
Abstract
We retrospectively analyzed the bone marrow (BM) smears of 10 children with mitochondrial cytopathies. Light microscopic examination showed large and coalescent cytoplasmic vacuolization of some BM precursors in nine cases, including two children with normal peripheral blood counts and four with sideroblastic anemia. BM ultrastructural study showed abnormal mitochondria in the erythroid lineage in all three children studied. Ultrastructural studies in two cases revealed a population of giant mitochondria with abnormal ultrastructure coexisting with a population of normal mitochondria in proerythroblasts, basophil erythroblasts, and less commonly in more mature erythroblasts. In a third child, mitochondria were normal in size with cristae either absent or exhibiting abnormal longitudinal orientation. Heteroplasmic segregation of mitochondria during cell division could account for the finding of a double population of cells on ultrastructural examination. These features suggest that cytologic and ultrastructural BM examination could be useful for the diagnosis of mitochondrial disorders. That is, when large and coalescent cytoplasmic vacuoles of BM precursor cells are present, the clinician should search for mitochondrial cytopathy in a child with unexplained cytopenia(s).
Similar content being viewed by others


Main
Constitutional mitochondrial disorders result in a wide variety of diseases with heterogeneous biochemical and clinical expression. Neuromuscular symptoms usually predominate(1), but recent reports have highlighted potential hematologic involvement in such diseases: Pearson's syndrome(2), which is due to an mt DNA deletion(3), includes pancreatic insufficiency and a sideroblastic anemia with vacuolization of BM precursors and is the model of hematologic expression in mitochondrial cytopathies. A few reports have also described the occurrence of sideroblastic anemia during the course of other mitochondrial cytopathies: Kearns-Sayre syndrome (external ophthalmoplegia, retinopathy, and complete heart block)(4), DIDMOAD syndrome (diabetes insipidus, diabetes mellitus, optic atrophy, and deafness)(5), and Leigh syndrome(6). We have shown that isolated cytopenias with macrocytic anemia and RS could be the sole expression of such disorders for several years before any other nonhematologic manifestations(7). The aim of this retrospective study was to emphasize the importance of certain characteristics of BM cells in this disorder that should lead the clinician to search for a mitochondrial disorder when hematologic anomalies remain perplexing.
METHODS
In our department, during the years 1990-1996, 21 patients were diagnosed with mitochondrial disorder because of hematologic, hepatologic, and/or neurologic dysfunction. At initial presentation, BM examination was performed for diagnostic purposes because of cytopenia and/or to search for storage cells. We retrospectively studied all the children with mitochondrial cytopathy and available CBC and BM smears.
The diagnosis of mitochondrial cytopathy was by enzymatic evidence of respiratory chain complexes (six patients) and/or the presence of a mutation in mt DNA in at least one affected tissue (four patients). Mitochondria from a vastus lateralis muscle obtained under general anesthesia were prepared as reported by Rustin et al.(8), and liver specimens were obtained by needle biopsies. Respiratory chain activities were also measured by use of lymphocytes, fibroblasts, and BM cells(8). NADH cytochrome c reductase (complex I and III), succinate ubiquinone DCPIP reductase (complex II), succinate cytochrome c reductase (complex II and III), ubiquinol cytochrome c reductase (complex III), and cytochrome c oxydase (complex IV) were measured with spectrophotometric and polarographic analyses(8).
Mt DNA was analyzed by Southern blotting by use of a full-length mt DNA probe: total DNA (5 µg) was digested, separated by agarose gel (0.7%) electrophoresis, and transferred into nylon filters (Hybond N+, Amersham). The filters were hybridized with [32P]dCTP-labeled mt DNA probes (2 × 106 cpm/mL). For characterization of the nucleotide sequence at the boundaries of the deletion, 1 µg total DNA was submitted to PCR amplification (30 cycles) with two oligonucleotide primers (primer A: nt 8335-nt 8353, primer B: nt 16190-nt 16170). DNA was amplified by mixing 100 pmol of each primer with 2.5 U Taq polymerase (Perkin Elmer Cetus). Amplified DNA was purified on 2% low melting-point agarose gel and recovered by heating for 5 min at 65°C. Direct sequencing was performed on an automatic DNA sequencer (Applied Biosystems 373 A sequencer) using 3.2 pmol of primer A, 100 ng of DNA, and 9.5 µL of sequencing reaction mixture (Applied Biosystems) and compared with the wild-type sequence.
BM smears were reviewed by the same cytologist. Peripheral blood and BM smears were evaluated using May-Grunwald-Giemsa staining. Perl's stain was performed on all BM aspirates; RS was calculated as the percentage of erythroblasts, and the diagnosis of RARS was considered when RS were present in >15% of erythroid cells(9). BM ultrastructural morphologic study was performed in three patients: thin sections were examined with a Philips CM10 electron microscope (Philips, Eindoven, the Netherlands).
RESULTS
CBC and BM slides were available for 10 children. The remaining 11 patients, who were excluded because BM smear examination was not available, had normal CBC. Abnormal hematologic features had been found in nine of the 10 children (Table 1): one child had had Leigh syndrome (case 9), and none of the other eight children had any syndrome known to be associated with mitochondrial disease. At presentation, four patients (patients 1-4) had only hematologic abnormalities, three had evidence of hepatic dysfunction (patients 5-7), and two had neurologic dysfunction (patients 8 and 9). The tenth patient, a boy with Kearns-Sayre syndrome and mt DNA deletion in muscle and lymphocytes, had a normal CBC and BM smears.
Aregenerative macrocytic anemia occurred in seven children from birth to 3 y of age and was associated with mild neutropenia in two cases (cases 4 and 6) and with thrombocytopenia in case 5. Two other patients who were referred because of neurologic dysfunction had normal CBC. No patient had vacuoles in the peripheral lymphocytes.
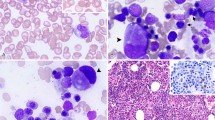
Abnormal BM cytologic and cytogenetic findings are summarized in Table 1. In all cases but one, initial BM smears showed large and confluent cytoplasmic vacuoles in erythroblasts, mostly proerythroblasts (eight cases), myeloblasts (eight cases), promyelocytes (seven cases), histiocytes (two cases), and megakaryocytes (two cases) (Fig. 1). They were present in only 1 to 6% of the cells of the affected hematopoietic lineage. RARS was present in three patients who expressed anemia as the initial manifestation of the disease. Erythrophagocytosis was observed in three cases.

Large cytoplasmic vacuoles in a promyelocyte.
Four BM karyotypes were performed (cases 1-4) and all were normal.
BM ultrastructural morphology was studied in patients 2, 3, and 7. In patients 2 and 7, two distinct populations of mitochondria were seen in some proerythroblasts and basophil erythroblasts but rarely in more mature erythroblasts (Fig. 2). One population was normal (mean length, 0.3 μ) whereas the other was very large (mean length, 2.5-3.0 μ). The ultrastructure of these giant mitochondria was abnormal with some containing longitudinal parallel stacks of cristae with a clear zone in the matrix; in others, very short cristae persisted only in contact with the inner membrane with a matrix full of particles smaller than cytoplasmic ribosomes. In more mature erythroblasts, three types of mitochondria could be distinguished: normal, normal and iron-loaded mitochondria, and a third type in which all mitochondria contained iron micelles. In patient 3, the size of mitochondria was normal, but the cristae were absent or exhibited abnormal longitudinal orientation.

BM electron microscopic examination. This figure shows a proerythroblast. The rhopheocytosis vesicles (a ferritin-coated invagination) (R) affirm the erythroid origin of the cell. In the middle of the figure, a giant mitochondrion shows a disorganized architecture with disappearance of the normal cristae, which are divided into fragments (arrow heads); the matrix is full of particles smaller than cytoplasmic ribosomes. The irregularity of one side of the membrane of the giant mitochondrion is due to a bias of cut. At the bottom, there is a normal mitochondrion (Mi-n). Magnification × 29 400.
CLINICAL AND HEMATOLOGIC COURSE
In patients 1-4, the hematologic dysfunction was initially the sole expression of the mitochondrial disease. Unrelated symptoms appeared 6 and 1 y later, respectively, in patient 1 (ptosis, deafness, and hypoparathyroidism) and in patient 2 (mild mental retardation), whereas no other symptoms have been observed in patients 3 and 4 with a follow-up of 15 mo and 2 1/2 y, respectively. In all of them, anemia led to repeated transfusions during the initial 6-mo period after diagnosis but, surprisingly, the anemia spontaneously resolved later. BM was reassessed after 6 and 2 y, respectively, in patients 1 and 2, when CBC and mean corpuscular volume were normal. The number of vacuolated cells increased in patient 1, whereas BM smears were near normal in patient 2 with few vacuolated cells and only 2% RS.
Mild pancytopenia developed in patient 6 at 3 y of age, but red blood cell transfusion did not appear to be necessary. The follow-up was only 2 mo in patient 7. Patient 5 died at age 6 mo because of hepatic failure. CBC remained normal in the three other patients during a 2- to 10-y follow-up.
DISCUSSION
In nine of the 10 children, including two with normal blood cell counts, BM precursor cells contained large and coalescent cytoplasmic vacuoles. These vacuoles were not stained by the May-Grunwald-Giemsa stain, and their nature remains unknown. On the basis of results of a large retrospective study highlighting the presence of large cytoplasmic vacuoles in childhood MDS only when associated with mitochondrial disorders(10), we assumed that this feature was pathologic. Large cytoplasmic vacuolization also occurs in leukemic cells and are associated with enlargement of mitochondria in leukemic cells cultured in the presence of tumor necrosis factor, which suggests that the mitochondrion is one of the primary targets of tumor necrosis factor(11). In contrast, moderate vacuolization can be observed along the process of maturation of all BM lineage cells, but vacuoles are small, sparse, and rarely coalesce in the absence of a mitochondrial disorder.
These cytologic features suggest that BM examination is useful because the presence of large and coalescent cytoplasmic vacuoles of BM precursor cells must lead the clinician to search for a mitochondrial cytopathy when a child presents with unexplained cytopenia(s). In children suspected of a mitochondrial cytopathy who have a normal CBC, a BM aspirate is usually not done unless its potential value is recognized prospectively, particularly when the aspiration can be performed when general anesthesia is needed for muscle and/or liver biopsy.
Additionally, sideroblastic anemia, diagnosed as RARS, was found in three patients in whom the anemia was the initial and isolated manifestation of the mitochondrial disorder. Sideroblastic anemias are a group of genetic or acquired disorders characterized by the presence of RS and ineffective erythropoiesis. RARS is a rare form of childhood MDS. In four large series including a total of 130 children with MDS, it has been observed only in the seven children who had mitochondrial cytopathy(12–15). Constitutional sideroblastic anemia constitutes a bundle of rare diseases, including disorders of various origins such as X-linked pyridoxine-responsive congenital sideroblastic anemia(16), autosomal recessive pyridoxine-refractory congenital sideroblastic anemia(17), and some mitochondrial cytopathies: Pearson's disease(3), Kearns-Sayre syndrome(4), Leigh syndrome (subacute necrotizing encephalomyelopathy)(6), and the rare thiamine-responsive sideroblastic anemia described in association with the DIDMOAD syndrome(5), recently identified as a mitochondrial enzyme defect resulting in an abnormality of either the mitochondrial or the nuclear DNA(18). These features suggest that the association of RARS with unrelated symptoms must lead to the search for a mitochondrial disorder with appropriate biochemical, histologic, and molecular investigations. Some of the cases described herein illustrate the difficulty in diagnosis when patients present with solely hematologic signs. In the etiologic diagnosis of anemia with reticulocytopenia in the first year of life, defects in the mitochondrial respiratory chain should be considered.
We observed spontaneous regression of the anemia in four girls with macrocytic anemia, associated in one case with mild neutropenia at onset of the disease. In contrast, Rötig et al.(3) have reported five children presenting with severe pancytopenia at onset of Pearson's marrow-pancreas syndrome, which persisted until rapid multivisceral failure and death. These different outcomes could depend on the initial percentage of deleted mt DNA in the BM cells and on its change with time. There has been a recent report of two patients in hepatic failure related to disorders of oxidative phosphorylation, one with early onset and a rapidly fatal course and the other with delayed onset and a milder clinical course, including spontaneous recovery from the liver disease(19).
Deletions of mt DNA may have a role in the pathogenesis of some forms of marrow dysfunction. Impairment of mt DNA can affect hematopoiesis, as illustrated by the suppression of mitochondrial protein synthesis by chloramphenicol(20). A number of authors have suggested that some cases of IASA could be due to a deletion in mt DNA(21,22). It has been proposed that, in IASA, the inability to incorporate iron into protoporphyrin IX to form heme could be due to failure of the mitochondrial respiratory chain to reduce Fe3+ to Fe2+, the form required for incorporation into protoporphyrin IX. Large deletions of mt DNA have not been observed in IASA(22) but a heteroplasmic point mutation of mt tRNA in nonlymphoid hematopoietic cell lineages from a patient with IASA has recently been reported(23). The observations in our patients suggest that heteroplasmic segregation of mitochondria during cell division accounts for the finding of a double population of cells on ultrastructural examination.
These results emphasize that the occurrence of large and coalescent cytoplasmic vacuoles in a low percentage of various BM precursor cells is a frequent cytologic finding in children with mitochondrial cytopathy, which must lead the clinician to search for a respiratory chain defect in children with unexplained cytopenia(s). The spectrum of hematologic disease is wide and varies from spontaneous remission to the occurrence of severe aplastic anemia.
Abbreviations
- BM:
-
bone marrow
- CBC:
-
complete blood count
- MDS:
-
myelodysplastic syndrome
- RARS:
-
refractory anemia with ring sideroblasts
- IASA:
-
idiopathic acquired sideroblastic anemia
- mt DNA:
-
mitochondrial DNA
- RS:
-
ring sideroblasts
References
Wallace DC 1989 Mitochondrial DNA mutations and neuromuscular diseases. Hum Genet Dis 5: 9–13
Pearson HA, Lobel JS, Kocoshis SA, Naiman LL, Windmiller J, Lammi AT 1979 A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J Pediatr 95: 976–984
Rötig A, Cormier V, Blanche S, Bonnefont JP, Ledeist F, Romero N, Schmitz J, Rustin P, Fisher A, Saudubray JM, Munnich A 1990 Pearson's marrow-pancreas syndrome. J Clin Invest 86: 1601–1608
Nelson I, Bonne G, Degoul F, Marsac C, Ponsot G, Lestienne P 1992 Kearns-Sayre syndrome with sideroblastic anemia: molecular investigations. Neuropediatrics 23: 199–205
Borgna-Pignatti C, Marradi P, Pinelli L, Monetti N, Patrini C 1989 Thiamine-responsive anemia in DIDMOAD syndrome. J Pediatr 114: 405–410
Yamadori I, Kurose A, Kobayashi S, Ohmori M, Imai T 1992 Brain lesions of the Leigh-type distribution associated with a mitochondriopathy of Pearson's syndrome: light and electron microscopic study. Acta Neuropathol 84: 337–341
Bader-Meunier B, Miélot F, Rötig A, Lavergne JM, croisille L, Rustin P, Landrieu P, Dommergues JP, Munnich A, Tchernia G 1994 Refractory anaemia and mitochondrial cytopathy in childhood. Br J Haematol 87: 381–385
Rustin P, Chretien D, Gérard B, Bourgeron T, Rötig A, Saudubray JM, Munnich A 1994 Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta 228: 35–51
Goasguen JE, Bennett JM 1992 Classification and morphologic features of the myelodysplastic syndromes. Semin Oncol 19: 4–13
Miélot F, Buisine J, Duchayne E, Fenneteau O, Goasguen J, Guitard AM, Maier-Redelsperger M, Malet M, Manel AM 1998 Myelodysplastic syndromes in childhood: is the FAB classification relevant?. Leuk Lymphoma 28: 531–40
Munker R, Greither L, Darsow M, Ellwart JW, Mailhammer R, Wilmanns W 1993 Effects of tumor necrosis factor on primary human leukemia cells ultrastructural changes. Acta Haematol 90: 77–83
Creutzig U, Cantu-Rajnoldi A, Ritter J, Romitti L, Odenwald E, Conter V 1987 Myelodysplastic syndromes in childhood. Am J Pediatr Hematol Oncol 9: 324–330
Brandwein JM, Horsman DE, Eaves AC, Eaves CJ, Massing BG, Wadsworth LD, Rogers PCJ, Kalousek DK 1990 Childhood myelodysplasia: suggested classification as myelodysplastic syndromes based on laboratory and clinical findings. Am J Pediatr Hematol Oncol 12: 63–70
Hasle H, Jacobsen BB, Pedersen NT 1995 Childhood myelodysplastic syndrome in Denmark: incidence and predisposing conditions. Leukemia 9: 1569–1572
Bader-Meunier B, Miélot F, Tchernia G, Buisine J, Delsol G, Duchayne E, Lemerle S, Leverger G, de Lumley L, Manel AM, Nathanson M, Plantaz D, Robert A, Schaison G, Sommelet D, Vilmer E 1996 Myelodysplastic syndromes in childhood: report of 49 patients from a French multicentre study. Br J Haematol 92: 344–350
Losowsky MS, Hall R 1965 Hereditary sideroblastic anemia. Br J Haematol 11: 70–85
Jardine PE, Cotter PD, Johnson SA, Fitzsimons EJ, Tyfield L, Lunt PW 1994 Pyridoxine-refractory congenital sideroblastic anaemia with evidence for autosomal inheritance: exclusion of linkage to ALAS2 at Xp 11:21 by polymorphism analysis. J Med Genet 31: 213–218
Rötig A, Cormier V, Chatelain P, Francois R, Saudubray JM, Rustin P, Munnich A 1993 Deletion of mitochondrial DNA in a case of early-onset diabetes mellitus, optic atrophy, and deafness (DIDMOAD, Wolfram syndrome). J Inher Met Dis 16: 527–530
Cormier-Daire V, Chretien D, Rustin P, Rötig A, Dubuisson C, Jacquemin E, Hadchouel M, Bernard O, Munnich A 1997 Neonatal and delayed onset liver involvement in disorders of oxidative phosphorylation. J Pediatr 130: 817–822
Manyan DR, Armura GK, Yunis AA 1972 Chloramphenicol-induced erythroid suppression and bone marrow ferrochelatase in dogs. J Lab Clin Med 79: 137–141
Hatfill SJ, Bohm L, Downing TG, Kirby R 1992 Mitochondrial DNA mutations in the myelodysplastic syndrome. [letter] Lancet 340: 370
Gattermann N, Aul C, Schneider W 1993 Is acquired sideroblastic anemia (AISA) a disorder of mitochondrial DNA?. Leukemia 7: 2069–2076
Gattermann N, Retzlaff S, Wang YL, Berneburg M, Heinisch J, Wlaschek M, Aul C, Schneider W 1996 A heteroplasmic point mutation of mitochondrial tRNALeu(CUN) in non-lymphoid haematopoietic cell lineages from a patient with acquired sideroblastic anaemia. Br J Haematol 93: 845–855
Acknowledgements
The authors thank Dr. Rötig for mitochondrial DNA studies, Professor Bernard for referring the patients, and Monique Dehan for her help in preparing the manuscript.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Bader-Meunier, B., Miélot, F., Breton-Gorius, J. et al. Hematologic Involvement in Mitochondrial Cytopathies in Childhood: A Retrospective Study of Bone Marrow Smears. Pediatr Res 46, 158–162 (1999). https://doi.org/10.1203/00006450-199908000-00005
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199908000-00005
This article is cited by
-
Pearson syndrome: a multisystem mitochondrial disease with bone marrow failure
Orphanet Journal of Rare Diseases (2022)
