
Abstract
The pyruvate dehydrogenase (PDH) complex is situated at a key position in energy metabolism and is responsible for the conversion of pyruvate to acetyl CoA. In the literature, two unrelated patients with a PDH complex deficiency and splicing out of exon 6 of the PDH E1α gene have been described, although intronic/exonic boundaries on either side of exon 6 were completely normal. Analysis of exon 6 in genomic DNA of these patients revealed two exonic mutations, a silent and a missense mutation. Although not experimentally demonstrated, the authors in both publications suggested that the exonic mutations were responsible for the exon skipping. In this work, we were able to demonstrate, by performing splicing experiments, that the two exonic mutations described in the PDH E1α gene lead to aberrant splicing. We observed a disruption of the predicted wild-type pre-mRNA secondary structure of exon 6 by the mutated sequences described. However, when we constructed mutations that either reverted or disrupted the wild-type predicted pre-mRNA secondary structure of exon 6, we were unable to establish a correlation between the aberrant splicing and disruption of the predicted structure. The mutagenic experiments described here and the silent mutation found in one of the patients suggest the presence of an exonic splicing enhancer in the middle region of exon 6 of the PDH E1α gene.
Similar content being viewed by others

Main
PDH complex deficiency is one of the most common defined genetic defects of mitochondrial energy metabolism(1). It is a severe disease, which results in early death in the majority of affected patients(2). Most of the cases are sporadic and result from a new mutation arising within the germ cells of one of the parents. However, familial cases have also been described(3–5).
The majority of the molecular defects of the PDH complex have been localized in the E1α subunit gene at chromosome Xp22.1 [gene symbol PDHA1; MIM# 312170;(6)]. More than 120 patients with PDH E1α deficiency have been described, and at least 75 different mutations in the coding region have been reported(7). Two cases of splicing out of exon 6 from the PDH E1α mRNA have been found(8, 9). De Meirleir et al.(8), studying a family with Leigh's encephalomyelopathy, found a partial skipping of the entire exon 6 in two affected boys. Analysis of the genomic DNA of these individuals revealed a silent mutation at nucleotide 45 of exon 6 [cDNA position 660 according to Lissens et al.(7)]. Chun et al.(9) found a deletion of exon 6 in all of the cDNAs examined in a female patient who died of severe neonatal lactic acidosis. At the genomic level, this patient was heterozygous for a missense mutation at nucleotide 13 of exon 6 (cDNA position 628). Although not experimentally demonstrated, the authors of both studies suggested that the exonic mutations were responsible in some way for the transcript with the missing exon.
It is estimated that up to 15% of all point mutations causing human genetic disorders do so by resulting in aberrant mRNA splicing. The majority of these mutations are located in the consensus splice sites at the intron/exon borders(10). However, a small proportion of these mutations affect exon sequences that are distinct from the consensus sites(8, 9, 11–20), but it is not clear how these exonic mutations affect splicing. It was suggested(8, 11, 18) that the exonic mutations may lead to aberrant splicing owing to a disruption of the pre-mRNA secondary structure. Although the exact role of the secondary structure of the nuclear-encoded pre-mRNA is not well defined, an influence of this structure on splicing has already been observed(21–24). Exonic mutations can also influence splicing when they are located in ESEs. ESEs are sequences that are internal to exons and function to enhance the specific recognition of splice sites during constitutive and alternative splicing(25). Most of the ESEs identified are purine-rich, although other classes of enhancer, such as AC-rich splicing enhancers, have been identified(26). They contain binding sites for SR-rich proteins that can activate splicing by binding to enhancers and recruiting the splicing machinery to the adjacent intron(27).
In the present study, we have used exon trap analysis to confirm that the exonic mutations described in the PDH E1α gene(8, 9) are responsible for the aberrant splicing of exon 6. Furthermore, we observed a disruption of the wild-type mfe secondary structure of exon 6, predicted by a computer program, in the mutant sequences. Therefore, several other mutant sequences were prepared by in vitro mutagenesis and studied by exon trap analysis to verify whether a possible correlation between the disruption of the predicted secondary structure of exon 6 and aberrant splicing could be found, but no correlation was observed. However, a purine-rich sequence in the middle region of exon 6 could possibly act as an exonic splicing enhancer.
METHODS
Exonic mutations in the PDH E1α gene leading to aberrant splicing.
De Meirleir et al.(8) described patient G.D. who had symptoms suggestive for Leigh's encephalomyelopathy. Fifty to 80% of his mRNAs from cultured fibroblasts coding for the PDH E1α gene lacked exon 6. Sequencing of his genomic DNA showed an A to G substitution at nucleotide 45 of exon 6 (660A→G;Fig. 1). The glycine at this position (G185) is not affected by the substitution (GGA→GGG). Analysis of parts of the boundary introns (32 bp of intron 5 and 61 bp of intron 6) revealed a normal sequence. Chun et al.(9) described patient 2641, a girl who died of severe neonatal lactic acidosis within the first 2 wk of life. Studies in cultured fibroblast showed a deletion of exon 6 in all cDNAs examined. Amplification of this exon from her genomic DNA revealed two alleles: a normal one and one with a G to A substitution at nucleotide 13 of exon 6 (628G→A) that causes an amino acid change from alanine to threonine (A175T). Sequencing of part of the boundary introns revealed normal sequences. From patient G.D., genomic DNA from leukocytes was available for study. From patient 2641, material was not available and site-directed mutagenesis was performed using the wild-type PDH E1α exon 6 as a template (further described) to create the described mutation.

Sequence of exon 6 of the PDH E1α gene. Bases 1 and 93 correspond to cDNA positions 616 and 708, respectively. The purine-rich region is localized between bp 43 and 50. When mutated, the outlined bases showed the ability to enhance the inclusion of exon 6 in the final mRNA; the bases inside the boxes caused exon skipping, and the base in italic does not interfere in the splicing process. The underlined sequence represents the probable regulatory splicing region.
Splicing experiments.
We used the pET01 exon trap vector from Mobitec (Göttingen, Germany). Part of the PDH E1α gene from bp 10,325 to 10,700(28) from a normal control subject and patient G.D. (mutation 660A→G) were PCR amplified using the following primers: sense, EXTR65: 5′-CAGAGAGATAGGGTTCGAAGG-3′ and antisense, EXTR63: 5′AAGTCAACCAAATTTGAGCTCGCC-3′. These primers introduced sites for Sac I and Taq I restriction enzymes in the 5′ and 3′ ends of the fragment, respectively, that were used for ligation of the PCR fragments into the plasmid. The PCR amplifications were performed in standard 100-μL reactions, containing 1 μg of genomic DNA, as described previously(8). After digestion with Sac I and Taq I restriction enzymes, the expected length of the fragment was 342 bp (130 bp of intron 5, 93 bp of exon 6, and 119 bp of intron 6). These fragments were then inserted into the pET01 vector digested with Sac I and Cla I restriction enzymes, and transformed into Escherichia coli strain HB101(29). The colonies containing the recombinant vectors were selected by restriction enzyme treatment with Kpn I and were further sequenced (T7-sequenase kit V 2.0, Amershan Life Science, Roosendaal, The Netherlands) using primers ETS1: 5′-GTGAGAATCCTTCCTTAAGTC-3′ and ETS3: 5′-CCTACACATCCTGTTACCA-3′. For the vectors containing the correct sequence, a large plasmid preparation was made using Qiagen Plasmid Purification Maxi Kit (Hilden, Germany). Purified plasmids were further used for transfections in COS-7 cells. Transfections were performed using the DEAE-Dextran method as described in the Exon Trap manual. After 72 h, the cells were harvested and prepared for total RNA extraction. All transfections were performed in triplicate.
RNA extraction and RT-PCR.
Total RNA was isolated from COS-7 cells using SV Total RNA Isolation System (Promega, Madison, WI, U.S.A.). The reverse transcription was performed with 2.5 μg of total RNA in a 33-μL reaction using the First-Strand cDNA Synthesis Kit (Amersham Pharmacia Biotech, Roosendal, The Netherlands) according to the instructions of the manufacturer. As a control for contamination with genomic DNA, duplicate samples were used for reverse transcription, in which reverse transcriptase was not added to the mixture in one sample. The primer used for the first-strand cDNA synthesis was primer 3: 5′-CTCCCGGGCCACCTCCAGTGCC-3′. For the PCR reaction, the sense primer 2: 5′-GAGGGATCCGCTTCCTGCCCC-3′ and the antisense primer 3 were used. The amplification took place in 25-μL reactions, containing 2.5 μL of cDNA, 10 mM Tris-HCl, 50 mM KCl, 1 mM MgCl2, 200 μM dNTPs, 300 nM of each primer, and 1 unit of Ampli Taq Gold (Perkin Elmer, Zaventem, Belgium). The conditions were 95°C for 12 min and 20 cycles of 94°C for 45 s, 59°C for 45 s, and 72°C for 1 min, with a final extension of 10 min at 72°C. The products were visualized on 2.5% agarose gels stained with ethidium bromide; part of the products were used for sequencing. When a PCR reaction resulted in two fragments, these fragments were separated on a 15% polyacrylamide gel and cut out to be sequenced.
To determine the optimal conditions, PCRs were run for 16, 18, 20, 22, 24, 26, and 28 cycles. Using 20 cycles, the PCR was still in the logarithmic phase. All the RT-PCRs were performed three times using RNA from different transfections.
Calculation of the mfe pre-mRNA secondary structures.
The mfe pre-mRNA secondary structures were calculated using the program RNAdraw V1.1(30). The calculations were imported from the RNA fold program included in the Vienna RNA package(31).
Site-directed mutagenesis.
Site-directed mutagenesis was performed using the Quick Change Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, U.S.A.). The double-stranded vector containing wild-type exon 6 of the PDH E1α gene was used as a template for all mutageneses with an exception for mutation 708+118T→C, for which the plasmid containing mutation 628G→A was used as a template. Primers used are listed in Table 1. Positions of mutations in exon 6 are shown in Fig. 1. The results of all the mutagenesis studies were confirmed by sequence analysis.
RESULTS
Cloning and expression of normal and mutant exon 6 E1α sequences.
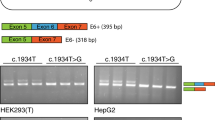
To verify whether the exonic mutation found in patient G.D. [660A→G,(8)] was responsible for the splicing out of exon 6, we used the exon trap approach. Plasmids containing wild-type or mutant 660A→G exon 6 were transfected in COS-7 cells. RT-PCR from total RNA was performed and resulted in almost equal quantities of amplification products of 339 bp and 246 bp for the wild-type (Fig. 2A, lane 3), and a product of 246 bp and a barely detectable product of 339 bp for mutant 660A→G (Fig. 2A, lane 4). Sequencing revealed that the product of 339 bp contained the PDH E1α exon 6 plus insulin exonic sequences (derived from the vector) and that the product of 246 bp contained only the insulin exonic sequences without exon 6.

RT-PCR analysis of the PDH E1α gene exon 6. Total RNA was isolated from transiently transfected COS-7 cells and used for RT-PCR analysis. Representative results for triplicate experiments are shown. In all the figures, lanes 1 to 3 correspond to mock-transfected cells, cells transfected with empty vector and wild-type, respectively. A, lane 4, mutant 660A→G. The wild-type constuct (lane 3) shows two bands: the upper band (339 bp) contains exon 6, whereas in the lower band (246 bp), exon 6 is lost. In the mutant sequence of patient G. D. (lane 4), we observed mainly the 246-bp band, indicating that most of the mRNA has lost exon 6. B, lanes 4 and 5, mutant 690C→A and mutant 628G→A, respectively. Mutant 628G→A (lane 5) contains only the band of 246 bp, indicating that all of the mRNA skipps exon 6. Mutant 690C→A (lane 4) maintains the same pattern as wild-type. C, lanes 4–6, mutants 666T→C, 671T→C and 708+118T→C, respectively. Mutants 666T→C and 671T→C (lanes 4 and 5, respectively) show the same pattern of splicing as wild-type. The ratio between the intensities of the fragments retaining (339 bp) and having lost exon 6 (246 bp) were higher in these mutants than in the wild-type. Mutant 708+118T→C (lane 6) had 100% skipping of exon 6. D, lanes 4 and 5, mutants 661A→C and 660delAA, respectively. Both mutants show the same pattern of splicing as wild-type, but the ratio between the intensity of the fragments retaining and skipping exon 6 is higher in the mutants than in the wild-type. Markers (M) for A, B, and C, 64 to 587 bp; marker for D, 124 to 2176 bp (Boehringer Mannheim, Brussels, Belgium).
The other exonic mutation leading to aberrant splicing of exon 6 of the PDH E1α gene was described in patient 2641 from the study by Chun et al.(9). To verify whether this mutation 628G→A (G to A substitution at nucleotide 13 of exon 6) was also responsible for the aberrant splicing, a site-directed mutagenesis in exon 6 from the wild-type PDH E1α gene was performed, mimicking the described mutation. After transfections in COS-7 cells, we observed complete splicing out of exon 6 for this sequence (Fig. 2B, lane 5).
An updated computer program, RNAdraw V1.1(30), was used to predict the mfe secondary structures of the 93 bp from exon 6 in wild-type, mutant 660A→G, and mutant 628G→A (data not shown). By comparison of the predicted structures, it was observed that both mutations disrupt the wild-type conformation.
Site-directed mutagenesis based on predicted mfe pre-mRNA secondary structures.
Because the exonic mutations in the PDH E1α gene affect splicing and disrupt the predicted wild-type secondary structure, we decided to test whether there is a relationship between the disruption of the predicted secondary structure and the splicing out of exon 6. For this purpose, site-directed mutagenesis in this exon was performed based on the RNAdraw program.
The first mutagenesis was designed in a way such that disruption of the mfe secondary structure of exon 6 was obtained without changing the amino acid glycine (G195). It was a C to A substitution at nucleotide 75 of exon 6 (690C→A). Transient transfection of the mutated construct in COS-7 cells showed, by RT-PCR, two amplification products of 339 bp and 246 bp (Fig. 2B, lane 4). The ratio between the intensity of these products was similar to that found in the wild-type construct.
When predicting the secondary structures above, we followed the same procedure as Chun et al.(9), Steinsgrimsdottir et al.(11), and Fukao et al.(12). These authors took into consideration only the exonic sequences for predicting the mfe pre-mRNA structures, and interactions between exonic/intronic sequences in the folding of the pre-mRNA were not considered. Again using the RNA draw program, we redrew the structures of wild-type and mutant (660A→G, 628G→A, and 690C→A) exon 6 of the PDH E1α gene, but this time taking into consideration part of the surrounding introns (Fig. 3, A – D ). We observed that at least 150 bp of each intron had to be included in the calculations to obtain a stable structure.

View of part of the predicted pre-mRNA mfe secondary structure of exon 6 and part of the boundary introns of the PDH E1α gene. A, wild-type. B, mutation 660G→A (patient G. D). C, mutation 628A→G (patient 2641). D, mutation 690C→A. E, mutation 666T→C. The exonic sequence is in bold, and the arrows indicate the mutated bases.
To test the hypothesis that the predicted mfe secondary structure of exon 6 influences the splicing process, we designed three new mutations, based on calculated structures including intronic sequences:1) a T→C substitution at nucleotide 51 of exon 6 (mutation 666T→C); this mutation does not change the amino acid asparagine (D187) but the wild-type pre-mRNA secondary structure is disrupted (Fig. 3E);2) a T→C substitution at nucleotide 56 of exon 6 (mutation 671T→C). The amino acid valine is changed into an alanine (V189A), and the pre-mRNA secondary structure is the same as for the wild-type;3) a mutation that does not affect the consensus splice sequences was made in intron 6 (T to C substitution 118 bp downstream of the 5′ splice site of intron 6 mutation 708+118T→C) in the construct containing mutation 628G→A. This mutation was designed in a way such that the disrupted secondary structure of exon 6 containing mutation 628G→A was reverted to the wild-type.
After mutagenesis and expression in COS-7 cells, we observed that the mutation 671T→C, with a conserved secondary structure, showed RT-PCR fragments of 339 bp and 246 bp (Fig. 2C, lane 5). The ratio between the intensity of fragments retaining and of those skipping exon 6 was higher in this construct when compared with the wild-type. However, the construct containing mutation 666T→C, with a disrupted secondary structure, also showed these two fragments and in the same proportions as mutation 671T→C (Fig. 2C, lane 4). The double construct with mutation 708+118 T→C showed 100% skipping of exon 6 (Fig. 2C, lane 6).
Mutation in a purine-rich region of exon 6 of the PDH E1α gene.
The mutation of patient G.D. is located in a purine-rich region that could be acting as an ESE (Fig. 1). We decided to test whether this is a conserved region for the recognition of exon 6 by the spliceosome complex. For this reason, we constructed two new mutations:1) an A→C substitution at nucleotide 46 of exon 6 (mutation 661A→C); the amino acid lysine is changed to glutamine (K186Q); and 2) a deletion was made to remove two of the four consecutive adenines of the purine-rich region (mutation 660delAA).
After transfection in COS-7 cells, we observed RT-PCR fragments of 339 bp and 246 bp for both mutations (Fig. 2D, lanes 4 and 5). The ratio between the intensity of the fragments retaining and of those skipping exon 6 was higher in these constructs when compared with the wild-type.
DISCUSSION
In the present study, we demonstrate by using exon trap analysis that the two exonic mutations described in exon 6 of the PDH E1α gene(8, 9) indeed cause aberrant splicing of this exon. Several cases of exonic mutations presumed to lead to aberrant splicing have been described in the literature, but in only three studies, splicing experiments were developed to demonstrate the causal relationship between both [(12, 13) and the present study].
In several instances, it has been suggested that disruption of the pre-mRNA secondary structure might be at the basis of the aberrant splicing. In general, computer programs are used to predict the pre-mRNA mfe secondary structures(18, 22, 32), some of which have been experimentally confirmed(21, 24). To test the hypothesis of pre-mRNA secondary structure disruption for exon 6 of the PDH E1α gene, we decided to use the calculations of the RNAdraw program(30) taken by the RNA fold program from the “Vienna RNA package” [RNA fold(31)], a program that has been shown to generate well-determined structures(33, 34). As a first approach, we decided to use only the 93 bp of exon 6 of the gene in the mfe calculations. However, as exonic sequences can interact with intronic sequences in the pre-mRNA(21, 23) and these interactions might be indispensable for obtaining a well-stabilized structure, we decided to change the strategy by including sequences from the surrounding introns. Nevertheless, the mutations did, in both cases, disrupt the predicted secondary structure of the wild-type pre-mRNA sequence. These results urged us to further study the hypothesis by designing several new mutations that retain, disrupt, or restore the predicted wild-type mfe secondary structure (690C→A, 666T→T, 671T→C, 628G→A + 708+118T→C). These mutations were created in the exon trap construct by site-directed mutagenesis and studied by exon trap analysis. However, no correlation could be found between the predicted structures and the splicing patterns.
A few reasons could be found to explain the lack of correlation between the predicted structures and the splicing patterns. First of all, the pre-mRNA secondary structure predicted by the RNAdraw program does not reflect the conformation of this RNA in vivo. Besides the fact that only part of the gene is taken into consideration for structure calculations, the computer program also does not take into account that these structures might be influenced by specific interactions of RNA–protein complexes such as the hnRNP A1 protein that might help to define the borders of mammalian introns(35, 36). A second possibility is that the predicted secondary structure exists but that it does not interfere in the splicing process. Indeed, Eperon et al.(37) have shown that the pre-mRNA secondary structure only sequesters splicing sites effectively when an alternative site is provided. When the alternative site is inactivated, the otherwise blocked site is used in vivo. Such sites might be revealed by an RNA duplex-unwinding activity, or by helix destabilization activity found in hnRNP preparations. In mammals, three putative helicases involved in mammalian pre-mRNA splicing have been identified, but their interactions with splicing sites have not yet been determined(38).
Nevertheless, we observed that the two mutations (660delAA and 661A→C) constructed in the purine-rich region of exon 6 were favoring the inclusion of this exon in the final mRNA, when compared with the wild-type construct. Several studies have demonstrated that splicing of pre-mRNAs containing weak 3′ splice site consensus sequences require splicing enhancer elements for activity(39–41). Nonsense mutations localized in ESEs are able to cause skipping of the respective exon, owing to a disruption of the interactions between an ESE and SR-rich proteins(27, 42). When comparing the purine-rich sequence of exon 6 (TGGAAAAGA) with ESE sequences published by Schaal and Maniatis(43), we observed that indeed the purine-rich sequence after mutation 660delAA (TGGAAGA) becomes more similar to a published ESE (YGGAGA, where Y is a pyrimidine), and the mutation 661A→C produced a sequence that was shown to be part of the IgM M2 enhancer and three other ESEs identified by the SELEX technique (GGAC AAG A—the underlined sequences are conserved and the three nucleotides in the middle are variable). It is possible that the mutation 660A→G is localized in an important site of specific protein binding, leading to aberrant splicing. The new mutations produced in this region could make this site more favorable for being recognized by this or other proteins. Mutagenic studies in ESEs of the human β-globin gene support this theory. It was shown for this gene that a point mutation and a double mutation in an ESE did not affect the normal splicing of exon 2. Indeed, these mutations lead to increased binding of a new, unidentified 70-kD protein. Interestingly, the authors found in another ESE that a point mutation, which does not change the amino acid sequence, had a dramatic effect on the mRNA splicing efficiency(27). Taking together the mutations of patient G.D., 660delAA, 661A→C, 666T→C, and 671T→C, we can delimit a region of 12 bp (between bp 42 and 56, Fig. 1) where the sequence changes are actively affecting splicing (inhibiting or enhancing the correct splicing of exon 6). This region could probably be a regulatory splicing region.
The in vivo splicing experiments in COS-7 cells result in either complete (mutation 628G→A) or almost complete (mutation 660A→G) skipping of the PDH E1α exon 6 for the mutant sequences, whereas approximately 50% skipping was observed for wild-type exon 6. The appearance of a faint band containing exon 6 for mutant 660A→G is in agreement with the in vivo situation in which only partial skipping of this exon is observed. The 50% skipping observed for the wild-type exon 6 sequence could possibly be explained on the basis that the weak 3′ splice site of exon 6 requires an ESE to be recognized by the spliceosome complex. This splice site indeed shows the lowest value of the entire PDH E1α gene when 3′ and 5′ splice site consensus values are calculated according to Shapiro and Senapathy(44). Absence or reduced production of a protein to interact with the ESE(s) in COS-7 cells could explain the partial skipping observed for this exon. The increased inclusion of exon 6 by the mutants could be explained by a better recognition of this protein or by the change of a site that is recognized by another protein that is present in these cells. Although specific SR-rich protein binding sites have been identified, individual SR-rich proteins are capable of recognizing a broad spectrum of weakly related sequences(27).
CONCLUSION
In conclusion, we could demonstrate for the first time that exonic mutations lead to aberrant splicing in the PDH E1α gene. We did not observe a relationship between disruption of the predicted secondary structure of the pre-mRNA and exon skipping. The mutagenic studies performed suggest the presence of a regulatory splicing region, most probably an ESE, localized in the middle region of exon 6 of the PDH E1α gene. Further experiments should be performed to define this regulatory element and its respective protein interactions.
Abbreviations
- ESE:
-
exonic splicing enhancer
- mfe:
-
minimum free energy
- PDH:
-
pyruvate dehydrogenase
- RT-PCR:
-
reverse transcriptase PCR
- SR:
-
serine-arginine
References
Robinson BH 1995 Lactic acidemia (disorders of pyruvate carboxylase, pyruvate dehydrogenase). In: Scriver CR, Beaudet AL, Sly WS, Valle D(eds) The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill, New York, 1479–1499.
Brown GK, Otero LJ, LeGris M, Brown RM 1994 Pyruvate dehydrogenase deficiency. J Med Genet 31: 875–879.
Dahl H-HM, Brown GK, Brown RM, Hansen LL, Kerr DS, Wexler ID, Patel MS, De Meirleir L, Lissens W, Chun K, MacKay N, Robinson BH 1992 Mutations and polymorphisms in the pyruvate dehydrogenase E1α gene. Hum Mutat 1: 97–102.
Lissens W, De Meirleir L, Seneca S, Benelli C, Marsac C, Poll-The BT, Briones P, Ruitenbeek W, van Diggelen O, Chaigne D, Ramaekers V, Liebaers I 1996 Mutation analysis of the pyruvate dehydrogenase E1α gene in eight patients with a pyruvate dehydrogenase complex deficiency. Hum Mutat 7: 46–51.
De Meirleir L, Specola N, Seneca S, Lissens W 1998 Pyruvate dehydrogenase E1α deficiency in a family: different clinical presentation in two sibilings. J Inherit Metab Dis 21: 224–226.
Kerr DS, Wexler ID, Tripatara A, Patel MS 1996 Human defects of the pyruvate dehydrogenase complex. In: Patel MS, Roche TE, Harris RA(eds) Alpha-keto Acid Dehydrogenase Complexes. Birkhauser Verlag, Basel, 249–269.
Lissens W, De Meirleir L, Seneca S, Liebaers I, Brown GK, Brown RM, Ito M, Naito E, Kuroda Y, Kerr DS, Wexler ID, Patel MS, Robinson BH, Seyda A 2000 Mutations in the X-linked pyruvate dehydrogenase (E1) α subunit gene (PDHA1) in patients with a pyruvate dehydrogenase complex deficiency. Hum Mutat 15: 209–219.
De Meirleir L, Lissens W, Benelli C, Ponsot G, Desguerre I, Marsac C, Rodriguez D, Saudubray J-M, Poggi F, Liebaers I 1994 Aberrant splicing of exon 6 in the pyruvate dehydrogenase-E1α mRNA linked to a silent mutation in a large family with Leigh's encephalomyelopathy. Pediatr Res 36: 707–712.
Chun K, MacKay N, Petrova-Benedict R, Federico A, Fois A, Cole DEC, Roberteson E, Robinson BH 1995 Mutations in the X-linked E1α subunit of pyruvate dehydrogenase: exon skipping, insertion of duplicate sequence, and missense mutations leading to the deficiency of the pyruvate dehydrogenase complex. Am J Hum Genet 56: 558–569.
Krawczak M, Reiss J, Cooper DN 1992 The mutational spectrum of single base pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet 90: 41–54.
Steingrimsdottir H, Rowley G, Dorado G, Cole J, Lehmann AR 208 208 1992 Mutations which alter splicing in the human hypoxanthine-guanine phosphoribosyltransferase gene. Nucleic Acids Res 20: 1201–1208.
Fukao T, Yamaguchi S, Wakazono A, Orii T, Hoganson G, Hashimoto T 1994 Identification of a novel exonic mutation at −13 from 5′ splice site causing exon skipping in a girl with mitochondrial acetoacetyl-coenzyme A thiolase deficiency. J Clin Invest 93: 1035–1041.
Liu W, Qian C, Francke U 1997 Silent mutation induces exon skipping of fibrillin-1 gene in Marfan syndrome. Nat Genet 16: 328–329.
Pie J, Casals N, Casale CH, Buesa C, Mascaro C, Barcelo A, Rolland MO, Zabot T, Haro D, Eyskens F, Divry P, Hegardt FG 1997 A nonsense mutation in the 3-hydroxy-3-methylglutaryl-CoA lyase gene produces exon skipping in two patients of different origin with 3-hydroxy-3-methylglutaryl-CoA lyase deficiency. Biochem J 323: 329–335.
Babcock D, Gasner C, Francke U, Malsen C 1998 A single mutation that results in an Asp to His substitution and partial exon skipping in a family with congenital contractual arachnodactyly. Hum Genet 103: 22–28.
Sakuntabhai A, Hammami-Hauasli N, Bodemer C, Rochat A, Prost C, Barrandon Y, de Prost Y, Lathrop M, Wojnarowska F, Bruckner-Tunderman L, Hovnanian A 1998 Deletions within COL7A1 exons distant from consensus splice sites alter splicing and produce shortened polypeptides in dominant dystrophic epidermolysis bullosa. Am J Hum Genet 63: 737–748.
Mendez M, Sorkin L, Rossetti MV, Astrin KH, del C Batlle AM, Parera VE, Aizencang G, Desnick RJ 1998 Familial porphyria cutanea tarda: characterization of seven novel uroporphyrinogen decarboxylase mutations and frequency of common hemochromatosis alleles. Am J Hum Genet 63: 1363–1375.
Hoffmeyer S, Nürnberg P, Ritter H, Fahsold R, Leistner W, Kaufmann D, Krone W 1998 Nearby stop codons in exons of the neurofibromatosis type 1 gene are disparate splice effectors. Am J Hum Genet 62: 269–277.
Kanai N, Yanai F, Hirose S, Nibu K, Izuhara K, Tani T, Kubota T, Mitsudome A 1999 A G to A transition at the last nucleotide of exon 6 of the γc gene (868G→A) may result in either a splice or missense mutation in patients with X-linked severe combined immunodeficiency. Hum Genet 104: 36–42.
Lorson CL, Androphy EJ 2000 An exonic enhancer is required for inclusion of an essential exon in the SMA-determining gene SMN. Hum Mol Genet 9: 259–265.
Clouet d'Orval BC, d'Aubenton Carafa YA, Sirand-Pungnet P, Gallego M, Brody E, Marie J 1991 RNA secondary structure repression of a muscle-specific exon in HeLa cell nuclear extracts. Science 252: 1823–1828.
Libri D, Piseri A, Fiszman MY 1991 Tissue-specific splicing in vivo of the β-tropomyosin gene: dependence on an RNA secondary structure. Science 252: 1842–1845.
Coleman TP, Roesser JR 1998 RNA secondary structure: an important cis-element in rat calcitonin/CGRP pre-messenger RNA splicing. Biochemistry 37: 15941–15950.
Muro AF, Caputi M, Pariyarath R, Pagani F, Buratti E, Baralle FE 1999 Regulation of fibronectin EDA exon alternative splicing: possible role of RNA secondary structure for enhancer display. Mol Cell Biol 19: 2657–2671.
Cooper TA, Mattox W 1997 The regulation of splice site selection, and its role in human disease. Am J Hum Genet 61: 259–266.
Coulter LR, Landree MA, Cooper TA 1997 Identification of a new class of exonic splicing enhancers by in vivo selection. Mol Cell Biol 17: 2143–2150.
Schaal TD, Maniatis T 1999 Multiple distinct splicing enhancers in the protein-coding sequences of a constitutively spliced pre-mRNA. Mol Cell Biol 19: 261–273.
Koike K, Urata Y, Matsuo S, Koike M 1990 Characterization and nucleotide sequence of the gene encoding the human pyruvate dehydrogenase α subunit. Gene 93: 307–311.
Hanahan D 1983 Studies on transformation of E. coli with plasmid. J Mol Biol 166: 557–580.
Matzura O, Wennborg A 1996 RNAdraw: an integrated program for RNA secondary structure calculation and analysis under 32-bit Microsoft Windows. Comput Appl Biosci 12: 247–249.
Hofacker IL, Fontana W, Stadler PF, Bonhoeffer LS, Tacker M, Schuster P 1994 Fast folding and comparison of RNA secondary structures. Monatsh Chem 125: 167–188.
Staffa A, Acheson NH, Cochrane A 1997 Novel exonic elements that modulate splicing of the human fibronectin EDA exon. J Biol Chem 272: 33394–33401.
Huynen MA, Perelson A, Vieira WA, Stadler PF 1996 Base pairing probabilities in a complete HIV-1 RNA. J Comput Biol 2: 253–274.
Huynen M, Gutell RR, Konings DA 1997 Assessing the reliability of RNA fold using statistical mechanisms. J Mol Biol 267: 1104–1112.
Solnick D, Lee SI 1987 Amount of RNA secondary structure required to induce an alternative splice. Mol Cell Biol 7: 3194–3198.
Blanchete M, Chabot B 1999 Modulation of exon skipping by high-affinity hnRNP A1-binding sites and by intron elements that repress splice site utilization. EMBO J 18: 1939–1952.
Eperon LP, Graham IR, Griffiths AD, Eperon IC 1988 Effects of RNA secondary structure on alternative splicing of pre-mRNA: is folding limited to a region behind the transcribing RNA polymerase?. Cell 54: 393
Will CL, Lührmann R 1997 Protein functions in pre-mRNA splicing. Curr Opin Cell Biol 9: 320–328.
Lopez AJ 1998 Alternative splicing of pre-mRNA: developmental consequences and mechanisms of regulation. Annu Rev Genet 32: 279–305.
Dye BT, Buvoli M, Mayer SA, Lin C-H, Patton JG 1998 Enhancer elements activate the weak 3′ splice site of α-tropomyosin exon 2. RNA 4: 1523–1536.
Yue B-G, Akusjärvi G 1999 A downstream splicing enhancer is essencial for in vitro pre-mRNA splicing. FEBS Lett 451: 10–14.
Watakabe A, Tanaka K, Shimura Y 1993 The role of exon sequences in splice site selection. Genes Dev 7: 407–418.
Schaal TD, Maniatis T 1999 Selection and characterization of pre-mRNA splicing enhancers: identification of novel SR protein-specific enhancer sequences. Mol Cell Biol 19: 1705–1719.
Shapiro MB, Senapathy P 1987 RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res 15: 7155–7174.
Author information
Authors and Affiliations
Additional information
Supported by the National Fund for Scientific Research (NFWO) grant 3.0065.87, the Fund for Scientific Research Flanders (FWO Vlaanderen) grant G0181.98, and the Research Council of the Vrije Universiteit Brussel (VUB), Brussels, Belgium.
Rights and permissions
About this article
Cite this article
Kupper Cardozo, A., De Meirleir, L., Liebaers, I. et al. Analysis of Exonic Mutations Leading to Exon Skipping in Patients with Pyruvate Dehydrogenase E1α Deficiency. Pediatr Res 48, 748–753 (2000). https://doi.org/10.1203/00006450-200012000-00008
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200012000-00008
This article is cited by
-
Laboratory approach to mitochondrial diseases
Journal of Physiology and Biochemistry (2001)
