
Abstract
We report effects of gene transfer and liver transplantation on urea synthesis in ornithine transcarbamylase deficiency (OTCD). We measured the formation of [15N] urea after oral administration of 15NH4Cl in two girls with partial OTCD before and after liver transplantation. Ureagenesis was less than 20% of that observed in controls before transplantation, and was normalized afterward. Studies performed on the OTCD sparse fur (spf/Y) mouse showed discordance between OTC enzyme activity and ureagenesis with modest increases in OTC enzyme activity after gene transfer resulting in significant improvement in ureagenesis. This study suggests that both liver transplantation and gene therapy may be effective in improving ureagenesis in OTCD.
Similar content being viewed by others
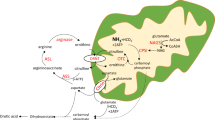


Main
Ornithine transcarbamylase deficiency (OTCD) is the most common inborn error of urea synthesis with a prevalence of about 1 in 40,000 births (1). It is an X-linked disorder (2) in which approximately 60% of hemizygous males present in the newborn period with a catastrophic illness of hyperammonemic coma (500–2000 μM; normal <30 μM) (3). Undetected or untreated, these infants usually die in the 1st week of life (4). Even with heroic treatment using dialysis, survival beyond the newborn period is only about 50%. Five-year survival rate using long-term nitrogen restriction and alternate pathway therapy is about 25% (4, 5). Virtually all surviving children with neonatal onset disease have developmental disabilities (6, 7). OTC activity in liver is absent to trace (4), and there is no appreciable urea synthesis as measured by stable isotope studies (8). Mutations causing neonatal disease usually are single base substitutions that affect amino acid residues in the interior of the enzyme, close to the active site (9).
In 40% of hemizygous males, symptoms develop later in childhood or even in young adulthood. Symptoms include episodes of vomiting, lethargy and bizarre behavior, which may progress to coma (10). During acute episodes plasma ammonium levels rise to 150–500 μM. OTC activity in liver is generally below 20% of control but above zero (4). Urea synthesis is also deficient but less so than would be predicted by OTC enzyme activity (11). Mutation analysis usually shows single base substitutions affecting amino acid residues on the surface of the protein (9).
Approximately 90% of females heterozygous for OTCD remain asymptomatic throughout life, while the remaining 10% present a clinical and biochemical picture similar to that described for partially deficient hemizygous males (12, 13). Hyperammonemia presumably results from adverse lyonization (14). Ureagenesis, as measured by stable isotope studies, is indistinguishable from control in asymptomatic heterozygotes and 15–40% of control in symptomatic heterozygotes (8). Long-term survival for both groups is around 80% but patients remain at risk for potential fatal hyperammonemic crises throughout life and must continue to restrict their protein intake and receive alternate pathway therapy (4). They are also at significant risk for having developmental disabilities, although these are generally less severe than in the neonatal onset hemizygous males (15, 16).
Because of the generally poor prognosis for affected individuals, there has been considerable effort placed on developing new treatment approaches. The current focus is on liver transplantation and gene therapy. A number of centers have started performing liver transplantation both on neonatally affected and partially deficient OTCD children. A recent review by Whitington (17) reports that 10 orthotopic liver transplants have been performed on children with OTCD in four centers. Of these, eight children are currently surviving 1–6 y post-transplant without the need for ongoing dietary restriction or alternate pathway therapy. No data were provided regarding metabolic correction. Neurologic outcome has correlated closely with clinical status before transplantation.
Gene therapy for OTCD remains a potential treatment rather than a reality. However, in many ways OTCD is an ideal target for human gene therapy. 1) The gene has been cloned;2) it is a relatively common inherited metabolic disease;3) the current clinical outcome is poor; and 4) there is a faithful animal model of the disorder, the sparse fur (spf/Y) mouse. The evidence that liver transplantation may improve outcome further suggests that liver directed gene therapy may be effective. Finally, the spectrum of disease manifestations found in OTCD indicates that even partial restoration of OTC activity may convert severe disease to a milder or even asymptomatic form. Studies in the spf/Y mouse with an adenoviral vector have resulted in metabolic improvement for up to 2–3 mo. OTC activity increased, plasma glutamine decreased, and urinary excretion of orotate decreased (18).
In the present study, we use the stable isotope 15NH4Cl as a probe to evaluate the effects of liver transplantation on ureagenesis in two girls with partial OTCD. We also report on the effects of gene transfer on ureagenesis in the spf/Y mouse. Our results suggest that both liver transplantation and gene therapy result in improved urea synthetic capacity in OTCD.
METHODS
Human subjects.
Two girls with partial OTCD were studied. The first, BS, was diagnosed as a manifesting heterozygote at age 6 y when she developed hyperammonemia (peak 200 μM). She was delirious but improved with i.v. alternate pathway therapy (sodium benzoate/sodium phenylacetate) followed by long-term treatment with a protein-restricted diet, sodium phenylbutyrate, and citrulline supplements. BS was subsequently found to have an arg-92-gln mutation in exon 3 of the OTC gene, which has been found in other neonatal onset OTCD families (9).
She did well on alternate pathway treatment until age 8 y when she began having increasingly severe hyperammonemic episodes that necessitated multiple hospitalizations despite appropriate dietary restriction and medication compliance. She also had marked anorexia requiring gastrostomy tube feedings.
At 9 y of age her parents opted for a liver transplant. She received a blood-matched full left lobe from an adult donor. The operation was completed without incident, and she was discharged without complications on postoperative d 11. Her liver function tests rapidly returned to normal. Since transplantation she has not required alternate pathway therapy or protein restriction. She has been clinically well without elevations in plasma ammonia levels for 9 mo, and her appetite has returned to normal so that she no longer requires tube feedings. Plasma arginine levels remained low after transplantation (12.3 μM, nl >30) so she has been supplemented with citrulline 330 mg/kg/d.
The second patient, EM, developed episodes of bizarre behavior beginning at 3 y of age, and at 4 y had an acute hyperammonemic episode (250 μM) that was diagnosed as Reye Syndrome. Over the next 9 mo she had episodes of irritability, abdominal pain, and vomiting occurring several times a week. The diagnosis of OTCD was made at 5 y of age based on orotic aciduria, elevated glutamine level and low citrulline level in plasma. It was confirmed by DNA analysis showing an arg-330-gly mutation in exon 9 of the OTC gene. She was treated with protein restriction and alternate pathway therapy using sodium phenylbutyrate and citrulline. Despite careful monitoring of the diet and metabolic therapy, she continued to have multiple hyperammonemic episodes (up to 300 μM) requiring hospitalizations for i.v. alternate pathway therapy. She also developed a feeding disorder and required gastrostomy tube feedings. Although her early development had been normal, it has lagged since the beginning of the hyperammonemic episodes. She repeated kindergarten and has entered a special education placement. Her cognitive functioning was in the borderline-normal range (IQ 70–75), and there was evidence of attention deficit disorder.
Because of her deteriorating status, her parents elected to undertake a liver transplant at 8 y of age. The surgical procedure was uncomplicated, but on d 6 she developed evidence of rejection, which was treated medically. She subsequently regained her appetite and at 9 mo after transplant received an unrestricted diet. Her only metabolic treatment was oral arginine supplements (150 mg/kg/d) for a low plasma arginine level (14 μM, nl>30). Plasma ammonium (32 μM) and glutamine (499 μM) were normal. Hypoargininemia has been reported in other transplanted urea cycle disorder patients (19). There were no further hyperammonemic episodes, and no appreciable change in her neurodevelopmental profile.
Spf/Y mouse colony.
The spf mouse colony used in these studies was originally obtained from Jackson Laboratories and has been backcrossed to C3HeB/J (C3H) mice for more than 20 generations. The colony was maintained on normal rodent diet (Teklad sterilizable rodent diet (W) 8656 North Penn Feeds Inc., Lansdale, PA). Animals had access to food ad libitum before and during the study. To produce spf/Y and littermate controls, heterozygous females were mated with wild type C3HeB/J. Spf/Y and male C3HeB/J progeny from this cross (6–10 wk of age) were used in this study.
Construction and propagation of the recombinant adenoviruses.
A second-generation recombinant adenovirus-carrying mouse OTC cDNA was used in the spf studies. It was constructed as described previously (18). Briefly, mouse OTC cDNA was generated by reverse transcription-polymerase chain reaction, cloned into a pGEM-T vector (Promega, Madison, WI), and restricted with SpeI and Sac II. A 1.5-kb fragment containing mouse cDNA was isolated, blunted, and cloned into the Eco RV site of an adenoviral vector pAd.CMV-linkI (18). The new plasmid, designated pAd.CMVmOTC, was lineralized with Eco RI and cotransfected into 293 cells with Cla I/ Xba I-restricted Ad5 viral DNA containing the temperature sensitive ts125 mutation in E2a (20) and the sub360 mutation in E3 (21). The resulting recombinant virus, designated H5.110CMVmOTC [for nomenclature of recombinant adenoviruses generated in the Institute for Human gene Therapy, University of Pennsylvania, see Englehardt et al. (22)] was purified through three rounds of plaque isolation. The second-generation β-galactosidase (lacZ) virus (H5.110CMV/lacZ) was constructed as described previously (23) and purified through three rounds of plaque isolation. Each virus was titered by a standard plaque-forming assay. The batches of virus used in the current study had a particle/pfu ratio of 40:1. The dose of virus used in the current study is based on our previous analysis of the dose-response relationship for this virus (18). The dose used in the current study (1 × 1011) particles/mouse) was one-half that used for many of the previous metabolic studies to minimize possible toxicity.
OTC enzyme activity.
Liver tissue was homogenized in mitochondria lysis buffer with a Polytron homogenizer. The homogenate was centrifuged in a microfuge at the maximum speed for 5 min, the supernatant was transferred to a new tube, and OTC enzyme activity was measured as described by Lee and Nussbaum (24) with modifications. Briefly, 2–10 μg of total cellular protein was added to 700 μL of reaction mixture (5 mM ornithine, 15 mM carbamyl phosphate, and 270 mM triethanolamine, pH 7.7), which was incubated at 37°C for 30 min. Reactions were stopped by adding 250 μL of 3:1 phosphoric acid/sulfuric acid (by volume). Citrulline production was then determined by adding 50 μL of 3% 2,3-butanedione monoxime, incubating at 95–100°C in the dark for 15 min, and measuring absorptance at 490 nm.
Orotic acid measurement.
Mouse urine was collected by housing the animals in metabolic cages overnight. Urinary orotic acid levels were determined by HPLC with UV detection according to Brusilow and Hauser (25) and expressed as a molar ratio to creatinine.
Stable isotope studies.
The two OTCD girls underwent a stable isotope study to measure ureagenesis before transplant and then 9 mo after liver transplant. There were no complications from the studies; specifically, there was no vomiting, nausea, or hyperammonemia (NH4 <40 μM). This study was approved by local IRB committees. Parental informed consent also was obtained.
15NH4Cl (>98.0 atom % excess) was purchased from Cambridge Isotope Laboratories (Woburn, MA). Reagents for derivatization of urea and amino acids were obtained from Pierce Chemical Co. (Rockford, IL). The studies were done in the morning, after an overnight fast. After obtaining baseline samples of blood, each subject received a single oral dose of 15NH4Cl (0.37 mmol/kg; 20 mg/kg) in chilled water. Heparinized venous blood specimens were taken at 0, 30, 60, 90, 120, 180, and 240 min after isotope administration. After centrifugation, plasma samples were kept frozen until they were analyzed. At 4 h the study was concluded.
The procedure used for the analysis of [15N]urea in blood has been described previously (8). Derivatization was done with the t-butyldimethylsilyl derivative (26). Blood urea was determined with the indophenol-hypochlorite method.
In the spf/Y mice the first stable isotope study was performed before administration of the adenovirus (d −3). On d 0, animals received virus (1 × 1011) particles) suspended in 0.1 mL of PBS via the tail vein. Stable isotope studies were again performed on d 7 or 14 after infusion. The animals were then killed and liver OTC enzyme activity was measured. On the day before the stable isotope study, mice were placed in metabolic cages for a 24-h urine collection for orotic acid and fasted overnight before the stable isotope study. On the day of the study, the animals received intraperitoneal 15NH4Cl (0.093 mmol/kg) at T = 0. Blood was sampled retro-orbitally at T = 15 and 30 min to measure 15N incorporation into urea. Heparinized blood samples were treated with 10% trichloroacetic acid and centrifuged to remove the precipitated proteins.
Isotopic enrichment (atom % excess) was calculated according to Rosenblatt et al. (27). This value represents the incremental label (%) over the baseline (T0) value. The concentration of labeled [15N]urea (mmol/L) is:MATH

Statistical analysis.
Statistical analyses were performed on results of the studies in the spf/Y mouse. Analysis of variance was used to compare OTC enzyme activity in virus-treated groups (7 and 14 d after gene transfer) versus untreated spf/Y and C3H controls. Analyses of orotate excretion and 15N incorporation into urea in the spf/Y mouse were designed to determine whether treatment altered levels from the animal's pre-gene transfer measure. Urinary orotate on d 7 or 14 was compared with each animal's pre-gene transfer level by an unpaired t test. Analysis of variance was used to compare 15N incorporation into urea (15 and 30 min post-challenge) on d 7 or 14 to each animal's pre-gene transfer level.
RESULTS
Ureagenesis after liver transplantation in patients with partialOTCD.
Both OTCD patients underwent a stable isotope study of 15N incorporation into urea before and 9 mo after liver transplantation. Before liver transplant they received a nitrogen-restricted diet (1.0–1.5 g protein/kg/d), sodium phenylbutyrate (2–3.5 mmol/kg/d) and citrulline (1 mmol/kg/d). After liver transplant they received an unrestricted diet and immunosuppressant therapy. Before gene therapy the girls had remarkably similar incorporation of 15N into urea, 0.18 (BS), and 0.19 (EM) mmol [15N]urea/240 min. This represents 16% and 17% of controls, respectively (8). After liver transplantation, urea incorporation increased 5–10 fold to 0.85 and 1.91 mmol [15N)]urea/240 min, respectively (Fig. 1).

Stable isotope studies of 15N incorporation into urea in two girls (A, EM;B, BS) with partial OTCD. Data were obtained before and 9 mo after liver transplantation. There was normalization of ureagenesis after transplant. (□) control ± SEM; (•) pretreatment; (▪) post-treatment.
Ureagenesis after gene transfer in the spf/Y mouse.
Adult spf/Y mice received 1 × 1011 particles of a second generation recombinant adenovirus (E1 deleted with ts125 mutation in E2a) containing mouse OTC cDNA through the tail vein 7 and 14 d before the performance of stable isotope studies. As controls we injected a second group of spf/Y mice with the adenovirus containing the lacZ gene (H5.110CMVlacZ) and a third group remained untreated. There were three similar control groups of unaffected C3H mice. There were at least six mice in each of the six treatment groups.
Figure 2 shows OTC activity in liver. At 7 d after gene transfer (Fig. 2A), there was no significant difference between OTC activity in OTC treated spf mice (77.5 ± 8.4 μmol citrulline/mg protein/h; mean ± SEM) and C3H controls (85.2 ± 12.2). Both had approximately 20-fold higher enzyme activity than found in the untreated (3.1 ± 0.2) and lacZ treated (1.4 ± 0.3) spf mice. The OTC cDNA containing adenovirus had no significant effect on OTC activity in the C3H mice, but the lacZ adenovirus may have suppressed OTC activity in the C3H mice. In the mice studied 14 d after gene transfer (Fig. 2B), the findings were similar, although the increase in OTC activity in the OTC-treated spf mice was less prominent, and the lacZ virus showed no effect on the OTC activity in C3H mice.

OTC activity (μmol cit/mg protein/h) in mouse liver obtained 7 (A) and 14 d (B) after gene transfer. spf/Y and control (C3H) mice received either no treatment (spf; C3H), 1 × 1011 particles of recombinant adenovirus containing mouse OTC cDNA (spf/OTC; C3H/OTC), or 1 × 1011 particles of recombinant adenovirus containing the β-galactosidase gene (spf/lacZ; C3H/lacZ). Data are the mean ± SEM of six independent observations. ANOVA was used to compare enzyme activity in treated groups to that in untreated spf/Y and C3H control groups. = denotes comparison to C3H control group. # denotes comparison with untreated spf/Y group. (*p= 0.01, **p= 0.0001).
Figure 3 demonstrates changes in orotate excretion in the six animal groups (spf, spf/lacZ, spf/OTC, C3H, C3H/lacZ, C3H/OTC). Urinary orotates are typically 20-fold elevated in spf/Y mice compared with C3H mice (28). The results of gene transfer mirror the finding in OTC enzyme activity. There were significant differences (p< 0.001) in orotate levels at both 7 d (0.78 ± 0.27 μmol/mg creatinine) and 14 d (1.83 ± 1.01 μmol/mg creatinine) after gene transfer in OTC treated spf mice compared with the untreated group (8.74 ± 4.88 and 7.44 ± 2.01 μmol/mg creatinine at d 7 and 14, respectively). These levels were not significantly different from control C3H mice (0.60 ± 22 and 0.41 ± 0.13 μmol/mg creatinine at d 7 and 14, respectively). There was a more modest decline in the lacZ-treated spf mice (2.36 ± 1.31 and 3.31 ± 1.72 μmol/mg creatinine at d 7 and 14, respectively) compared with the untreated group (p= 0.04 at d 7, p= 0.03 at d 14). There was no significant change in orotate levels in the C3H mice groups.

Urinary orotate levels (expressed as percent of pre-gene transfer levels) in spf/Y (A) and C3H (B) mice in three treatment groups 7 and 14 d after gene transfer. Spf/Y or control (C3H) mice received either no treatment or 1 × 1011 particles of a recombinant adenovirus containing either mouse OTC cDNA or the β-galactosidase gene. Data are the mean ± SEM of six independent observations. An unpaired t test was used to compare orotate on d 7 or 14 to the pre-gene transfer level. (*p= 0.001). (▪) spf; (▴) spf/lacZ; (•) spf/OTC; (□) C3H; (▵) C3H/lacZ; (○) C3H/OTC.
Figure 4 illustrates 15N incorporation into urea at 15 and 30 min. There was a 2- to 2.5-fold increase in ureagenesis after gene transfer at both 7 and 14 d in the spf/Y mice (Fig 4A and 4 C). For 15-min measurements, 0.35 ± 0.09 (baseline), 0.71 ± 0.19 (d 7), and 0.99 ± 0.32 (d 14) μmol/h; for 30-min measurements, 0.46 ± 0.13, 0.80 ± 0.14, and 0.89 ± 0.31 μmol/h. As urea synthesis in spf mice is approximately 60% of control C3H mice at baseline (28), this represented normalization of ureagenesis. As expected there were no changes in ureagenesis in the C3H mice treated with OTC or lacZ virus (Fig 4B and 4 D).

15N incorporation into urea (expressed as percent of pre-gene transfer levels) 7 and 14 d after transfer in spf/Y (A and C) and C3H (B and D) mice. Spf/Y or control (C3H) mice received either no treatment or 1 × 1011 particles of a recombinant adenovirus containing either mouse OTC cDNA or the β-galactosidase gene. Samples were obtained 15 and 30 min after ip injection of 5 mg/kg 15NH4 Cl. Data are the mean ± SEM of six independent observations. ANOVA was used to compare 15N incorporation into urea on d 7 and 14 to each group's pretreatment value. (*p= 0.05, **p= 0.01, ***p= 0.001, ****p= 0.0001).(▪) spf; (▴) spf/lacZ; (•) spf/OTC; (□) C3H; (▵) C3H/lacZ; (○) C3H/OTC.
DISCUSSION
Current therapy for OTCD relies on nitrogen restriction combined with the stimulation of alternative pathways of waste nitrogen excretion (4). Because of the poor outcome of this therapy, novel approaches to treatment are being proposed, focusing on liver transplantation and gene therapy. This paper presents evidence that both liver transplantation and gene transfer corrects urea synthetic capacity in OTCD.
In a sense the liver transplantation data provides a control for the gene therapy studies. The stable isotope studies in the two girls with partial OTCD show deficient ureagenesis before transplantation and normal ureagenesis post-transplant, demonstrating that the urea cycle in the transplanted livers is functioning effectively. The low plasma arginine levels in both patients are probably due to the fact that the partial ureagenesis capacity in intestine and kidneys is not restored by the liver transplantation.
The results in the OTCD spf mice mirror those found in humans with OTCD. The gene transfer study showed that a dose of 1 × 1011 particle/mouse (4 × 1012 particles/kg) resulted in correction of the OTC enzyme activity, normalization of ureagenesis as measured by stable isotopes, and metabolic correction of orotate excretion. There was also evidence of a modest nonspecific increase in ureagenesis stimulated by the lacZ virus as has been previously reported (29).
This study suggests that gene therapy using a recombinant adenovirus containing the OTC cDNA is capable of correcting urea synthetic activity in OTCD. It further supports the use of stable isotope studies as markers for efficacy of gene therapy in human trials. Finally, it suggests that relatively low doses of gene transfer may have a significant functional benefit without completely correcting enzyme activity.
Abbreviations
- OTC:
-
ornithine transcarbamylase
- OTCD:
-
ornithine transcarbamylase deficiency
- Spf:
-
sparse fur
- CMV:
-
cytomegalovirus
- lacZ:
-
β-galactosidase
- Pfu:
-
plaque forming unit
- MOI:
-
multiplicity of infection
- HDM:
-
hormone defined medium
- FBS:
-
fetal bovine serum
References
Nagata N, Matsuda I, Matsuura T, Oyanagi K, Tada K, Narisawa K, Kitagawa T, Sakiyama T, Yamashita F, Yoshino M 1991a Retrospective survey of urea cycle disorders: Part 2. Am J Med Genet 40: 477–481
Ricciuti GC, Gelehrter TD, Rosenberg LE 1976 X-chromosome inactivation in human liver: confirmation of X-linkage of ornithine transcarbamylase. Am J Hum Genet 28: 332–338
Matsuda I, Nagata N, Matsuura T, Oyanagi K, Tada K, Narisawa K, Kitagawa T, Sakiyama T, Yamashita F, Yoshino M 1991 Retrospective survey of urea cycle disorders: Part 1. Am J Med Genet 38: 85–89
Brusilow SW, Horwich AL 1995 Urea cycle enzymes. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds) The Metabolic and Molecular Basis of Inherited Disease, 7th ed. McGraw-Hill Information Services Co, New York, pp 1187–1233
Feillet F, Leonard JV 1998 Alternate pathway therapy for urea cycle disorders. J Inherit Metab Dis 21: 101–111
Msall M, Batshaw ML, Suss R, Brusilow SW, Mellits ED 1984 Neurologic outcome in children with inborn errors of urea synthesis. N Engl J Med 310: 1500–1505
Nagata N, Matsuda I, Oyanagi K 1991b Estimated frequency of urea cycle enzymopathies in Japan. Am J Med Genet 39: 229–229
Yudkoff M, Daikhin Y, Nissim I, Jawad A, Wilson JM, Batshaw ML 1996 In vivo nitrogen metabolism in ornithine transcarbamylase deficiency. J Clin Invest 98: 2167–2173
Tuchman M, Morizono H, Rajagopal BS, Plante RJ, Allewell NM 1998 The biochemical and molecular spectrum of ornithine transcarbamylase deficiency. J Inherit Metab Dis 21: 40–58
Finkelstein JE, Hauser ER, Leonard CO, Brusilow SW 1990 Late-onset ornithine transcarbamylase deficiency in male patients. J Pediatr 117: 897–902
Yudkoff M, Daikhin Y, Ye X, Wilson JM, Batshaw ML 1998 In vivo measurement of ureagenesis with stable isotopes. J Inherit Metab Dis 21: 21–29
Arn PH, Hauser ER, Thomas GH, Herman G, Hess D, Brusilow SW 1990 Hyperammonemia in women with a mutation at the ornithine carbamoyltransferase locus. N Engl J Med 322: 1652–1655
Batshaw ML, Msall M, Trojak J 1986 The risk of serious illness in carriers of ornithine transcarbamylase deficiency. J Pediatr 108: 236–241
Belmont JW 1996 Genetic control of X inactivation and processes leading to X-inactivation skewing. Am J Hum Genet 59: 1101–1108
Uchino T, Endo F, Matsuda I 1998 Neurodevelopmental outcome of long-term therapy of urea cycle disorders in Japan. J Inherit Metab Dis 21: 151–159
Msall M, Monahan PS, Chapanis N, Batshaw ML 1988 Cognitive development in children with inborn errors of ure synthesis. Acta Paediatr 30: 435–441
Whitington PF, Alonso EM, Boyle JT, Molleston JP, Rosenthal P, Emonds JC, Millis JM 1998 Liver transplantation for the treatment of urea cycle disorders. J Inherit Metab Dis 21: 112–118
Ye X, Robinson MB, Batshaw ML, Furth EE, Smith I, Wilson JM 1996 Prolonged metabolic correction in adult ornithine transcarbamylase-deficient mice with adenoviral vectors. J Biol Chem 271: 3639–3646
Tuchman M 1989 Persistent acitrullinemia after liver transplantation for carbamylphosphate synthetase deficiency. N Engl J Med 320: 1498–1499
Ensinger MJ, Ginsberg HS 1972 Selection and preliminary characterization of temperature-sensitive mutants of type 5 adenovirus. J Virol 10: 328–339
Logan J, Shenk T 1984 Adenovirus tripartite leader sequence enhances translation of mRNAs late after infection. Proc Natl Acad Sci USA 81: 3655–3659
Englehardt J, Ye X, Doranz B, Wilson J 1994 Ablation of E2a in recombinant adenoviruses improves transgene persistence and decreases inflammatory response in mouse liver. Proc Natl Acad Sci USA 91: 6196–6200
Yang Y, Nunes FA, Berencsi K, Gonczol E, Engelhardt JF, Wilson JM 1994 Inactivation of E2a in recombinant adenoviruses limits cellular immunity and improves the prospect for gene therapy of cystic fibrosis. Nat Genet 7: 363–369
Lee JT, Nussbaum RK 1989 An arginine to glutamine mutation in residue 109 of human ornithine transcarbamylase completely abolishes enzymatic activity in Cos1 cells. J Clin Invest 84: 1762–1766
Brusilow SW, Hauser E 1989 Simple method of measurement of orotic acid and orotidine in urine. J Chromatogr 493: 388–391
Frederick WS, Berg PJ, Miles AJM, Haymond MW 1984 Use of t-butyldimethylsilylation in the GC-MS analysis of physiologic compounds found in the plasma using electron-impact ionization. Anal Biochem 141: 101–109
Rosenblatt J, Chinkes D, Wolfe M, Wolfe RR 1992 Stable isotope tracer analysis by CE-MS, including quantification of isotopomer effects. Am J Physiol 263:E584–E596
Batshaw ML, Yudkoff M, McLaughlin BA, Gorry E, Anegawa NJ, Smith IAS, Hyman SL, Robinson MB 1995 The sparse fur mouse as a model for gene therapy in ornithine carbamoyltransferase deficiency. Gene Ther 2: 743–749
Castell JV, Hernandez D, Gomez-Foix AM, Guillen I, Donato T, Gomez-Lechon MF 1997 Adenovirus-mediated gene transfer into human hepatocytes: analysis of the biochemical functionality of transduced cells. Gene Ther 4: 455–464
Author information
Authors and Affiliations
Additional information
Supported by Grants P30-HD32649, HD26979, NS37915 and FD-R-001529 from the Food and Drug Administration.
Rights and permissions
About this article
Cite this article
Batshaw, M., Robinson, M., Ye, X. et al. Correction of Ureagenesis after Gene Transfer in an Animal Model and after Liver Transplantation in Humans with Ornithine Transcarbamylase Deficiency. Pediatr Res 46, 588 (1999). https://doi.org/10.1203/00006450-199911000-00016
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199911000-00016
This article is cited by
-
AAV2/8-mediated Correction of OTC Deficiency Is Robust in Adult but Not Neonatal Spfash Mice
Molecular Therapy (2009)
-
Evidence for forebrain cholinergic neuronal loss in congenital ornithine transcarbamylase deficiency
Metabolic Brain Disease (2000)
