
Abstract
We investigated the adverse effect of hypocapnia on the neonatal rabbit brain. Two-week-old Japanese white rabbits were assigned to three groups, hyperventilation (H group), ischemia (I group), or hypocapnia with ischemia (HI group) and then subjected for 1.5 h with simultaneous measurement of the mean arterial blood pressure (MABP) and intracranial Hb concentration changes. Marked reductions of Paco2 and MABP were induced in the hyperventilation-loaded groups and the ischemia-loaded groups, respectively. The intracranial oxyhemoglobin and total Hb concentrations decreased slightly in the H group and markedly in the I and HI groups after the start of experimemtal protocols, although there were no statistical differences between the I and HI groups. Animals were killed at 24 h after experiments and then subjected to pathologic examination. Damaged neurons with shrunken cell bodies and nuclear changes were found on light microscopic examination, mainly in the pyramidal cell layer of the subiculum and cornu ammonis 1. The numerical density of damaged neurons was significantly higher in the HI group than those in the H or I groups (p < 0.05). These damaged neurons were positive on DNA nick end labeling. A DNA ladder was detected on electrophoresis with a DNA sample extracted from hippocampal tissue in the HI group, but not in the other two groups. On electron microscopic examination, not only condensation of the nucleus but also disruption of mitochondria and the cell membrane were detected. These results suggested that hypocapnia under hypotension might cause neuronal cell death in the hippocampus of neonatal rabbit. Not only ischemia but also a metabolic change induced by hypocapnia might contribute to this apoptotic neuronal cell damage.
Similar content being viewed by others

Main
Hyperventilation is often performed as a therapy for cardiopulmonary care, especially for persistent pulmonary hypertension of the newborn in neonatal care unit. However, it has been suggested that prolonged deep hypocapnia is one of the risk factors for periventricular leukomalacia (1–6) and pontosubicular neuron necrosis (PSN) (7) in neonates. Many experiments have shown that hypocapnia induces constriction of cerebral vessels and decreases cerebral blood flow (8–15). Furthermore, it is suggested that hypocapnia exacerbates hypoxic-ischemic brain damage in neonatal rat pups and hypercapnia due to inhalation of CO2 prevents it (16, 17). Thus, hyperventilation, after hypocapnia, may have an adverse effect on neonatal brains. However, the pathomechanisms of hypocapnia-associated brain damages are not enirely understood. To clarify these issues, we conducted animal experiments using neonatal rabbits subjected to hyperventilation and evaluated the intracranial Hb and the brain damage.
MATERIALS AND METHODS
Surgical preparation.
Twenty-six female rabbits (2 wk old, Japanese White), weighing 225–310 g, were allowed free access to food and water until the experiments. The animals were anesthetized with urethane (1 g/kg i.p.). After tracheostomy and intubation, the animals were paralyzed with pancronium bromide (0.2 mg/kg i.v.) and then subjected to mechanical ventilation (Harvard Rodentventilator Model 683; Harvard Apparatus Co., Natick, MA, U.S.A.) with room air. The initial respirator settings were as follows: tidal volume, 10 mL/kg, and respiratory rate, 30 cycles/min. The right femoral artery was cannulated to continuously monitor the mean arterial blood pressure (MABP) (Nihon Kohden, Tokyo, Japan) and to obtain blood samples (200 μL) for blood gas analysis (Ciba-Corning model 288; CHIRON, Emeryville, CA, U.S.A.). Body temperature was maintained at 37 ± 0.2°C with a heat controller (Small animals heat controller; Unique Medical, Tokyo, Japan).
Experimental protocols.
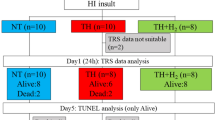
Animals were divided into three groups: hyperventilation group (H group;n = 8, for pathologic examination;n = 2, for DNA sample), ischemia group (I group;n = 8, for pathologic examination;n = 2, for DNA sample), and hyperventilation and ischemia group (HI group;n = 8, for pathologic examination;n = 2, for electon microscopy;n = 2, for DNA sample). After stabilization and blood gas analysis, hyperventilation was performed at a respiratory rate of up to 100 cycles/min with a fixed tidal volume in the H group, and MABP of less than 50 mm Hg was achieved by withdrawal of blood from a femoral artery in the I group, respectively. Hyperventilation and blood withdrawal were combined in the HI group. Those were maintained for 1.5 h in all three groups. Second blood gas analysis was performed at 30 min after the start of protocol. At the end of experiments, the respiratory rate returned to 30 cycles/min and blood returned to the veins.
Measurement of intracranial Hb.
The optodes of a near-infrared spectroscopy (NIR-1000; Hamamatsu Photonics, Hamamatsu, Japan) were attached on the temporal scalp bilaterally to measure changes in the intracranial Hb concentration with near-infrared light, of which the wavelengths were 780, 808, 830, 847, 867, and 911 nm.
Pathologic examination.
Animals were killed under deep anesthesia at 24 h after experiments. They were perfused through the left ventricle with heparinized saline, followed by 4% paraformaldehyde in 0.1 M phosphate buffer, pH 7.4. Then each brain was removed and preserved in 4% paraformaldehyde overnight at 4°C. Coronal sections of the cerebrum 3 mm thick, initially sectioned at the optic chiasma, were obtained. After the sections were embedded in paraffin and cut into 4-μm sections, pathologic examination was performed with hematoxylin and eosin. The incidence of damaged neurons [= (damaged neurons) × 100/(damaged neurons + intact neurons)] in the hippocampus was calculated in three separate areas per specimen under high power.
DNA nick end labeling.
Terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP)-biotin nick end labeling (TUNEL) was performed on hippocampal sections. The sections were deparaffinized and incubated with 20 μg/mL proteinase K (Sigma Chemical Co., St. Louis, MO, U.S.A.) for 20 min at 37°C, followed by washing with PBS (PBS, pH 7.4) for 2 min, four times. Then the sections were covered with equilibation buffer (30 mM Trizam base, pH 7.4, 140 mM sodium cacodylate, 1 mM cobalt chloride). TdT and dUTP in equilibration buffer were added to the sections followed by incubation at 37°C for 20 min. The reaction was stopped by washing with PBS. After internal peroxidase block, the sections were covered with peroxidase-conjugated streptoavidine for 30 min. Color development was performed with 0.02 M diaminobenzidine tetrahydrochloride as the chromogen in 0.05 M Tris buffer, pH 7.4, containing 0.006% H2O2.
Electon microscopy.
Specimens of the hippocampal formation in the HI group were sectioned and fixed with 2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4, for 2 h. Then specimen was postfixed with 2% OsO4 for 2 h, dehydrated with ethanol, and then embedded in Epon. A semithin section was stained with toluidine blue and an ultrathin section (60 nm) was stained with uranyl acetate and lead citrate. Electron microscopic examination was performed with an H-7000 (Hitachi Co., Ltd., Tokyo, Japan), followed by photography.
DNA electrophoresis.
Fresh brain samples of the hippocampus in each group and unoperated controls were removed rapidly and stored at –80°C until DNA extraction. Then 80 mg of the tissues were suspended in 50 μL of hypotonic lysis buffer (0.1% Triton X-100, 5 mM Tris-HCl, pH 8.0, 20 mM EDTA) for 30 min, and then centrifuged at 2500 rpm for 5 min, the supernatant being removed two times. The samples were incubated with 10 μL of ribonuclease, 20 μL of an enzyme activator solution, and 10 μL of protein digestion enzyme (Apotosis Ladder Detection Kit; Wako Pure Chemical Industries, Ltd., Tokyo, Japan) at 50°C for 30 min. Then, to the samples were added 300 μL of a DNA extraction solution (Apotosis Ladder Detection Kit) and 300 μL of isopropanolum. After shaking of the tubes, the samples were stood at room temprature for 15 min and then centrifuged in a 10,000 gradient for 5 min. The precipitates were washed with 70% ethanol two times and then dissolved in 20 μL of Tris-CL-EDTA buffer. Then 10-μL samples were separated electrophoretically on 1.5% agarose gels and stained with SYBRTM Green I (Molecular Probe, Inc., Eugene, OR, U.S.A.). DNA images were visualized under UV light with a AlphaImager 2000 (α Inotech Corporation, San Leandro, Berkeley, CA, U.S.A.).
Statistical analysis.
A one-way factorial ANOVA, followed by a post hoc test, was used for intergroup comparisons. Comparisons between intragroup values were performed by means of one-way repeated-measures ANOVA. Statistic software, Stat View (Abacus Concepts, Inc., CA, U.S.A.), was used. The results were expressed as means ± SEM.
Approval of the animal experiment.
This experiment was approved by the Animal Experiment Committee of the National Institute of Neuroscience, NCNP, Japan.
RESULTS
The physiologic parameters are listed in Table 1. There were no statistically significant differences in body weight, blood gas values, or MABP at 30 min before the start of experimental protocol among the three groups. Hyperventilation significantly reduced Paco2, and increased pH and Pao2 at 30 min after the start of experimental protocol. Blood withdrawal significantly reduced MABP to below 50 mm Hg. There were statistically significant differences among the three groups in pH, Paco2, MABP (p < 0.01), and Pao2 (p < 0.05) at 30 min after the start of experimental protocol.
The intracranial deoxyhemoglobin concentration slightly increased in all three groups after the start of experimental protocol. The intracranial oxyhemoglobin and total Hb concentrations decreased markedly in the I and HI groups, and slightly in the H group (Fig. 1). There were statistically significant differences between the H group and the other two groups, but not between the I and HI groups at 30, 60, and 90 min after the start of experimental protocol.

Comparison of the parameters of a near-infrared spectroscopy. Deoxyhemoglobin (HbR) increased slightly in all three groups. Oxyhemoglobin (HbO2) and total Hb (tHb) markedly decreased in the I and HI groups, and slightly decreased in the H group. There were statistically significant differences between the H group and the other two groups at 30–90 min, but not between the I and HI groups. Squares, H group; circles, I group; triangles, HI group. Error bars indicate SEM. *p < 0.05.
On pathologic examination, damaged neurons with shrunken cell bodies with eosinophilia, condensation of nucleus, and karyorrhexis were detected mainly in the hippocampal subiculum and cornu ammonis 1(CA1), being especially prominent in the HI group (Fig. 2, A–C). These damaged neurons were positively labeled with the TUNEL method (Fig. 2, D). On electron microscopic examination, damaged neurons showed central condensation and fragmentation of nuclear chromatin, and vacuoles within the cytoplasm (Fig. 2, E). These vacuoles comprised swollen organelles such as mitochondria, Golgi apparatus, and endoplasmic reticulum (Fig. 2, E, F). There was partial disruption of cell membrane (Fig. 2, G) but no apoptotic bodies.

Histologic change in the subicular neurons. On HE staining (A) H group; (B) I group; (C) HI group shrunken cell bodies with condensation of the nucleus (arrows) and karyorrhexis (arrowheads) were found in all three groups. The incidence of damaged neurons was most prominent in the HI group. These damaged neurons were positive with the TUNEL method (D). On electron microscopic examination, typically damaged neuron showed central aggregation and fragmentation of chromatin (E, N1 and N2) in the HI group. Many vacuoles were found in the cytoplasm, suggesting swelling of the endoplasmic reticulum (arrow in N1) and several organelles (arrowhead in N2) Under high power, swollen mitchondria (arrow) and a broken crista (arrowhead) were found (F). The cell membrane was also disrupted (arrowhead in G)
Electrophoresis of DNA extracted from the hippocampus showed an oligonucleosomal ladder in the HI group, but not in the control or the other two groups (Fig. 3).

On electrophoresis, an oligonucleosomal ladder was detected for the HI group, but not for the other two groups. M, marker of 123 bp repeat; lane 1, noninstrumental control; lane 2, H group; lane 3, I group; lane 4, HI group.
The incidences of damaged neurons were significantly higher in the HI group than in the H or I group in the subiculum and CA1 (p < 0.05) (Fig. 4).

The incidence of damaged neurons was significantly higher in the HI group than in the other two groups in both the subiculum and CA1. Open bar, H group; hatched bar, I group; filled bar, HI group. Error bar indicates SEM. *p < 0.05.
DISCUSSION
The present study firstly indicated that prolonged deep hypocapnia under hypotension induces hippocampal neuronal death in neonatal rabbit brains. In our results, hyperventilation or blood withdrawal alone seldom caused hippocampal neuron injury in healthy neonatal rabbits. However, the combination of hypocapnia and hypotension at the same time caused marked injury to hippocampal pyramidal neurons. These findings suggest that hypocapnia itself may not cause brain injury under healthy conditions, but may adversely affect the neonatal brain under pathologic conditions.
The precise pathomechanism regarding why the hypocapnia affects neuronal tissue is not entirely understood. However, several adverse effects are described in previous studies. Prolonged hypocapnia reduces the cerebral blood flow (8–15) and tissue oxygen consumption (16–18). These findings suggest that prolonged hypocapnia may induce hypoxia and ischemia in the CNS. However, in our results, reductions of intracerebral oxyhemoglobin and total Hb in the H group were less than those in the I group, but there were only a few damaged hippocampal neurons without any statistically significant differences between those two groups. Therefore, the reduction of only CBF is not enough to explain the neuronal damages in the present study.
Another explanation for the adverse effect is an increase in the extracellular pH. Although the extracellular pH in the brain was not measured in this study, it has been reported that the cerebrospinal fluid pH increases transiently as the arterial blood pH increases (21). In several experiments, an increase in the extracellular pH increases neuronal cell excitability through the activation of excitatory amino acid receptors (22), and a decrease in the extracellular pH inhibits these receptors and voltage-dependent Ca channels to prevent neuronal injury (23, 24). Furthermore, an increase in pH causes the production of reactive oxygen species and the release of cytochrome c as a result of mitochondrial transmembrane voltage disruption (25), this step being essential for the induction of apoptosis (26–28). These findings suggest that hypocapnia alone may adversely affect neuronal cells and induce apoptotic cell death.
In the present study, damaged neurons showed TUNEL positivity (29). In addition, DNA extraction from hippocampal tissue of the HI group revealed an oligonucleosomal ladder (30). These findings suggest that apoptosis contributes to neuronal death (31). Although hallmarks of apoptosis were observed, disruption of mitochondria and the cell membrane, and no apoptotic bodies, as opposed to apoptosis, were also detected on electron microscopy (32). These results support the continuity of apoptotic and necrotic morphologic changes detected in the process of programed cell death (33, 34), and features of both apoptosis and necrosis exist in the same cell of ischemic brain injury (35). Thus, apoptotic as well as necrotic processes might contribute to the neuronal death detected in our study.
Pontosubicular neuron necrosis (PSN) is one of the pathologic conditions frequently detected in autopsied neonatal brains (36, 37). PSN is characterized by selective neuron necrosis distributed in the pontine nuclei and subiculum of the hippocampus. Necrotic neurons represent the morphologic changes of nuclear condensation and karyorrhexis, and have some apoptotic aspects (38, 39). Furthermore, hypocapnia is thought to be a risk factor for PSN (7). Although necrotic neurons in our results were distributed mainly in the subiculum and rarely in the pontine nuclei, there were some similar morphologic changes to apoptotic one. Therefore, our experiment might become a model of PSN.
In conclusion, hypocapnia with hypotension causes hippocampal neuronal death, representing both apoptotic and necrotic neuronal degeneration. The induction of neuronal damage depended not only on hypotension and the cerebral circulation but also on hypocapnia. Because hypocapnia may adversely affect the neonatal brain, it should be important to avoid prolonged deep hypocapnia in sick neonates.
Abbreviations
- CA1:
-
cornu ammonis 1
- CBF:
-
cerebral blood flow
- PSN:
-
pontosubicular neuron necrosis
- MABP:
-
mean arterial blood pressure
- TUNEL:
-
TdT-mediated dUTP-biotin nick end labeling
References
Calvert SA, Hoskins EM, Fong KW 1987 Etiological factors associated with the development of periventricular leukomalacia. Acta Paediatr Scand 76: 254–259
Fujimoto S, Togari H, Yamaguchi N, Mizutani F, Suzuki S, Sobajima H 1994 Hypocarbia and cystic periventricular leukomalacia in premature infants. Arch Dis Child 71: F107–F110
Graziani L, Spitzer AR, Mitchell DG, Merton DA, Stanly C, Robinson N, McKee L 1992 Mechanical ventilation in preterm infants: neurosonographic and developmental studies. Pediatrics 90: 515–522
Greisen G, Munck H, Lou H 1987 Severe hypocapnia in preterm infants and neurodevelopmental deficit. Acta Pediatr Scand 86: 401–404
Iida K, Takashima S, Takeuchi Y 1992 Etiologies and distribution of neonatal leukomalacia. Pediatr Neurol 8: 205–209
Ikonen RS, Janas MO, Koivikko MJ, Laippala P, Kuusinen EJ 1992 Hyperbilirubinemia, hypocarbia and periventricular leukomalacia in preterm infants: relationship to cerebral palsy. Acta Paediatr 81: 802–807
Hashimoto K, Takeuchi Y, Takashima S 1991 Hypocarbia as a pathogenic factor in pontosubicular necrosis. Brain Dev 13: 155–157
Gleason CA, Short BL, Jones MDJ 1989 Cerebral blood flow and metabolism during and after prolonged hypercapnia in newborn lambs. J Pediatr 115: 309–314
Hansen NB, Brubakk AN, Bartlid D, Oh W, Stonestreet BA 1984 The effect of variations in PaCO2 on brain blood flow and cardiac output in the newborn piglet. Pediatr Res 18: 1132–1136
Hansen NB, Nowicki PT, Miller RR, Malone T, Brikers R, Menke JA 1986 Alteration in cerebral blood flow and oxygen consumption during prolonged hypocarbia. Pediatr Res 20: 147–150
Kamei A, Ozaki T, Takashima S 1994 Monitoring of the intracranial hemodynamics and oxygenation during and after hyperventilation in newborn rabbits with near-infrared spectroscopy. Pediatr Res 35: 334–338
Reuter JH, Disney TA 1986 Regional cerebral blood flow and cerebral metabolic rate of oxygen during hyperventilation in the newborn dog. Pediatr Res 20: 1102–1106
Rosenberg AA 1992 Response of the cerebral circulation to hypocarbia in postasphyxia newborn lambs. Pediatr Res 32: 537–541
Wyatt JS, Edwards AD, Cope M, Delpy DT, McCormick DC, Potter A, Reynolds EOR 1991 Response of cerebral blood volume to changes in arterial carbon dioxide tension in preterm and term infants. Pediatr Res 29: 553–557
Young RSK, Yagel SK 1984 Cerebral physiological and metabolic effects of hyperventilation in the neonatal dog. Ann Neurol 16: 337–342
Vannucci RC, Towfighi J, Heitjan DF, Brucklacher RM 1995 Carbon dioxide protects the perinatal brain from hypoxic-ischemic damage: an experimental study in the immature rat. Pediatrics 95: 868–874
Vannucci RC, Brucklacher RM, Vannucci SJ 1997 Effect of carbon dioxide on cerebral metabolism during hypoxia-ischemia in the immature rat. Pediatr Res 42: 24–29
Kennealy JA, McLennan JE, Loudon RG, McLaurin RL 1980 Hyperventilation induced cerebral hypoxia. Am Rev Respir Dis 122: 407–412
Sugioka K, Davis DA 1960 Hyperventilation with oxygen: a possible cause of cerebral hypoxia. Anaethesiology 21: 135–143
Wilson DF, Pastuzka A, DiGiacomo JE, Pawlowski M, Schneiderman R, Delivoria-Papadopoulos M 1991 Effect of hyperventilation on oxygenation of the brain cortex of newborn piglets. J Appl Physiol 70: 2691–2696
Davies DG, Nolan WF 1982 Cerebral interstitial fluid acid-base status follows arterial acid-base perturbations. J Appl Physiol 53: 1551–1555
Giffard RG, Weiss JH, Choi DW 1992 Extracellular alkalinity exacerbates injury of cultured cortical neurons. Stroke 23: 1817–1821
Giffard RG, Monyer H, Christine CW, Choi DW 1990 Acidosis reduces NMDA receptor activation, glutamate neurotoxicity, and oxygen-glucose deprivation neuronal injury in cortical cultures. Brain Res 506: 339–342
Ou-Yang Y, Kristian T, Mellergard P, Siesjo BK 1994 The influence of pH on glutamate- and depolarization-induced increases of intracellular calcium concentration in cortical neurons in primary culture. Brain Res 646: 65–72
Majima HJ, Oberley TD, Furukawa K, Mattson MP, Yen HC, Szweda LI, St Clair DK 1998 Prevention of mitochondrial injury by manganese superoxide dismutase reveals a primary mechanism for alkaline-induced cell death. J Biol Chem 273: 8217–8224
Zamzami BN, Marchetti P, Castedo M, Zanin C, Vayssiere JL, Petit PX, Kroemer G 1995 Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo. J Exp Med 181: 1661–1672
Zamzami N, Susin SA, Marchetti P, Hirsch T, Gometz-Monterrey I, Castedo M, Kroemer G 1996 Mitochondrial control of nuclear apoptosis. J Exp Med 183: 1533–1544
Zamzami BN, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B, Kroemer G 1995 Sequential reduction of mitochondrial transmembrane potential and generation of reactive oxygen species in early programmed cell death. J Exp Med 182: 367–377
Gavrieli Y, Sherman Y, Ben-Sasson SA 1992 Identification of programed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol 119: 493–501
Bortner CD, Oldenburg NBE, Cidlowski JA 1995 The role of DNA fragmentation in apoptosis. Trends Cell Biol 5: 21–26
Choi DW 1996 Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol 6: 667–672
Deshpande J, Bergstedt K, Linden T, Kalimo H, Wieloch T 1992 Ultrastructural changes in the hippocampal CA1 region following transient cerebral ischemia: evidence against programmed cell death. Exp Brain Res 88: 91–105
Portera-Cailliau C, Price DL, Martin LJ 1997 Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol 378: 70–87
Portera-Cailliau C, Price DL, Martin LJ 1997 Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J Comp Neurol 378: 88–104
MacManus JP, Linnik MD 1997 Gene expression induced by cerebral ischemia: an apoptotic prespective. J Cereb Blood Flow Metab 17: 815–832
Friede RL 1972 Ponto-subicular lesion in perinatal anoxia. Arch Pathol 94: 343–354
Mito T, Kamei A, Takashima S, Becker LE 1993 Clinicopathological study of pontosubicular necrosis. Neuropediatrics 24: 204–207
Bruck Y, Bruck W, Kretzschmar HA, Lassmann H 1996 Evidence for neuronal apoptosis in pontosubicular neuron necrosis. Neuropathol Appl Neurobiol 22: 23–29
Stadelmann C, Bruck W, Bancher C, Jellinger K, Lassmann H 1998 Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. J Neuropathol Exp Neurol 57: 456–464
Author information
Authors and Affiliations
Additional information
This study was supported by grants from the Ministry of Health and Welfare, and the Ministry of Education, Science and Culture, Japan.
Rights and permissions
About this article
Cite this article
Ohyu, J., Endo, A., Itoh, M. et al. Hypocapnia Under Hypotension Induces Apoptotic Neuronal Cell Death in the Hippocampus of Newborn Rabbits. Pediatr Res 48, 24–29 (2000). https://doi.org/10.1203/00006450-200007000-00007
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200007000-00007
This article is cited by
-
Activation of the Central Histaminergic System is Involved in Hypoxia-Induced Stroke Tolerance in Adult Mice
Journal of Cerebral Blood Flow & Metabolism (2011)
-
Cannabinoid as a neuroprotective strategy in perinatal hypoxic-ischemic injury
Neuroscience Bulletin (2011)
-
Hypercapnia and hypocapnia in neonates
World Journal of Pediatrics (2008)
