
Abstract
Adhesion molecules play a major role in the recruitment of neutrophils to the site of inflammation. Neutrophils' localization is dynamic and involves multiple steps. In each step a different family of adhesion molecules takes part. The rolling phase is mediated by the selectin family, the E-, L-, and P- selectins, and their ligand, sialyl Lewis X. The next step, the activation and firm adhesion of the neutrophils to the endothelium, is regulated by the integrin family and their ligand, the Ig superfamily. The final step of transendothelial migration is again mediated by these two families of adhesion molecules. Although many in vitro studies were able to show the role of these molecules, their real importance was demonstrated in rare disease states where one of the adhesion molecule was absent. Two adhesion molecule deficiencies were described, both characterized by recurrent infections, defect in wound healing, and marked leukocytosis. Leukocyte adhesion deficiency (LAD) I is caused by a defect in the β subunit of the integrin molecule, whereas in LAD II, the ligand for the selectin, the sialyl Lewis X is markedly decreased. Further insight was also gained with the generation of strains of mice deficient in one or another adhesion molecules (knock-out mice) Exploiting current knowledge on adhesion molecules and their role in health and disease, several trials have been designed to assess the effect of blocking their activity in conditions associated with increased expression of various adhesion molecules.
Similar content being viewed by others
Main
Adhesion molecules play major role in many fields of medicine including embryology, immunology, malignancy, and have been referred to as “the glue of life.” In the last 5 y the structure, regulation, and function of many adhesion molecules have been elucidated, and their contribution to physiologic and pathologic conditions has been demonstrated. This new information enables us to implement the basic science to the bed side, and the therapeutic effects of several “anti-adhesion” agents are currently under clinical investigation.
This review focuses mainly on the role of adhesion molecules in leukocyte-endothelial interaction, crucial for the inflammatory and immune responses. The clinical significance of this interaction will be discussed. The basic characteristics of the various adhesion molecules have recently been reviewed in depth(1, 2).
The process of leukocyte accumulation at sites of inflammation is a dynamic one and involves multiple steps. The orchestration of these steps must be precisely regulated to ensure a rapid response, with only minimal damage to healthy tissue. Leukocyte interaction with vascular endothelial cells is essential in guiding the inflammatory response and is mediated by several families of adhesion molecules. These include the integrins, the selectins, and members of the Ig superfamily. Each is involved in a different phase of leukocyte emigration through the endothelium, and the synchronization of their expression and function is crucial for the normal recruitment of leukocytes from the blood stream to the tissue.
LEUKOCYTE INTEGRINS
Integrins (Table 1) are transmembrane cell surface proteins that bind to cytoskeletal proteins and communicate extracellular signals. Each integrin consists of a noncovalently linked, heterodimericα and β chains. Integrins have been arranged in subfamilies according to the β subunits, and each β subunit may have from one to eight different α subunits associated with it(3). The specificity of binding to various cell adhesion factors appears to depend primarily on the extracellular portion of the α subunit for all the integrins, although both subunits are required for functional activity of each molecule(3).
The β chains have characteristic features. Tandem repeats of four cysteine-rich regions that are thought to be essential for tertiary structure are conserved among the various β chains. Approximately 100 amino acids from the NH2 terminus are additionally conserved units that are critical for maintenance of the α/β heterodimer(4). Within the integrin family of adhesion receptors, only five members have so far been shown to be involved in leukocyte adhesion to endothelium. Three belong to the β2 leukocyte integrins (CD18), one to the β1 (CD27), and the last to the β7 subfamily.
β2 Integrins. This subfamily comprises three membrane glycoproteins with a common β subunit, designated β2 (CD18). The α subunits of each of the three heterodimer members-LFA-1, Mac-1, and p150,95-are designated CD11a, CD11b, and CD11c, respectively. Both theα and β subunits have a relatively small cytoplasmic domain which contains regions capable of binding to cytoskeletal elements(5). The extracellular domain is much larger, and the aminoterminal portion of the β subunit, which contains the ligand binding region, is folded into a loop which is stabilized by disulfide bonds. Theα and β subunits are initially assembled in the cytoplasm, and only then is the complete β2 integrin heterodimer expressed extracellularly. Both subunits are required for expression, and if CD18 is absent, CD11 is not detected on the cell surface, although the synthesis of the CD11 molecule is normal(6).
β2 integrin expression is restricted to leukocytes, but among subsets of leukocytes the distribution of CD11/CD18 differs. LFA-1 is expressed by lymphocytes, monocytes, and neutrophils. Mac-1 and p150,95 expression is restricted primarily to myeloid cells, although they are also expressed by some lymphocytes and natural killer cells. They participate in many leukocyte adhesion-related functions in addition to migration through the endothelial cells, such as phagocytosis, killing of bacteria, and antibody-dependent, cell-mediated cytotoxicity(7). Neutrophil and monocyte adhesion to endothelium relies mainly on LFA-1 and Mac-1 with only a minor role for p150,95. Lymphocyte adhesion involves the interaction of LFA-1 with its endothelial ligands(8).
An important characteristic of the leukocyte integrins is that under baseline conditions they exist in a relatively inactive conformation, rendering the leukocyte nonadhesive. One of the key events at the adhesion cascade is the activation and deactivation of these integrins at the proper times and places. Activation of leukocytes by a variety of mediators(e.g. C5a, platelet-activating factor, or IL-8) results in a transient increase in adhesion by CD11/CD18-dependent mechanisms. This increased adhesive ability occurs through qualitative changes, transformation of LFA-1 and Mac-1 from low to high avidity state, and, in phagocytes, up-regulation of the surface expression of Mac-1 and p150,954(1, 2). Leukocytes mediate adhesion through binding to their ligands, members of the Ig superfamily integrins, which are expressed on all cells and may affect leukocyte adherence and interactions with all cells in the body, particularly during inflammatory conditions.
β1 and β7 Integrins. The largest number of integrins are members of the β1 or VLA subfamily. They comprise a series of cellular receptors for extracellular matrix proteins including fibronectin, collagen, and laminin. One member of this subfamily,α4β1 (VLA-4, CD49d/CD29), has been shown to be involved in lymphocyte, eosinophil, basophil, NK cell, and monocyte adhesion to fibronectin and to cytokine-activated endothelial cells by binding to an induced endothelial cell surface protein, VCAM-1(9). The site of VLA-4 involved in binding to VCAM-1 is distinct from that involved in fibronectin binding. Because blood neutrophils do not express VLA-4, they cannot use this pathway to adhere to stimulated endothelium.
The α4β7 integrin is expressed mainly on lymphocytes and is the homing receptor that binds to the mucosal vascular addressin MAdCAM-1(2).
IG SUPERFAMILY
The ICAM were originally defined functionally as LFA-1 ligands. This family includes three molecules, ICAM-1, -2, and -3. The gene for ICAM-1 is located on chromosome 19, and the molecule has five Ig-like domains, with a short hinge region separating the third and fourth Ig-like domain(10). It is a ligand for both LFA-1 and Mac-1. The binding sites for the integrins are distinct; LFA-1 binds to domains 1 and 2, whereas Mac-1 binds to domain 3. Human ICAM-2 is a single-copy gene located on chromosome 17. It has only two extracellular Ig-like domains, and the binding site for LFA-1 is located in these domains(11). Other integrin molecules do not bind ICAM-2. Recently a third ICAM molecule was described. ICAM-3 also consists of five Ig-like domains with a high degree of homology with ICAM-1, and binds only to LFA-1(12). The distribution and regulation of the ICAMs are quite distinct. ICAM-1 is expressed only at low levels on some vascular endothelial cells and on lymphocytes under normal conditions and is dramatically up-regulated by endotoxin, IL-1, and TNF. This increased expression which starts 4-6 h after contact with the cytokines lasts for several days. ICAM-2 expression, in contrast, is constitutive and is not regulated by the various cytokines(13). ICAM-3 is constitutively and strongly expressed on resting lymphocytes but not on endothelial cells. Once the cell is activated, ICAM-3 will accumulate in the uropod of the cell, thus facilitating interaction and aggregation with more leukocytes(14), and recruiting cells to the inflammatory area. Recently, a soluble form of ICAM-1 has been reported. It retains its ability to bind to LFA-1 and was found to be elevated in the serum of patients with various pathologic conditions which are characterized by a prominent inflammatory response. ICAM-2 and -3 have not yet been shown to exist as soluble forms(15).
VCAM-1 also belongs to the Ig superfamily. It contains seven Ig-like domains, the ligand binding site for the integrin VLA-4, and is located within the NH2-terminal first domain. VCAM-1 is inducible by cytokines on endothelial cells with a time course similar to ICAM-1. A single VCAM-1 gene gives rise through alternative splicing to distinct isoforms that differ in the number of integrin binding sites(16). A soluble form has also been described and has been found to correlate with disease activity in several inflammatory disorders(15). Soluble forms of members of the Ig superfamily may play a role in activation of cells at local sites of inflammation or at distant sites via distribution through the circulation.
SELECTINS
This family of adhesion molecules was discovered in 1989, when the cDNA sequences of three distinct cell surface glycoproteins found on endothelium(E-selectin, CD62E), platelets (P-selectin, CD62P), and leukocytes(L-selectin, CD62L) were reported(17). These molecules were previously designated ELAM-1, PADGEM or GMP-140, and MEL-14 or LECAM-1, respectively. The genes for the selectins family are closely linked on chromosome 1, reflecting their common evolutionary origin. All three members have common structural features, most prominently an NH2-terminal lectin-like domain, which is central to the carbohydrate-binding properties of all three selectins. The lectin domain is followed by a domain homologous to the epidermal growth factor and a discrete number of molecules similar to those found in certain complement-binding proteins(18). The term selectin was proposed to highlight the amino-terminal lectin domain and to indicate the selective function and expression of these molecules.
All three selectins are involved in the recruitment of leukocytes to sites of tissue injury, but there are fundamental differences in their distribution, activation, and mode of expression.
E-selectin. E-selectin is restricted to endothelial cells, and its expression is induced when the cells are activated by IL-1 or TNF. These substances activate the transcription of E-selectin, resulting in peak cell surface expression at 4-6 h, decreasing to basal levels by 24 h(19). Although E-selectin expression in vitro is typically transient, it is chronically expressed in certain inflammatory conditions and is also detected in the serum. The role of soluble E-selectin is still unclear, and its serum level does not correlate with disease activity.
P-selectin. P-selectin is expressed on platelets as well as on endothelial cells. Its expression does not necessarily require de novo synthesis because it is stored in the α granules in platelets and in secretory granules (Weibel-Palade bodies) in endothelial cells. Thus, within minutes of activation of either cell type by thrombin or histamine, P-selectin is rapidly redistributed to the surface of the cells. Its expression in vitro is typically very short-lived, up to 15 min. However, studies in vivo suggest that endothelial P-selectin may also be regulated at the level of protein synthesis, providing a mechanism for more prolonged expression(20).
L-selectin. In contrast to the E- and P-selectins, L-selectin is constitutively expressed on leukocytes but not on endothelial cells. Although originally described as a lymphocyte homing receptor, it was subsequently shown to be expressed on most other leukocytes. After a transient increase of this selectin during activation, it is shed rapidly due to a proteolytic cleavage near the membrane insertion(21). Soluble circulating L-selectin can be measured in the plasma of normal individuals as well as in several pathologic conditions(22). This soluble form can partially inhibit leukocyte adhesion to cytokine stimulated endothelium, but its in vivo role is still unknown.
All three selectins have a major role in leukocyte adherence to endothelial cells, which is mediated through binding to ligands on the leukocyte (E and P) or endothelial cell (L)(23). These ligands are carbohydrate groups which are typically found as a terminal structures of one or more glycoproteins and/or glycolipids. The lectin and the epidermal growth factor domains in the selectins play a crucial role in mediating this binding. One major selectin ligand is a member of a class of sialylated and fucosylated tetrasaccharides related to the sialylated Lewis X blood group (SLeX, CD15)(23). SLeX is heavily expressed on neutrophils and monocytes. Peripheral lymphocyte express SLeX only after activation. Both the sialic acid and the fucose linkages were shown to be critical for efficient binding. MAb against SLeX block the binding of neutrophils to activated endothelial cells(23). Furthermore, cell lines which do not express this ligand show no binding, but after induction of SLeX by transfection of appropriate fucosyltransferases, binding is observed.
THE ADHESION CASCADE
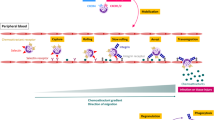
The migration of leukocytes from the blood stream to the tissue occurs in several steps (Fig. 1)(24). First, loose adhesion to the vessel wall, primary in postcapillary venules, under conditions of flow causes the leukocytes to roll on the endothelium. This transient and reversible step is a prerequisite for the next stage, the activation of leukocytes. This is followed by firm adhesion after which migration occurs. Each of these steps involves different adhesion molecules, and can be differentially regulated (Fig. 1).

The various steps of the adhesion cascade. After endothelial activation by cytokines derived from the inflammatory tissue, selectins are expressed on the endothelium. The interaction between the selectins and their leukocyte ligands will start the rolling process. This will lead to leukocyte activation with increased expression of the integrins which will cause the sticking of the cells to the endothelium. In the last phase, transmigration is mediated mainly by binding of platelet-endothelial cell adhesion molecule-1 (PECAM-1) to integrins.
Step 1: Rolling, selectin-dependent. Leukocytes in the circulation must resist tremendous shear forces to stop along the vascular endothelium. Under normal conditions, leukocytes move rapidly and do not adhere to the endothelium. The phenomenon of leukocyte rolling has been known for more than a century, but its molecular basis was delineated only recently. Several studies(25, 26) showed that, although CD18 MAb block sticking, they do not effect rolling. MAb to selectins, however, markedly reduced the rolling process in vivo and in vitro(27). Moreover, rolling is dramatically diminished in P-selectin-deficient mice(28). The selectins expressed on the endothelial cells, will bind to the leukocytes through their ligands, mainly SLeX. MAb to SLeX will also significantly reduce rolling and patients who lack SLeX molecules will show defective in vivo rolling(29). The presentation of SLeX to E- and P-selectins may be partially mediated by L-selectin which is expressed on the leukocytes. The rolling phase is transient in part because L-selectin is shed quickly from the leukocytes, and P-selectin on endothelial cells is internalized. Furthermore, at the site of inflammation, free SLeX appears and may compete for binding to the selectins(30).
Step 2: Activation, integrin-dependent. The transition from selectin-mediated adhesion to integrin-mediated adhesion occurs rapidly and involves the activation of the integrin receptors. During rolling, the slowly moving cells are exposed to diverse mediators, which are generated at the site of inflammation and are capable of activating integrins. These mediators may be secreted as soluble molecules and then bind to adjacent endothelium. The molecular interaction of E-selectin with its leukocyte cell surface ligand may also stimulate β2 integrin function(31). Increased adhesive capacity is mediated through both qualitative(conformational) and quantitative (up-regulation of surface expression) changes in the integrin receptors.
Step 3: Firm adhesion and transendothelial migration, integrins and Ig-like ligands. Activation of integrins results in increased affinity for their Ig-like ligands on the endothelial cells. This ensures that binding is firm enough to withstand the continuous shear forces in the blood vessels. MAb to both integrins and their Ig-like ligands block the firm adhesion of leukocyte to endothelial cells. The final event, the transendothelial migration of the leukocytes to the site of inflammation, is in part dependent on integrins/Ig-like interactions and can be blocked by MAb to these molecules. Another member of the endothelial Ig-superfamily PECAM-1 (CD31), was found to be important mainly in transmigration. CD31 is constitutively expressed on endothelial cells and in contrast to ICAM-1 that is expressed over the entire surface of the endothelial cell, CD31 is located at intercellular junctions(32). Heterotypic interaction between leukocyte and endothelial CD31 was found to be crucial for neutrophil and monocyte diapedesis between endothelial cells. Anti PECAM-1 MAb blocked leukocyte migration across cytokine activated endothelial monolayers(32).
ADHESION MOLECULE DEFECTS
Studies of genetic deficiency syndromes have provided important insights into the molecular basis and biology of leukocyte emigration. Both animal and human deficiency syndromes have been described.
Animals models. A spontaneous mutation in the gene coding for the β2 integrin has been described both in dog and cattle. In the cattle model two point mutations were identified within the gene encoding bovine CD18. These animals suffered from recurrent infections and persistent mature neutrophilia associated with poor growth performance(33). The carrier frequency of the defective gene among Holstein cattle is approximately 15% in bulls and 6% in cows.
Using methods of homologous recombination in embryonic stem cells, it is possible to generate strains of mice deficient in specific adhesion molecules. A “knock-out” mouse with partial CD18 deficiency was found to be viable and fertile, with mild granulocytosis. The mutant mice showed an impaired inflammatory response to chemical peritonitis and delayed rejection of cardiac transplants(34).
In another model, ICAM-1-deficient mice were generated. The animals developed normally and had mild granulocytosis. Deficient mice exhibited abnormalities of inflammatory response, especially in impaired neutrophil emigration(35). In addition, leukocytes from these mice provide negligible stimulation for the mixed lymphocyte reaction. In a recent study using the ICAM-1-deficient mice, mutant mice were resistant to lethal effects of high dose of endotoxin which correlated with significant decrease in neutrophil infiltration in the liver. As production of various inflammatory cytokines is normal in these mice, the protective effects appear to be related to a diminution in critical leukocyte-endothelial interactions(36).
Mice lacking P-selectin develop normally. However, they exhibit striking leukocytosis, diminished rolling of leukocytes in mesenteric vessels, and delayed recruitment of neutrophils to the peritoneal cavity after experimentally induced peritonitis(28). E-selectin-deficient mice have a milder defect in neutrophil recruitment, and do not have leukocytosis. Still, administration of anti P-selectin MAb to the E-selectin knock-out mice completely blocks neutrophil recruitment, suggesting that in this model some redundancy of function may exist between the two endothelial cell selectins(37). L-selectin knock-out mice were also generated. These mice had a severe reduction in the number of lymphocytes localized to peripheral lymph nodes and showed significant defects is leukocyte rolling(38).
Human LAD syndromes. The best way to appreciate the in vivo importance of the various adhesion molecules is by looking at those rare “experiments of nature” in which a specific defect in adhesion molecule exists. Currently, two such syndromes are described(Table 2)(39). In the first, LAD I, the β2 integrin family is deficient, whereas in the second, LAD II, the fucosylated ligands for selectins are absent.
LAD I. This syndrome was described more than 10 y ago and is clinically characterized by severe life-threatening infections, chronic neutrophilia, and impaired wound healing. Delayed separation of the umbilical cord, chronic gingivoperiodontitis, and lack of pus formation complete the clinical picture(40). The syndrome is a result of a defect in the expression of the β2 subunit (CD18) of the integrin molecule. Heterogeneous mutations in the common β2 chain, splicing, frame shift, missense, and initiation codon have been demonstrated to be the molecular basis of LAD I(41).
As a consequence, neutrophils are unable to emigrate from the blood vessels to the tissue, because firm adhesion and transmigration is severely impaired(42). Biopsies from infected lesions in these patients demonstrated inflammatory infiltrates totally devoid of neutrophils. This histopathologic feature is particularly striking considering that marked peripheral granulocytosis (5-20-fold higher than normal) during episodes of infections is a constant finding(43). Although the defect in neutrophil adherence accounts for the major clinical manifestations, cellular and humoral immune function are impaired in vitro. However, the in vivo significance of these abnormalities is still unclear.
Clinically, LAD I has been divided into two groups, a severe phenotype and a moderate one(44). In the severe form no expression of CD18 can be demonstrated even with leukocyte activation, and the patients suffer from life-threatening infection. The severely affected patients die early in life if definitive therapy is not instituted. In the moderate form, some surface expression (around 5%) exists, and these patients have fewer serious infections and survive into adulthood(44). Although antibiotics have some beneficial effect with infectious complications, the definitive approach to the treatment of the severe form of the disease is by bone marrow transplantation. Because the syndrome is a monogenic disorder involving hematopoietic cells, it is obviously an attractive candidate for curative treatment by gene therapy(45).
LAD II. This new adhesion molecule deficiency syndrome was described in 1992. It is also characterized by recurrent infections, failure to form pus, gingivitis, and pronounced neutrophilia(46). The severity of the infectious complications resembles that of the moderate type of LAD I. In contrast to LAD I, this syndrome is also characterized by mental and growth retardation, and it exhibits the rare Bombay blood group phenotype(47). LAD II is due to a congenital defect of endogenous fucose metabolism. This results in an inability to synthesize fucosylated carbohydrate structures, such as the selectin ligand, sialyl Lewis X. Neutrophils from the LAD II patients express normal levels of β2 integrins and L-selectin, but lack SLeX or other fucose-containing surface antigens. Neutrophils from these patients do not bind to E-selectin expressed on cytokine-activated cultured endothelial cells, or to purified E- or P-selectin(48), and exhibit a marked decrease in migration to skin chambers or skin window in vivo(49). Still, under static conditions the cells adhere normally through integrin-ICAM interactions and can also migrate across endothelial monolayers(59). Compelling evidence in support of the current paradigm of leukocyte-endothelial interaction was obtained by using intravital microscopy to study neutrophil behavior from LAD I and LAD II patients. Fluorescein-labeled cells were observed during interactions with venules in rabbit peritoneum treated with IL-1 to induce E-selectin. Neutrophils from LAD I patients showed normal rolling, but were unable to stick and emigrate upon chemotactic stimulation. Neutrophils from LAD II patients rolled poorly, and failed to stick and emigrate under the shear forces provided by flow. However, when flow was reduced the cells adhered and emigrated in response to a chemoattractant(30). This study clearly showed that selectin-carbohydrate ligand interactions initiate low affinity adhesion manifested by rolling under condition of flow, and subsequent activation induces engagement of integrins with endothelial Ig-like ligands promoting firm adhesion and emigration into the tissue.
ADHESION MOLECULES IN DISEASE STATES-THERAPEUTIC APPLICATIONS
As well as being involved in host defense, leukocyte-endothelial interactions can generate pathologic inflammation in a variety of conditions. Diseases characterized by acute inflammation with infiltration of neutrophils are often associated with increased expression of E- and P-selectins, whereas in chronic conditions ICAM-1 or VCAM-1 expression predominates(50). An increase in local expression as well as in serum-soluble adhesion molecules has been reported in diverse pathologic conditions including arteriosclerosis, vasculitis, arthritis, renal and hepatic diseases, ischemia reperfusion conditions, organ rejection, metastasis, and many more pathologic conditions(51).
The characterization of the molecular basis of leukocyte-endothelial interactions has suggested several avenues for inhibiting the adhesion process. MAb against the various adhesion molecules have been developed and tested in many animal models with beneficial effects. It should be noted that these MAb are mainly of murine origin and thus are not practical for long-term use in humans. Furthermore, they may cause neutropenia and may have a deleterious effect on several immune responses(52). Multiple other “anti-adhesion” strategies have been proposed, including soluble adhesion receptor proteins that compete for the ligands, small peptides that are specific for the binding site of the adhesion receptors, and saccharides that block selectin-mediated rolling. Other potential targets include blocking the signaling pathways.
It is beyond the scope of this review to describe the many animal models used to evaluate the effect of anti-adhesion therapy. However, several disorders of diverse etiology will be presented as examples of their novel approach to treatment of inflammatory diseases.
Ischemia and reperfusion injury. Tissue and organ damage resulting from ischemia and reperfusion has been described in a variety of clinical disorders, including stroke, myocardial infarction, organ transplantation, and organ hypoperfusion. Reperfusion after a period of arrested blood flow is known to enhance tissue damage in animal models. Reperfusion injury is associated with activation of the leukocytes and endothelial cells, resulting in increased adherence of leukocytes which emigrate to the tissue. Reperfusion-induced leukocyte adherence may thus damage both the vessel wall and surrounding tissue. Using the rabbit ear as a model it was found that both edema and necrosis were significantly decreased by MAb to β2 integrin and to P-selectin(53).
Asthma. The late phase reaction in asthma is due to an inflammatory response with accumulation of neutrophils and eosinophils. Airway hyperreactivity and bronchoalveolar eosinophilia that accompany repeated allergen challenges were effectively reduced by i.v. infusion or inhalation of anti-ICAM-1 MAb. Other anti-adhesion MAb, such as anti-VLA-4, may also be of beneficial effect(54).
Diabetes. Nonobese diabetic mice develop diabetes which is insulin-dependent at the age of 3-6 mo after an autoimmune process which is very similar to insulin-dependent diabetes mellitus in man. Insulitis with lymphocyte infiltration occurs at the β cells islets several months before overt diabetes appears. At this early stage there is an increased expression of several adhesion molecules in the pancreatic islets. Studies have shown that antibodies to VLA-4, which is specific for lymphocytes, reduce T cell infiltration and slow disease progression(55).
Bacterial infections. Adhesion molecules are critical for the accumulation of inflammatory leukocytes at site of bacterial infection. In some bacterial infections, such as meningitis, these leukocytes contribute to irreversible tissue damage. Polysaccharide fucoidin, a blocking agent of the L-selectin, was found to cause a dramatic decrease in the accumulation of leukocytes in the cerebrospinal fluid in experimental meningitis in the rabbit(56).
Graft rejection. Increased expression of endothelial adhesion molecules has been observed during human renal, cardiac, and liver transplant rejection. In monkeys, anti-ICAM-1 MAb reduce lymphocyte infiltration and prolong kidney allograft survival. Preliminary conclusions of a phase I clinical trial conducted on human recipients of kidney allografts suggest that therapy with anti ICAM-1 MAb may increase graft survival(57).
Our understanding of the mechanisms of leukocyte emigration into areas of inflammation has increased dramatically in the last decade. The physiologic and pathophysiologic roles played by the various adhesion molecules are rapidly being determined, along with the realization that certain subsets of adhesion molecules may be important in specific forms of inflammation(58). These finding open new therapeutic avenues for many pathologic conditions which are currently associated with high morbidity and mortality.
Abbreviations
- LFA-1:
-
lymphocyte-associated antigen-1
- Mac-1:
-
macrophage antigen-1
- VLA:
-
very late antigen
- VCAM:
-
vascular cell adhesion molecule
- ICAM:
-
intercellular adhesion molecule
- SLeX:
-
sialyl Lewis X
- LAD:
-
leukocyte adhesion deficiency
- TNF:
-
tumor necrosis factor
References
Carlos TM, Harlan JM 1994 Leukocyte endothelial adhesion molecules. Blood 84: 2068–2101
Springer TA 1994 Traffic signals for lymphocyte recruitment and leukocyte emigration: the multistep paradigm. Cell 76: 301–314
Hynes RO 1992 Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69: 11–25
Smyth SS, Joneckis CC, Parise LV 1993 Regulation of vascular integrins. Blood 81: 2827–2843
Hibbs ML, Xu H, Stacker SA, Springer TA 1991 Regulation of the adhesion of ICAM-1 by the cytoplasmic domain of LFA-1 integrin subunit. Science 251: 1611–1613
Springer TA, Thompson NS, Miller LJ, Schmalsstieg FC, Anderson DC 1984 Inherited deficiency of the MAC-1, LFA-1,p150,95 glycoprotein family and its molecular basis. J Exp Med 160: 1901–1918
Kishimoto TK, Rothlein R 1993 Adhesion molecules which guide neutrophil endothelial cell interaction at site of inflammation. In: Gupta S, Griscelli C (eds) New Concepts in Immunodeficiency Diseases. John Wiley & Sons, New-York, pp 131–152
Dustin ML, Springer TA 1988 Lymphocyte function associate antigen-1 (LFA-1) interaction with intecellular adhesion molecule-1 (ICAM-1) is one of at least three mechanisms for lymphocyte adhesion to culture endothelial cell. J Cell Biol 107: 321–331
Lobb RR, Hemler ME 1994 The pathophysiology role of4 integrins in vivo. J Clin Invest 94: 1722–1728
Diamond MS, Staunton DE, Marlin SD, Springer TA 1991 Binding of the integrin MAC-1 (CD11b/CD18) to the third immunoglobulin like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell 65: 961–971
Staunton DE, Dustin ML, Springer TA 1989 Functional cloning of ICAM-2, a cell adhesion ligand for LFA-1 homologous to ICAM-1. Nature 339: 61–64
de Fougerolles AR, Springer TA 1992 Intracellular adhesion molecule 3, a third adhesion counter-receptor for lymphocyte function associated molecule 1 on resting lymphocytes. J Exp Med 175: 185–190
Etzioni A 1994 Adhesion molecules in host defense. Clin Diagn Lab Immunol 1: 1–4
Campanero MR, Sanchez-Matios P, del Pozo MA, Sanchez-Madrid F 1994 ICAM-3 regulates lymphocyte morphology and integrin mediated T cell interaction with endothelial cell and extracellular matrix ligands. J Cell Biol 127: 867–878
Gearing AJH, Newman W 1993 Circulating adhesion molecules in disease. Immunol Today 14: 506–512
Pepinsky B, Hession C. Chen LL, Moy P, Burkly L, Jakubowski A, Show EP, Benjamin C 1992 Structure/function studies in vascular cell adhesion molecule-1. J Biol Chem 267: 17820–17826
Bevilacqua MP 1993 Endothelial leukocyte adhesion molecules. Annu Rev Immunol 11: 767–804
Bevilacqua MP, Nelson RM 1993 Selectins. J Clin Invest 91: 379–387
Cotran RS, Gimbrone MA, Bevilacqua PM, Mendrick DL, Pober JS 1986 Induction and detection of a human endothelial activation antigens in vivo. J Exp Med 164: 661–666
Gotsch U, Jager U, Dominis M, Vestweber D 1994 Expression of P-selectin on endothelial cells is upregulated by LPS and TNF- in vivo. Cell Adhes Commun 2: 7–14
Jung TM, Dailey MO 1990 Rapid modulation of homing receptors (gp90 mel-14) induced by activators of protein kinase C: receptor shedding due to accelerated proteolytic cleavage at the cell surface. J Immunol 144: 3130–3136
Schleiffenbaum B, Spertini O, Tedder TF 1992 Soluble L-selectin is present in human plasma at high levels and retains functional activity. J Cell Biol 119: 229–238
Varki A 1994 Selectin ligands. Proc Natl Acad Sci USA 91: 7390–7397
Butcher EC 1991 Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell 67: 1033–1036
Lawrence MB, Springer TA 1991 Leukocyte roll on a selectin at physiologic flow rates: distinction from and prerequisite for adhesion through integrins. Cell 65: 859–873
von Andrian UH, Chambers JD, McEvoy LM, Bargatze REF, Arfors KE, Butcher EC 1991 Two step model of leukocyte-endothelial cell interaction in inflammation: distinct role of LECAM-1 and the leukocyte2 integrins in vivo. Proc Natl Acad Sci USA 88: 7538–7542
Olofsson AM, Arfors KE, Ramezani L, Wolitzky BA, Butcher EC, von Andrian UH 1994 E-selectin mediated leukocyte rolling in interleukin-1 treated rabbit mesentery venules. Blood 84: 2749–2758
Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD 1993 Leukocyte rolling and extravasation are severely compromised in P-selectin deficient mice. Cell 74: 541–554
von Andrian UH, Berger EM, Ramezani L, Chambers JD, Ochs HD, Harlan JM, Paulson JC, Etzioni A, Arfors KE 1993 In vivo behavior of neutrophils from two patients with distinct inherited leukocyte adhesion deficiency syndromes. J Clin Invest 91: 2893–2897
DeGraaf TW, Von der Stelt ME, Anbergen MG, van Dijk W 1993 Inflammation induced expression of sialyl Lewis X containing glycan structures on A acid glycoprotein (oromucid) in human sera. J Exp Med 177: 657–666
Zimmerman GA, Prescott SM, McIntyre TM 1992 Endothelial cell interactions with granulocytes: tethering and signaling molecules. Immunol Today 13: 93–100
Muller WA, Weigl SA, Deng X, Phillips DM 1993 PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med 178: 449–457
Shuster DE, Kehrli ME, Ackermann MR, Gilbert RO 1992 Identification and prevalence of a genetic defect that causes leukocyte adhesion deficiency in Holstein cattle. Proc Natl Acad Sci USA 89: 9225–9229
Wilson RW, Ballantyne CM, Smith CW, Montgomery C, Bradley A, O'Brien WE, Beaudet AL 1993 Gene targeting yields a CD18 mutant mouse for study of inflammation. J Immunol 151: 1571–1578
Sligh JE, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL 1993 Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc Natl Acad Sci USA 90: 8529–8533
Xu H, Gonzalo JA, St. Pierre Y, Williams IR, Kupper TS, Cortan RS, Springer TA, Gutierrez-Ramos JC 1994 Leukocytosis and resistance to septic shock in intercellular adhesion molecule-1 deficient mice. J Exp Med 180: 95–109
Labow MA, Norton CR, Rumberger JM, Lombard-Gillooly KM, Shuster DJ, Hubband J, Bertko R, Knaas PA, Terry RW, Harbison ML, Kontgen F, Stewart CL, McIntyre KM, Will PC, Burns DK, Wolitzky BA 1994 Characterization of E-selectin deficient mice: demonstration of overlapping function of the endothelial selectins. Immunity 1: 709–720
Arbones ML, Ord DC, Ley K, Ratech H, Maynard-Curry C, Otten G, Capon DJ, Tedder TF 1994 Lymphocyte homing and leukocyte rolling and migration are impaired in L-selectin deficient mice. Immunity 1: 247–260
Etzioni A 1994 Adhesion molecule deficiencies and their clinical significance. Cell Adhes Commun 2: 257–260
Harlan JM 1993 Leukocyte adhesion deficiency syndrome: insights into the molecular basis of leukocyte emigration. Clin Immunol Immunopathol 67:s16–s24
Sligh JE, Hurwittz MY, Zhu C, Anderson DC, Beauder AL 1992 An initiation codon mutation in CD18 in association with the moderate phenotype of leukocyte adhesion deficiency. J Biol Chem 267: 714–718
Schwartz BR, Harlan JM 1991 Consequence of deficient granulocyte endothelium interaction. In: Gordon JL (ed) Vascular Endothelium Interactions with Circulating Cells. Elsevier Science Publishers B.V., Amsterdam, pp 231–252
Anderson DC, Springer TA 1987 Leukocyte adhesion deficiency: an inherited defect in the Mac-1, LFA-1, and p150,95 glycoproteins. Annu Rev Med 38: 175–194
Fisher A, Lisowska-Grospierre B. Anderson DC, Springer TA 1988 Leukocyte adhesion deficiency: molecular basis and functional consequences. Immunodefic Rev 1: 39–54
Wilson JM, Ping AJ, Krauss JC, Mayo-Bond L, Rogers CE, Anderson DC, Todd RF 1990 Correction of CD18-deficient lymphocytes by retrovirus mediated gene transfer. Science 248: 1413–1416
Etzioni A, Frydman M, Pollack S, Avidor I, Phillips ML, Pualsom JC, Gershoni-Baruch R 1992 Severe recurrent infections due to a novel adhesion molecule defect. N Engl J Med 327: 1789–1792
Etzioni A, Harlan JM, Pollack S, Phillips LM, Gershoni-Baruch R, Paulson JC 1993 Leukocyte adhesion deficiency (LAD) II: a new adhesion defect due to absence of sialyl Lewis X, the ligand for selectins. Immunodeficiency 4: 307–308
Etzioni A, Phillips LM, Paulson JV[undot]iC, Harlan RE 1995 Leukocyte adhesion deficiency (LAD) II. In: Cell Adhesion and Human Disease (Ciba Foundation Symposium 189). Wiley, Chichester, UK, pp 51–62
Price TH, Ochs HD, Gershoni-Baruch R, Harlan JM, Etzioni A 1994 In vivo neutrophil and lymphocyte function studies in a patient with leukocyte adhesion deficiency type II. Blood 84: 1635–1639
Adams DH, Shaw S 1994 Leukocyte endothelial interactions and regulation of leukocyte migration. Lancet 343: 831–836
Bevilacqua MP, Nelson RM, Mannori G, Cecconi O 1994 Endothelial leukocyte adhesion molecules in human disease. Annu Rev Med 45: 361–378
Albelda SM, Smith CW, Ward PA 1994 Adhesion molecules and inflammatory injury. FASEB J 8: 504–512
Mihelcic D, Schleiffenbaum B, Tedder TF, Sharar SR, Harlan JM, Winn RK 1994 Inhibition of leukocyte L-selectin function with a monoclonal antibody attenuates reperfusion injury to the rabbit ear. Blood 84: 2322–2328
Bochner BS, Schleimer RP 1994 Role of adhesion molecules in human eosinophil and basophil recruitment. J Allergy Clin Immunol 94: 427–438
Burkly LC, Jakubowski A, Hattori M 1994 Protection against adaptive transfer of autoimmune diabetes mediated through very late antigen-4 integrin. Diabetes 43: 529–534
Granert C, Raud J, Xie X, Lindquist L, Lindbom L 1994 Inhibition of leukocyte rolling with polysaccharide fucoidin prevents pleocytosis in experimental meningitis in the rabbit. J Clin Invest 93: 929–936
Haug CE, Colvin RB, Delmonico FL, Auchincloss H, Tolkoff-Rubin N, Prteffer FI, Rothlein R, Norris S, Scharschmidt L, Cosimi AB 1993 A phase I trail of immuno-suppression with anti ICAM-1 (CD54) mAb in renal allograft recipients. Transplantation 55: 766–773
Gorski A 1994 The role of cell adhesion molecules in immunopathology. Immunol Today 15: 251–255
Phillips LM, Schwartz BR, Etzioni A, Boyer R, Ochs HD, Paulson JC, Harlan JM 1995 Neutrophil adhesion in leukocyte adhesion deficiency syndrome type II (LAD II). J Clin Invest ( in press)
Acknowledgements
The author is indebted to John M. Harlan, M.D., and Hans D. Ochs, M.D., for their thoughtful comments.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Etzioni, A. Adhesion Molecules-Their Role in Health and Disease. Pediatr Res 39, 191–198 (1996). https://doi.org/10.1203/00006450-199602000-00001
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199602000-00001
This article is cited by
-
Peripheral blood BDNF and soluble CAM proteins as possible markers of prolonged disorders of consciousness: a pilot study
Scientific Reports (2024)
-
Population Pharmacokinetic Analysis of Rivipansel in Healthy Subjects and Subjects with Sickle Cell Disease
Drugs in R&D (2021)
-
Apparent activation energies of protein–protein complex dissociation in the gas–phase determined by electrospray mass spectrometry
Analytical and Bioanalytical Chemistry (2017)
-
Structural and functional analysis of cell adhesion and nuclear envelope nano-topography in cell death
Scientific Reports (2015)
-
Defects in the Leukocyte Adhesion Cascade
Clinical Reviews in Allergy & Immunology (2010)
