
Abstract
Nitric oxide (NO) produced by inducible NO synthase contributes to ischemic brain damage. However, the role of inducible NO synthase-derived NO on neonatal hypoxic-ischemic encephalopathy has not been clarified. We demonstrate here that aminoguanidine, a relatively selective inhibitor of inducible NO synthase, ameliorated neonatal hypoxic-ischemic brain damage and that temporal profiles of NO correlated with the neuroprotective effect of aminoguanidine. Seven-day-old Wister rat pups were subjected to left carotid artery occlusion followed by 2.5 h of hypoxic exposure (8% oxygen). Infarct volumes (cortical and striatal) were assessed 72 h after the onset of hypoxia-ischemia by planimetric analysis of coronal brain slices stained with hematoxylin-eosin. Aminoguanidine (300 mg/kg i.p.), administered once before the onset of hypoxia-ischemia and then three times daily, significantly ameliorated infarct volume (89% reduction in the cerebral cortex and 90% in the striatum;p < 0.001). NO metabolites were measured by means of chemiluminescence using an NO analyzer. In controls, there was a significant biphasic increase in NO metabolites in the ligated side at 1 h (during hypoxia) and at 72 h after the onset of hypoxia (p < 0.05). Aminoguanidine did not suppress the first peak but significantly reduced the second one (p < 0.05), and markedly reduced infarct size in a neonatal ischemic rat model. Suppression of NO production after reperfusion is a likely mechanism of this neuroprotection.
Similar content being viewed by others
Main
Perinatal HIE is the single most important problem in neonatal neurology, and some patients with HIE develop cerebral palsy and mental retardation (1). HI causes brain damage by activating a cascade of biochemical events (2–4). Calcium that enters neurons through the glutamate receptor channels triggers toxic events including the production of NO (3, 5). NO is formed by NOS (EC 1.14.13.39), which features three distinct forms (6). nNOS (NOS I) and endothelial NOS (NOS III) produce NO intermittently, only when the intracellular calcium concentration increases. iNOS (NOS II) is activated by certain stimuli, typically endotoxins and cytokines, and is not calcium-dependent; thereby, the enzyme can be continuously active and produce large amounts of NO (4).
NO produced by iNOS has been implicated in ischemic brain damage in an adult rodent model (7). Although a number of studies have tried to elucidate the role of NO produced by iNOS and the efficacy of the protective function of AG, a relatively selective iNOS inhibitor (8, 9), in adult ischemic rat models (10–16), little is known about the role of iNOS and the possible neuroprotective effects of AG on neonatal HI brain damage (17). We have previously described the neuroprotective effect of a nonselective NO synthesis inhibitor, NG-nitro-L-arginine (18), and the temporal profile of the number of neurons containing nNOS (19) and of NO metabolites (20) in a neonatal HI rat model. We hypothesized that iNOS is a major source of NO that is primarily neurotoxic in NHIE. In this study, we investigated the neuroprotective effect of AG on HI brain damage in neonatal rats and the temporal profiles of NO.
METHODS
HI insult.
HI brain damage was induced basically by means of previously described methods (21). All procedures for the care and use of the animals conformed to the Guidelines for Animal Experiments of Kyoto University. Briefly, 7-d-old Wistar rat (SLC, Inc., Kyoto, Japan) pups (day of birth = d 1) were prepared for surgery. Under ether anesthesia, the left carotid artery was doubly ligated and then severed between the two ligatures. After surgery, the rat pups were allowed to recover for 2 to 3 h, and at the same time were fasted for 3 h before hypoxic exposure by separating them from their dams. The hypoxic exposure was achieved by placing littermates (n = 8–10) in a 2.0-L airtight plastic box submerged in a 37.0°C water bath and flushed for 2.5 h with a humidified mixture of 8% oxygen and 92% nitrogen delivered at 1.1 L/min. After 0.5 h recovery in the 37.0°C water bath, they were returned to their dams until they were killed.
The littermates were randomly assigned to one of two groups, the AG or the vehicle group. There were no differences in sex and body weight between the two groups. AG sulfate was administered intraperitoneally at a dose of 300 mg/kg in 50 μL of PBS every 8 h, starting at 1 h before the hypoxic exposure (i.e. the second injection was given at 4.5 h after the end of the hypoxic exposure). The drug was administered for three consecutive days, for a total of nine times. Control pups received the vehicle (50 μL of PBS) according to the same schedule.
Determination of infarct volume.
The rat pups were perfusion-fixed with 10% formaldehyde buffered to pH 7.4 72 h after the HI insult at 10 d of age (control group, n = 13; AG group, n = 18). The pups were anesthetized with chloral hydrate intraperitoneally and were perfused with heparinized saline followed by a neutral-buffered formaldehyde solution through the left ventricle. Each pup was decapitated after perfusion, and the head was stored in the same fixative at 4°C for at least 24 h. Coronal blocks of the brain were processed in a graded series of ethanol and xylene. After embedding in paraffin wax, 6-μm-thick coronal slices were sectioned and stained with hematoxylin-eosin.
Infarcted areas were calculated at five coronal levels selected according to the stereotaxic atlas of the 10-d-old rat brain (22). The following coronal levels with longitudinal coordinates and anatomic structures were chosen: A 5.6 mm at the caudate-putamen and anterior commissure; A 4.4 mm at the globus pallidus; A 3.2 mm at the anterior tip of the dorsal hippocampus; A 2.0 mm at the middle of the dorsal hippocampus; and A 0.8 mm at the ventral hippocampus. All area measurements were performed on maps of these five coronal levels, by using an image analyzer (National Institutes of Health image) with a magnification factor of 33 800 pixels/mm2. The measurements included the cerebral cortex above the rhinal fissure in all five coronal planes and the striatum in two coronal planes at A 5.6 mm and A 4.4 mm. The percentage volume occupied by HI damage in the cortex and the striatum was obtained by dividing the sum of the damaged areas ipsilateral to the carotid ligation by the sum of the ipsilateral areas of each structure.
Measurement of NO.
We used a previously reported method (20) to measure NO metabolites at 0 h, 1 h (during hypoxia), 2.5 h (end of hypoxia), 6 h, 24 h, and 72 h after the start of hypoxia. At each time point, some rats were decapitated (AG-treated group, n = 5–8; vehicle-treated group, n = 5–12), and each side of the cerebral cortex was removed onto an ice-cooled glass plate. After adding 1.5 mL of buffer (0.1 M potassium phosphate, pH 7.5, 20 mM EDTA), the tissue was homogenized. The supernatant was obtained through centrifugation (14 000 rpm for 10 min, then for 20 min) and was filtered with Centricon 10 (Amicon, Beverly, MA), 5 ×g for 1 h to remove Hb. NO metabolites in the supernatant were measured by the chemiluminescence method (23), using an NO analyzer (FES-450, ScholaTech, Osaka, Japan). Samples were injected into a sealed vial containing a saturated ascorbic acid solution. All NO metabolites, most of which consist of nitrite and nitrate (6), are reduced to NO. NO was carried on a constant stream of argon gas to a reaction chamber. Light emitted from the chemiluminescence reaction of NO and ozone was detected by a photomultiplier tube. The value of NO metabolites were expressed as picomoles per milligram of soluble protein. Protein was assayed with the method of Bradford (24).
Data analysis.
The results are presented as mean ± SEM. The death rate of the animals and the sex differences were analyzed with the χ2 test, and the difference in body weight with the two-tailed unpaired Student's t test. The infarcted area in the cortex and striatum was not distributed normally, so differences were assessed with the two-tailed Mann-Whitney U test. The concentrations of NO metabolites in the cortices of ligated and unligated sides were compared by means of the two-tailed paired Student's t test. Comparisons of NO metabolites in each side of the cortex between the two groups were made by the unpaired Student's t test. Differences were considered significant at p < 0.05.
RESULTS
The mortality rates until planned time of killing (72 h after HI insult) did not differ significantly between the groups with the AG treatment (28%, eight of 29) and with the vehicle treatment (21%, five of 24). All but one (a vehicle-treated control) died during the hypoxic exposure. Body weight gain did not differ between the two groups, with the weight changing from 10.6 ± 0.2 g on d 7 to 12.5 ± 0.5 g on d 10 in the AG-treated pups (n = 18) and from 10.6 ± 0.1 g on d 7 to 12.3 ± 0.4 g on d 10 in the vehicle-treated controls (n = 13).
Infarct volume.
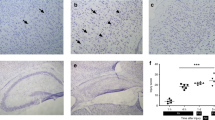
Table 1 shows that AG significantly protected against HI infarction both in the neocortex and in the striatum (p < 0.001). Cortical and striatal infarctions with HI changes were sharply demarcated and were easily delineated by using the image analyzer. Some scattered ischemic neurons were found outside the gross lesions. The mean percent volume of HI brain damage of the AG-treated pups showed 89% reduction in the cerebral cortex and 90% reduction in the striatum relative to that of the vehicle-injected controls. Figure 1 shows the typical distribution of macroscopic brain damage in this model, indicating that gross infarction was seen in the neocortex and the striatum, and was significantly reduced by AG treatment. The protective effect of AG on the brain was seen in all coronal sections both in the cerebral cortex and the striatum (Fig. 2;p < 0.001).

Coronal brain sections showing the middle of the dorsal hippocampus in the coronal plane A 2.0 mm selected with the aid of a 10-d-old rat brain atlas (22). A, vehicle-injected control. Arrowheads indicate HI infarction. B, rat pup given AG intraperitoneally 1 h before hypoxia and then every 8 h. Neuroprotective effect is evident. Hematoxylin-eosin stain, original magnification ×3.125.

Extent (mm2) of HI brain damage in neonates in different coronal planes of the cerebral cortex (A) and striatum (B) from caudal to rostral. There are significant differences (p < 0.001; Mann-Whitney U test) between vehicle-treated controls and AG-treated pups in all coronal planes in both cerebral cortex and striatum. Data represent mean ± SEM. (control group, n = 13; AG group, n = 18).
Changes in NO metabolites.
The results are shown in Figure 3. In the vehicle-treated controls, there were two significant peaks in NO metabolites in the ligated side compared with the unligated side. One peak occurred during hypoxia, at 1 h, and the other after hypoxia, at 72 h. In the AG-treated group, the concentration of NO metabolites in the ligated side substantially increased during the hypoxic phase, and although the peak was lower than that in the vehicle-treated controls, the difference was not statistically significant (p = 0.37). After hypoxia, the concentrations of NO metabolites in the ligated side did not increase and were significantly lower than those in the vehicle-treated controls at all three times. No obvious increase in NO metabolites was noted in the unligated side in either controls or the AG-treated pups. Except at 24 h, no concentrations of the NO metabolites in the unligated side differed at any time between the vehicle-treated and the AG-treated groups.

NO metabolites in the ligated (open circles) and the unligated side (closed circles). In the vehicle-treatment group (A), there were more NO metabolites in the ligated than in the unligated side at 1 (during hypoxic exposure), 6, 24, and 72 h (after hypoxia) (*p < 0.05). In the AG-treated group (B), NO metabolites in the ligated side increased only at 1 h (*p < 0.05). NO metabolites in the ligated side of the AG-treated group were significantly lower than those of the vehicle group at 6, 24, and 72 h, but not at 1 h (¶p < 0.05). NO metabolites in the unligated side of the AG-treated group did not differ from those in the vehicle group at any times except at 24 h, when the concentration of NO metabolites in the AG-treated group was lower than that in the vehicle group (†p < 0.05). Results are presented as mean ± SEM.
DISCUSSION
We demonstrated that AG significantly reduced infarct size in a neonatal HI rat model. The temporal profile of NO metabolites indicates that neuroprotection by AG is attributable to suppression of NO produced by iNOS after the end of the hypoxic period, during the reoxygenation phase. The results of this study imply that even delayed treatment with AG may ameliorate HI brain damage. iNOS continuously produces large amounts of NO, whereas nNOS produces NO intermittently, only when the intracellular calcium concentration increases (4). We have demonstrated that NO concentrations increase transiently during ischemia and rise again during reperfusion, and that the first increase derives from nNOS and the second one from iNOS (20). We consider that iNOS-derived NO is the main detriment in NHIE, inasmuch as the amount and duration of this type of NO production are much more pronounced than those of nNOS-derived NO. We also think that suppression of iNOS-derived NO is practically possible, because it increases later after the onset of HI than does nNOS-derived NO. We therefore believe that selective inhibition of iNOS is the best strategy for managing NHIE.
Two peaks of NO metabolites occurred in the lesioned side of the cortex in the neonatal rat, one during hypoxia and the other during the reoxygenation period. Under physiologic conditions, NO is degraded into nitrite and nitrate (6), and concentrations of nitrite and nitrate reflect NO production after cerebral ischemia (25). The same temporal changes in NO with a biphasic increase as seen in our model were reported for transient unilateral MCAO in an adult rat when measured by a microsensor placed in the cortex (26). In transient bilateral MCAO in an adult rat, the nitrite anion concentration in the striatum, monitored by means of in vivo microdialysis, transiently increases during the ischemia, and rises again immediately after the cessation of 21 min of ischemia (27).
The second peak in NO metabolites observed in the ligated side of the brain in control pups is thought to be introduced by iNOS. Our finding that AG, a relatively selective iNOS inhibitor, suppressed the second peak supports this notion. As further support for this hypothesis, transient expression of iNOS mRNA is noticed only from 12 to 60 h after the induction of hypoxia in the same HIE model of a neonatal rat (17), and expression of iNOS mRNA increases from 6 to 24 h after intrauterine HI insult (28). In addition, adult rat model experiments have shown that iNOS enzymatic activity increases in the postischemic brain. In transient ischemia, iNOS mRNA expression peaks at 12 h and returns to baseline at 4 d, whereas in permanent ischemia, iNOS mRNA is first observed at 12 h, peaks at 48 h, and disappears by 7 d (10, 11). On the other hand, expression of nNOS mRNA is reduced from 24 h after the intrauterine HI insult up to postnatal d 14 (28), and constitutive NOS activity from 24 h to 7 d after MCAO in an adult rat (29).
Although the first peak of NO metabolites was not suppressed by AG, the AG-treated pups showed a marked reduction of infarct volume. This indicates that delayed treatment may also be effective. Iadecola et al. (12) reported that treatment started even 24 h after permanent MCAO in adult rats could ameliorate cerebral ischemic damage. Cockroft et al. (13), however, reported that AG retained its neuroprotective activity when administered up to 2 h but not 3 h after the onset of cerebral ischemia in a triple-vessel model of an adult rat, in which two vessels were permanently, and the other was temporarily, occluded. These studies suggest that NO produced by iNOS can transform ischemic but potentially viable brain tissue into an irretrievably infarcted lesion, resulting in large strokes, and that AG can prevent the transformation. The implications of the observation that a 24-h delay in AG treatment can still reduce brain damage are extremely attractive, since asphyctic brain damage is unpredictable in most clinical situations and therapeutic drugs have to be administered after the episode. NOS activities in the rat forebrain 1 wk postnatally do not differ very much from those in an adult rat (30); therefore, delayed AG treatment may also protect against NHIE.
The concentration of NO metabolites peaked at 72 h in the ligated side in the vehicle-treated controls, and decreased slightly at 96 h in our preliminary investigation (data not shown). Therefore, administration of AG for up to at least 3 d is needed to have an ameliorative effect on brain damage. Administration of AG for up to 2 d after permanent MCAO in an adult rat does not, but treatment for 3 d or 4 d does, significantly reduce injury volume (14). Palmer et al. (31) reported that administration of AG twice a day for up to 2 d at a dose of 100 mg/kg failed to reduce atrophy at d 14 of recovery in a neonatal HI rat model. This negative result might be caused by the short duration of treatment, low dosage of AG, or a combination of the two.
Our treatment appears to be close to the optimal dosage and time course for AG treatment in immature rats, judging from the significant protective effect of AG and from the half-life of AG, which is estimated to be between 6 and 8 h in vivo (8), and approximately 4.4 h in human with normal renal function (32). In our preliminary study, administration of AG three times daily at a dose of 100 mg/kg did not show obvious neuroprotective effect (data not shown). Administration of AG twice a day at doses of 50 mg/kg does not, but that of 100 mg/kg does, show a significant decrease in infarct volume, and administration of 400 mg/kg shows the greatest reduction in an adult rat with permanent MCAO (15). Furthermore, a single administration of a dose of 320 mg/kg AG does, but that of 160 mg/kg AG does not, demonstrate a significant protective effect at 24 h after occlusion in a triple-vessel model in an adult rat (13).
Little is known about the influence of AG on neonatal physiologic conditions. Although changes in physiologic variables, such as arterial pressure, blood gases, plasma glucose, or rectal temperature, may affect brain damage, measuring them is not technically feasible in small 7-d-old pups. In the experimental model used for our study, cerebral blood flow is restored to normal from 30 min up to 24 h after the end of HI insult, and a moderate decrease in cerebral blood flow in the ipsilateral hemisphere at 3 and 6 d of recovery occurs as a result, rather than a cause, of tissue necrosis seen at recovery (33). Therefore, it is unlikely that alterations in cerebral blood flow, which are in part controlled by endothelial NOS, contribute substantially to the neuroprotective effect of AG. Previous experiments in adult ischemic models showed no significant differences in those variables between AG-treated animals and controls (13, 14).
The reduction in infarct size afforded by AG is reportedly antagonized by L-arginine, but not by D-arginine, indicating that the protective effect is mediated through the L-arginine–NO pathways (12, 16). Although AG is an inhibitor of tissue polyamine oxidase, and prevents the cytotoxic effect of spermine (34), this is an unlikely neuroprotective mechanism, because although increased polyamine synthesis and degradation began early after an ischemic insult (35), delayed AG treatment demonstrates a protective effect in an adult rat (15). AG is also an inhibitor of the formation of advanced glycation end products (36). Systematically administered advanced glycation end product–modified BSA increases cerebral infarct size in an adult rat, which is attenuated by AG (37). However, this is an unlikely neuroprotective mechanism of AG, inasmuch as normal basal concentrations of the products do not contribute significantly to the pathogenesis of stroke in nondiabetic animals (13).
Abbreviations
- HIE:
-
hypoxic-ischemic encephalopathy
- NHIE:
-
neonatal hypoxic-ischemic encephalopathy
- HI:
-
hypoxia-ischemia hypoxic-ischemic
- NO:
-
nitric oxide
- NOS:
-
nitric oxide synthase
- iNOS:
-
inducible nitric oxide synthase
- nNOS:
-
neuronal nitric oxide synthase
- AG:
-
aminoguanidine
- MCAO:
-
middle cerebral artery occlusion
References
Aicardi J 1998 Neurological diseases in the perinatal period. In: Aicardi J (ed) Diseases of the Nervous System in Childhood, 2nd Ed. Mac Keith Press, London, 32–66.
Hattori H, Wasterlain C 1990 Excitatory amino acids in the developing brain: ontogeny, plasticity, and excitotoxicity. Pediatr Neurol 6: 219–228.
Strijbos PJLM, Leach MJ, Garthwaite J 1996 Vicious cycle involving Na+ channels, glutamate release, and NMDA receptors mediates delayed neurodegeneration through nitric oxide formation. J Neurosci 16: 5004–5013.
Iadecola C, Ross ME 1997 Molecular pathology of cerebral ischemia: delayed gene expression and strategies for neuroprotection. Ann N Y Acad Sci 835: 203–217.
Dawson VL, Dawson TM, London ED, Bredt DS, Synder SH 1991 Nitric oxide mediates glutamate neurotoxicity in primary cortical cultures. Proc Natl Acad Sci U S A 88: 6368–6371.
Gross SS 1995 Nitric oxide: pathophysiological mechanisms. Annu Rev Physiol 57: 737–769.
Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME 1997 Delayed reduction of ischemic brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci 17: 9157–9164.
Corbett JA, McDaniel ML 1996 Selective inhibition of inducible nitric oxide synthase by aminoguanidine. Methods Enzymol 268: 398–408.
Southan GJ, Szabó C 1996 Selective pharmacological inhibition of distinct nitric oxide synthase isoforms. Biochem Pharmacol 51: 383–394.
Iadecola C, Zhang F, Xu S, Casey R, Ross ME 1995 Inducible nitric oxide synthase gene expression in brain following cerebral ischemia. J Cereb Blood Flow Metab 15: 378–384.
Iadecola C, Zhang F, Casey R, Clark HB, Ross ME 1996 Inducible nitric oxide synthase gene expression in vascular cells after transient focal cerebral ischemia. Stroke 27: 1373–1380.
Iadecola C, Zhang F, Xu X 1995 Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am J Physiol 268: R286–R292.
Cockroft KM, Meistrell M III, Zimmerman GA, Risucci D, Bloom O, Cerami A, Tracey KJ 1996 Cerebroprotective effects of aminoguanidine in a rodent model of stroke. Stroke 27: 1393–1398.
Zhang F, Iadecola C 1998 Temporal characteristics of the protective effect of aminoguanidine on cerebral ischemic damage. Brain Res 802: 104–110.
Nagayama M, Zhang F, Iadecola C 1998 Delayed treatment with aminoguanidine decreases focal cerebral ischemic damage and enhances neurologic recovery in rats. J Cereb Blood Flow Metab 18: 1107–1113.
Zhang F, Casey RM, Ross E, Iadecola C 1996 Aminoguanidine ameliorates and L -arginine worsens brain damage from intraluminal middle cerebral artery occlusion. Stroke 27: 317–323.
Muramatsu K, Kato I, Yamaguchi N, Morikawa I, Hyodo J, Suzuki S, Kobayashi M, Togari H, Wada Y, Fujimoto I, Hukuda A, Nishino H 1996 The expression of inducible nitric oxide synthase mRNA in the neonatal rat brain after hypoxic-ischemic insult. Acta Neonatal Jpn 32: 634–636.
Hamada Y, Hayakawa T, Hattori H, Mikawa H 1994 Inhibitor of nitric oxide synthesis reduces hypoxic-ischemic brain damage in the neonatal rat. Pediatr Res 35: 10–14.
Higuchi Y, Hattori H, Hattori R, Furusho K 1996 Increased neurons containing neuronal nitric oxide synthase in the brain of a hypoxic-ischemic neonatal rat model. Brain Dev 18: 369–375.
Higuchi Y, Hattori H, Kume T, Tsuji M, Akaike A, Furusho K 1998 Increase in nitric oxide in the hypoxic-ischemic neonatal rat brain and suppression by 7-nitroindazole and aminoguanidine. Eur J Pharmacol 342: 47–49.
Rice JE, Vannucci RC, Brierley JB 1981 The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141.
Sherwood NM, Timiras PS 1970 A Stereotaxic Atlas of the Developing Rat Brain. University of California Press, Berkeley, CA, 15–75.
Brien JF, McLaughlin BE, Nakatsu K, Marks GS 1991 Quantitation of nitric oxide formation from nitrovasodilator drugs by chemiluminescence analysis of headspace gas. J Pharmacol Methods 25: 19–27.
Bradford MM 1976 A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254.
Togashi H, Mori K, Ueno K, Matsumoto M, Suda N, Saito H, Yoshioka M 1998 Consecutive evaluation of nitric oxide production after transient cerebral ischemia in the rat hippocampus using in vivo brain microdialysis. Neurosci Lett 240: 53–57.
Malinski T, Bailey F, Zhang G, Chopp M 1993 Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J Cereb Blood Flow Metab 13: 355–358.
Shibata M, Araki N, Hamada J, Sasaki T, Shimazu K, Fukuuchi Y 1996 Brain nitrite production during global ischemia and reperfusion: an in vivo microdialysis study. Brain Res 734: 86–90.
Cai Z, Hutchins JB, Rhodes PG 1998 Intrauterine hypoxia-ischemia alters nitric oxide synthase expression and activity in fetal neonatal rat brains. Dev Brain Res 109: 265–269.
Iadecola C, Xu X, Zhang F, El-Fakahany EE, Ross ME 1995 Marked induction of calcium-independent nitric oxide synthase activity after focal cerebral ischemia. J Cereb Blood Flow Metab 15: 52–59.
Lizasoain I, Weiner CP, Knowles RG, Moncada S 1996 The ontogeny of cerebral and cerebellar nitric oxide synthase in the guinea pig and rat. Pediatr Res 39: 779–783.
Palmer C, Roberts RL 1997 Long term brain injury in the neonatal rat is reduced by nitric oxide synthase inhibition induced after a hypoxic-ischemic insult. Pediatr Res 41: 294Aabstr
Foote EF, Look ZM, Giles P, Keane WF, Halstenson CE 1995 The pharmacokinetics of aminoguanidine in end-stage renal disease patients on hemodialysis. Am J Kidney Dis 25: 420–425.
Mujsce DJ, Christensen MA, Vannucci RC 1990 Cerebral blood flow and edema in perinatal hypoxic-ischemic brain damage. Pediatr Res 27: 450–453.
Brunton VG, Grant MH, Wallace HM 1994 Spermine toxicity in BHK-21/C13 cells in the presence of bovine serum: the effect of aminoguanidine. Toxicol In Vitro 8: 337–341.
Paschen W, Cleef M, Röhn G, MŸller M, Pajunen AEI 1993 Ischemia-induced disturbances of polyamine synthesis. Prog Brain Res 96: 147–160.
Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A 1986 Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science 232: 1629–1632.
Zimmerman GA, Meistrell M III, Bloom O, Cockroft KM, Bianchi M, Risucci D, Broome J, Farmer P, Cerami A, Vlassara H, Tracey KJ 1995 Neurotoxicity of advanced glycation endproducts during focal stroke and neuroprotective effects of aminoguanidine. Proc Natl Acad Sci U S A 92: 3744–3748.
Author information
Authors and Affiliations
Additional information
Supported by a research grant (9B-1) for Nervous and Mental Disorders from the Japanese Ministry of Health and Welfare.
Rights and permissions
About this article
Cite this article
Tsuji, M., Higuchi, Y., Shiraishi, K. et al. Protective Effect of Aminoguanidine on Hypoxic-Ischemic Brain Damage and Temporal Profile of Brain Nitric Oxide in Neonatal Rat. Pediatr Res 47, 79 (2000). https://doi.org/10.1203/00006450-200001000-00015
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200001000-00015
This article is cited by
-
Nitric oxide and the brain. Part 2: Effects following neonatal brain injury—friend or foe?
Pediatric Research (2021)
-
Free radicals and neonatal encephalopathy: mechanisms of injury, biomarkers, and antioxidant treatment perspectives
Pediatric Research (2020)
-
Inflammatory responses in hypoxic ischemic encephalopathy
Acta Pharmacologica Sinica (2013)
-
Putative Agmatinase Inhibitor for Hypoxic-Ischemic New Born Brain Damage
Neurotoxicity Research (2013)
-
The Role of Erythropoetin in Ischemic Preconditioning, Postconditioning, and Regeneration of the Brain after Ischemia
Neuroscience and Behavioral Physiology (2011)
