 (Table 2). The unsaturated species C12:1, C14:1, C16:1, and C18:1 were also elevated. No significant changes were observed after treatment with carnitine, except for an increase in C2 levels and a decrease in the long-chain AC, probably age related. Results of AC measured 7 d after treatment with riboflavin showed that, on average, AC values were slightly lower than those obtained before treatment, but remained elevated compared with normal values (Table 2).
The oxidation of tritiated fatty acids in lymphocytes was apparently more sensitive than the 14C substrate oxidations (15– 20%versus 34– 41%, respectively;Table 3).
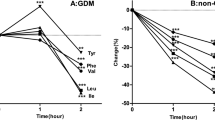
To compare the AC, FFA, NEFA, and insulin responses during the tests, we calculated the sum of the AC C6, C8, C10, C12, C14, C16, and C18 and that of the corresponding FFA for each fasting time. Results are shown in Table 4 and Figure 1. The tests, performed at age 3 mo, revealed that when the patient received BM (test number 1), the AC, FFA, and NEFA were elevated, whereas insulin levels tended to be low. When the patient was given F+GP (test number 2), AC, FFA, and NEFA markedly decreased. This response was associated with an increment in the insulin levels. Test number 3, performed at 6 mo with a similar feed (F+GP), showed slight elevations of FFA and NEFA at 2 and 4 h, with a clear increase of AC, FFA, and NEFA in the 6-h sample (≥100%versus the 4-h values). Insulin levels tended to decrease throughout the test and were lower than those of test number 2. In an attempt to avoid the increase of AC, FFA, and NEFA at 6 h, uncooked complex carbohydrates (CS) were gradually added to the diet, maintaining the total amount of carbohydrates unchanged. However, when the feed with F+GP+CS was tested (test number 4), the AC and NEFA showed mild elevations at 2 and 4 h, with a marked increase in the 6-h sample (near 200%versus the 4-h values). Levels of insulin in that test were lower than in the previous one for each fasting time. To determine whether this unexpected response was caused by the patient's inadequate A activity, the test was repeated giving the patient A right before the formula (test number 5, F+GP+CS+A). The results showed an improvement of AC, FFA, and NEFA at 2 and 4 h without increase at 6 h. Insulin levels were higher than those obtained in tests 3 and 4, at 2, 4, and 6 h of fasting. Levels of 3-hydroxybutyrate were slightly increased in test number 1 only. No hypoglycemia was detected during the tests. Total carnitine values were similar in all tests, but the percentage of free carnitine was low in the test with BM.

Results of the five tests. Test number 1 was performed while the patient was receiving BM. The following tests (numbers 2–5) were performed by giving the patient a bottle of F with the recommended nutrient composition for MADD: carbohydrates, 78%; protein, 7%; and fat, 15%. The formula was prepared mixing an infant formula, a low-fat milk, and different types of carbohydrates: GP for tests 2 and 3, and a mixture of GP+CS) for tests 4 and 5. Amylase was given in test number 5 immediately before the formula. Samples were obtained every 2 h in each test. More details are outlined in Table 1 and in the text (see Methods).
Statistical analysis showed a significant positive correlation among AC, FFA, and NEFA (AC versus FFA, r = 0.889, p < 0.001; AC versus NEFA, r = 0.84, p < 0.001; FFA versus NEFA, r = 0.81, p < 0.001).
Insulin values showed negative correlations with AC (r = −0.43, NS), FFA (r = −0.60, p < 0.05), and NEFA (r = −0.59, p < 0.05).
The results of individual AC values measured during the tests showed that C3, C16, and C18 remained within normal limits in all tests, whereas C4 and C5 remained abnormal despite the dietary changes. Levels of C2 remained at the low normal limits in all tests, regardless of fasting. Values of individual FFA showed a pattern similar to that of the corresponding AC (data not shown). Accordingly, significant correlations were found between the individual AC and the corresponding FFA, with p < 0.001 for C6 (r = 0.988), C8 (r = 0.882), C10 (r = 0.924), and C14 (r = 0.948) and p < 0.02 for C12 (r = 0.672) and C16 (r = 0.681;n =11 for each carbon length).
The patient is currently 3.9 years old. Her weight is slightly above the 97th percentile, length is at the 75th percentile, and head circumference is at the 95th percentile. She continues with a low-fat, low-protein, high-carbohydrate diet. CS is given four times a day, mixed with F, juice, or water. Amylase was discontinued. She has had a few intercurrent illnesses with no decompensations, and her neurologic examination and development are normal.
DISCUSSION
MADD is a disorder of fatty acid and protein metabolism for which diet is considered part of the treatment (1, 2). However, there is little information about the optimal composition of the diet, the type of carbohydrate to be given, the fasting periods that are safe for the patients at different ages, and the expected long-term clinical and biochemical outcome. Follow-up of the patients is complicated by the fact that some of the biochemical markers of the disease, like glucose levels or organic acids, will be abnormal only when patients are under stress. Therefore, there is a need for a biochemical marker that will be sensitive enough to reflect variations in diet or fasting, when the patients are not in crisis.
We report the diagnosis of a patient with MADD through a newborn screening program using MS-MS, and the biochemical response of this patient to different feeds to evaluate the usefulness of AC and FFA to monitor the response to these dietary changes.
It is known that patients with MADD can present with a wide range of severity (1, 2). The family history and the physical examination of our patient suggest an intermediate-to-severe form of the disease. It has been suggested that neonatal detection of MADD through newborn screening programs using MS-MS may not be reliable for all cases, and, to our knowledge, only one patient has been prospectively detected in such a program (9). However, in the present case, the levels of short-, medium-, and long-chain AC obtained on the sixth day of life, while the patient was asymptomatic, were clearly abnormal. The AC profile was similar to that reported for older children with MADD (7, 8). However, comparison of the actual AC values is difficult because AC levels in children with fatty acid oxidation defects vary with age, metabolic status, and fasting period, and these data were not provided in previous reports. Additionally, internal standards used in those studies for quantitation were different (7, 8).
Results of the oxidation of palmitic and myristic acids were similar to those reported by Brivet et al.(16) in a patient with MADD who presented clinically at 2 y of age. The oxidation of 14C-labeled fatty acids was also consistent with MADD. As riboflavin can modify the results of the oxidation tests performed in fibroblasts, (WR Rhead, personal communication, 1999), it is possible that the results of our studies in lymphocytes could have also been modified by the vitamin treatment.
Results of tests 1 and 2 showed that breast-feedings were associated with markedly increased levels of AC, FFA, and NEFA and that these values tended to normalize when a low-fat, low-protein, high-carbohydrate diet was given. This biochemical response shows that the recommended dietary treatment for MADD allows a good metabolic control and that breast-feedings should probably be avoided. When the patient was 6 mo old, test number 3 was performed to determine her biochemical response to a fasting period >4 h. Even though the patient was under good metabolic control, receiving the recommended diet and maintaining normal blood sugar levels during the test, there was a clear increase in AC, FFA, and NEFA levels after 6 h of fasting. These results are in agreement with those observed for MCAD deficiency (23), very long-chain acyl-CoA dehydrogenase deficiency (24), and long-chain hydroxy-acyl-CoA dehydrogenase deficiency (25) and highlight the importance of fasting control in children with fatty acid oxidation defects.
It has been suggested that CS, as used for glycogen storage diseases, could be used a source of slow-release carbohydrates to avoid the effects of fasting in children with fatty acid oxidation defects (13). We have documented that CS decreases the levels of octanoylcarnitine in children with MCAD deficiency (23). However, CS may not be useful in children <2 y of age because they may not have enough endogenous A activity (26). It has been suggested that giving CS in progressively increasing amounts over time may be helpful to overcome that problem (27). However, the results obtained in test number 4 suggested that in spite of giving CS progressively for a period of 4 mo, the patient's endogenous A activity was not enough to break down the CS given. When exogenous A was given before the formula (test number 5), levels of AC, FFA, and NEFA tended to normalize even at 6 h, suggesting that the exogenous enzyme allowed the proper carbohydrate breakdown These results are in agreement with the report of Schiffrin et al.(28) in a 9-mo-old patient with glycogen storage disease type I. This therapeutic approach may be an alternative to the use of continuous overnight feedings (through nasogastric tube or gastrostomy), which have been suggested for young children with severe fatty acid oxidation defects (13).
Dietary treatment of MADD is aimed at decreasing the use of fatty acids and protein as a fuel, which can result in the accumulation of toxic compounds. For that reason it is essential to avoid long periods of fasting, which would mobilize FFA from adipose tissue stores and activate the lipolytic pathway. Additionally, the high-carbohydrate diet is supposed to increase endogenous insulin secretion, which will suppress lipolysis and stimulate lipogenesis. We demonstrate that levels of insulin vary inversely with levels of AC, FFA, and NEFA. This negative correlation suggests that changes of those metabolites are secondary to changes in the insulin levels, which are induced by the different carbohydrate composition of the diet.
The use of AC for the diagnosis of fatty acid oxidation defects is now widely accepted; however, there is little information about the use of these compounds for the follow-up of the patients. For that purpose it is important to be sure that variations in AC levels reflect changes in the levels of fatty acids, which are likely to be the toxic compounds (29). The significant correlations found in our study between the different carbon length AC and the corresponding FFA are in agreement with our previous report in MCAD deficiency (23), and validate the usefulness of measuring the former as a marker of the metabolic status of the patient. Owing to the simple blood collection, monitoring of AC would be the method of choice for those centers with access to MS-MS technology coupled with a fast turn-around time. However, it is important to consider the time of the sample in relation to fasting, to be sure that normal levels of free carnitine are maintained and that valproic acid- or pivalic acid-containing drugs are not given (30).
As previously reported for MCAD deficiency (31), this patient's outcome and family history suggest that early diagnosis and treatment can change the natural history of MADD.
Abbreviations
- MADD:
-
multiple acyl-CoA-dehydrogenase deficiency
- AC:
-
acylcarnitines
- MS-MS:
-
tandem mass spectrometry
- BM:
-
breast milk
- F:
-
infant formula
- GP:
-
glucose polymers
- CS:
-
uncooked cornstarch
- NEFA:
-
total nonesterified fatty acids
- A:
-
amylase
- MCAD:
-
medium-chain acyl-CoA dehydrogenase
References
Frerman FE, Goodman SI 1995 Nuclear encoded defects of the mitochondrial respiratory chain, including glutaric acidemia type II. In: Scriver C. Beaudet L, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, 1611–1631.
Nyhan WL, Ozand PT 1998 Multiple acyl-CoA-dehydrogenase deficiency/glutaric aciduria type II/ethylmalonic-adipic aciduria. In: Nyhan WL, Ozand PT (eds) Atlas of metabolic diseases. Chapman & Hall Medical, London, 245–252.
Rhead WJ, Wolff JA, Lipson M, Falace P, Desai N, Fritchman K, Moon A, Sweetman L 1987 Clinical and biochemical variation and family studies in the multiple acyl-CoA dehydrogenation disorders. Pediatr Res 21: 371–376
Turnbull DM, Barlett K, Eyre JA, Gardner-Medwin D, Johnson MA, Fisher J, Watmough NJ 1988 Lipid storage myopathy due to glutaric aciduria type II: treatment of a potentially fatal myopathy. Dev Med Child Neurol 30: 667–672
Mongini T, Doriguzi C, Palmucci L, DeFrancesco A, Bet L, Manfredi L, Ponzetto C, Bresolin N 1992 Lipid storage myopathy in multiple acyl-CoA dehydrogenase deficiency: an adult case. Eur Neurol 32: 170–176
Rinaldo P 1994 Laboratory diagnosis of inborn errors of metabolism. In: Suchy FJ (ed) Liver disease in children. Mosby, St. Louis, 295–308.
Rashed MS, Bucknall M, Little D, Awad A, Jacob M, Alamoudi M, Alwattar M, Ozand P 1997 Screening blood spots for inborn errors of metabolism by electrospray tandem mass spectrometry with a microplate batch process and a computer algorithm for automated flagging of abnormal profiles. Clin Chem 43: 1129–1141
Vreken P, Van Lint AEM, Bootsma AH, Overmars H, Wanders RJA, Van Gennip AH 1999 Quantitative plasma acylcarnitines analysis using electrospray tandem mass spectrometry for the diagnosis of organic acidemias and fatty acid oxidation defects. J Inher Metab Dis 22: 302–306
Naylor EW, Chase DH 1999 Automated tandem mass spectrometry for mass newborn screening for disorders in fatty acid, organic acid, and amino acid metabolism. J Child Neurol 14( suppl): S4–S8
Green A, Marshall TG, Gray RGF, Pollit RJ 1985 Riboflavin-responsive ethylmalonic-adipic aciduria. J Inher Metab Dis 8: 67–70
Brivet M, Tardieu M, Khellaf A, Boutron A, Rocchiccioli F, Haengeli CA, Lemonnier A 1991 Riboflavin responsive ethylmalonic-adipic aciduria in a 9-month-old boy with liver cirrhosis, myopathy and encephalopathy. J Inher Metab Dis 14: 333–337
Mooy PD, Przyrembel H, Giesberts MAH, Scarbohydrateslte HR, Blom W, van Gelderen HH 1984 Glutaric aciduria type II: treatment with riboflavine, carnitine and insulin. Eur J Pediatr 143: 92–95
Treem WR 1994 Inborn defects in mitochondrial fatty acid oxidation. In: Suchy FJ (ed) Liver disease in children. Mosby, St. Louis, 852–887.
Millington DS, Terada N, Chace DH, Chen YT, Ding JH, Kodo N, Roe CR 1992 The role of tandem mass spectrometry in the diagnosis of fatty acid oxidation disorders. In: Coates P, Tanaka K. (eds) New developments in fatty acid oxidation. Wiley-Liss, New York, 339–354.
Millington DS, Chace DH, Hillman SL, Kodo N, Terada N 1994 Diagnosis of metabolic disease. In: Matsuo T, Caprioli RM, Gross ML, Seyana Y (eds) Biological mass spectrometry: present and future. John Wiley & Sons, New York, 559–579.
Brivet M, Slama A, Saudubray JM, Legrand A, Lemonnier A 1995 Rapid diagnosis of long chain and medium chain fatty acid oxidation disorders using lymphocytes. Ann Clin Biochem 32: 154–159
Saudubray JM, Coude F, Damaugre F, Johnson C, Gibson K, Nyhan W 1982 Oxidation of fatty acid in cultured fibroblasts: a model system for the detection and study of defects in oxidation. Pediatr Res 16: 877–881
Costa CG, Dorland L, Holwerda U, Tavares de Almeida I, Poll-The WT, Jacobs C, Duran M 1998 Simultaneous analysis of plasma free fatty acids and their 3-hydroxy analogs in fatty acid B-oxidation disorders. Clin Chem 44: 463–471
Ducombe W 1963 The colorimetric microdetermination of long chain fatty acids. Biochem J 88: 7–10
Buttery JE, Chamberlain BR, Pannall PR 1984 Colorimetric measurement of abnormal level of 3-hidroxibutirate in plasma. Clin Chem Acta 139: 167–171
Slocum RH, Cummings JG 1991 Amino acid analysis of physiological samples. In: Hommes FA (ed) Techniques in diagnostic human genetics. Wiley-Liss, New York, 87–126.
Hoppel CL 1991 Determination of carnitine. In: Hommes FA (ed) Techniques in diagnostic human genetics. Wiley-Liss, New York, 309–326.
Abdenur JE, Chamoles NA, Specola N, Schenone AB, Jorge L, Guinle A, Bernard CI, Levandowskiy V, Lavorgna S 1999 MCAD deficiency: acylcarnitines by tandem mass spectrometry are useful to monitor dietary treatment. Adv Exp Med Biol 466: 353–363
Costa CG, Dorland L, Tavares de Almeida I, Jakobs C, Duran M, Poll-The B 1998 The effect of fasting, long-chain triglyceride load and carnitine load on plasma long-chain acylcarnitine levels in mitochondrial very long-chain acyl-CoA dehydrogenase deficiency. J Inher Metab Dis 21: 391–399
Abdenur JE, Chamoles NA, García Alvarez M, Artero M, Schenone AB, Levanodwskiy V, Lavorgna S 1999 Deficiencia de 3-hidroxi-acil-CoA-dehidrogenasa de cadena larga: utilidad de las acilcarnitinas en el diagnóstico y tratamiento. II Congreso Latinoamericano de Errores Innatos del Metabolismo y Pesquisa Neonatal. Santiago, Chile, October 24–27, abstract book, 110
Chen YT 1995 Glycogen storage diseases. In: Scriver C, Beaudet L, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, 935–965.
Fernandes J, Chen YT 1995 Glycogen storage diseases. In: Fernandes J, Saudubray G, Van den Berghe G (eds) Inborn metabolic diseases. Springer-Verlag, Berlin, 71–85.
Schiffrin A, Polychronakos C, Abu-Srair H 1986 Glycogen storage disease type 1. Letter to the editor. N Engl J Med 315: 520–521
Trauner DA, Adams H 1981 Intracranial pressure elevations during octanoate infusion in rabbits: an experimental model of Reye's syndrome. Pediatr Res 15: 1097–1099
Abdenur JE, Chamoles NA, Guinle A, Schenone AB, Fuertes A 1998 Diagnosis of isovaleric acidemia by tandem mass spectrometry: false positive result due to pivaloylcarnitine in a newborn screening programme. J Inher Metab Dis 21: 624–630
Roe CR, Millington DS, Maltby DA, Kinnebrew P 1986 Recognition of medium-chain acyl-CoA dehydrogenase deficiency in asymptomatic siblings of children dying of sudden infant death or Reye-like syndromes. J Pediatr 108: 1–13
Acknowledgements
The authors thank Dr. S. I. Goodman for the revision of the manuscript, Dr. Caubet, Laboratorios Casasco, Argentina, who provided carnitine for the treatment of this patient, and the staff of the Department of Pediatric Endocrinology at the Hospital de Niños Pedro de Elizalde, who followed the patient during intercurrent illnesses.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Abdenur, J., Chamoles, N., Schenone, A. et al. Multiple Acyl-CoA-Dehydrogenase Deficiency (MADD): Use of Acylcarnitines and Fatty Acids to Monitor the Response to Dietary Treatment. Pediatr Res 50, 61–66 (2001). https://doi.org/10.1203/00006450-200107000-00013
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200107000-00013
