
Abstract
A copolymer of TEMPO- and aniline-substituted norbornene was prepared by ring-opening metathesis polymerization of the corresponding monomers. Electropolymerization of the pendant aniline groups in the copolymer gave a layer of polynorbornene populated with the redox-active TEMPO pendants, in which polyaniline chain was incorporated. Electroactivity of the TEMPO pendants throughout the layer and its excellent charging/discharging cyclability, in addition to the amorphous nature of the layer, suggested that the polyaniline chain was homogeneously dispersed in the layer and that each chain served as crosslinking moiety. The effect of the polyaniline chains was further enhanced when they were formed by in-situ electropolymerization of the aniline group in the preformed layer of the copolymer/polyaniline composite. The polyaniline chain served as a conducting path to reduce the charge-transfer resistance for redox mediation, which gave rise to an excellent rate performance for the charging/discharging process of the layer, compared with those for the composite layers of TEMPO-substituted polymers with polyaniline and other conductive additives prepared by the conventional grinding methods.
Similar content being viewed by others

Introduction
Charge transport by a redox mediation process is accomplished by an electron self-exchange reaction between redox-active sites.1, 2, 3, 4, 5 When the redox-active sites are electrochemically reversible6, 7, 8, 9 and are bound to a polymer chain, an electron self-exchange reaction between the neighboring sites effects the redox mediation through the polymer layer, which requires that electroneutralization by electrolyte ions is accomplished throughout the layer to maintain the electroactivity.10, 11, 12 Aliphatic, or non-conjugated polymers populated with organic robust radicals such as nitroxide,13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23 nitronyl nitroxide,24, 25 spirobisnitroxide26 and galvinoxyl27, 28 have been found to be the typical examples, and their ideal redox-mediating properties29 are different from those of conventional redox polymers such as poly(vinylferrocene),30, 31, 32, 33 which has been dominated by the ‘break-in effect’ of the electroneutralizing ions to give rise to limited charge capacity and hysteresis during the charging/discharging process. The ideal, that is, diffusion-limited behavior by the so-called ‘radical polymers’34, 35 has led to reversible and exhaustive charging with the simple redox diffusive process formulated by the finite diffusion equation36 for the layer with a discrete thickness on a current collector,37 and by the equation for semi-infinite diffusion36 that applies to wholly gelated cells with the radical polymer.38 Added support for the diffusional process has been obtained by the radical polymer layer sandwiched with two electrodes, which gave rise to the steady–state redox gradient across the layer to produce limiting current according to the layer thickness.39 In rechargeable devices such as batteries40, 41, 42, 43, 44, 45, 46 and electrochromic cells,47, 48, 49 such polymers must be partitioned into anode and/or cathode sides to avoid the redox shuttling between the two electrodes,50, 51, 52, 53, 54 and, for this purpose, the polymers are usually crosslinked to suppress dissolution into electrolytes and yet to maintain the swelling properties.37 In addition, conductive additives such as vapor-grown carbon nanofiber,29 carbon nanotube (CNT), graphene55 and carbon foam56 have been employed to fabricate polymer composite electrodes to reduce the resistivity of the organic layer for excellent rate performance,57, 58 which have spawned various methods to prepare the polymer/carbon composite layers with the minimum requisite amounts of the carbon additives to maintain the overall redox capacity, such as the CNT wrapping59 and surface-initiated polymerization of the radical monomers from carbon surface60 and other electroconductive material surfaces.61, 62, 63
We anticipated that the effect of the additives should be maximized when the two dissimilar materials for redox mediation and electrical conduction are compounded at a molecular level to afford an integrated composite material. However, attempts to use π-conjugated polymer backbones to bear the redox-active pendant groups as the organic electrode-active materials have met with failure, resulting in fluctuating redox potentials as a result of electronic intersite communication64, 65, 66 through the conjugated chain and a significant loss of the charge storage capacity. Reasoning that the mass-transfer process for electroneutralization might be disfavored within the conjugated polymer layers because of their inherent crystallinity, we turned to incorporate the conducting path within the preformed layer of the redox mediating polymer. Here we report our successful attempts to significantly reduce the charge-transfer resistance67, 68 of the composite electrode by incorporating polyaniline chain into the radical polymer both as the conducting path and the crosslinking moiety, which gave insights into the nature of charge propagation and storage processes in the polymer to allow excellent power-rate capability and charge-discharge cyclability as the organic electrode-active material.
Experimental Procedure
Materials
All solvents were purified by distillation before use. Tetrabutylammonium perchlorate (TBAClO4) was obtained from Tokyo Kasei (Tokyo, Japan) and purified by recrystallization. All other reagents were obtained from Kanto Chemical (Tokyo, Japan) or Tokyo Kasei and were used without further purification.
Synthesis of monomers
Diastereomeric mixtures of the endo-, endo- and endo-, exo-derivatives of bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid bis-(2,2,6,6-tetramethylpiperidin-1-oxyl-4-yl) ester (1) and bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylic acid 2-methyl 3-(2-oxo-2H-chromen-7-yl) ester (3) (Scheme 1) were prepared according to the method described in our previous report.37 2-(3-Aminophenyl) 3-methyl bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate (2) (Scheme 1) was prepared from norbornene monomethyl ester and 3-nitrophenol as follows. To a solution of norbornene monomethyl ester (0.50 g, 2.6 mmol) and 3-nitrophenol (0.41 g, 3.0 mmol) in CH2Cl2 (17 ml) was added 2-chloro-1-methylpyridinium iodide (0.76 g, 3.0 mol), 4-dimethylaminopyridine (0.12 g, 1.0 mmol) and triethylamine (0.69 ml, 4.95 mmol). The resulting mixture was stirred at room temperature for 15 h. Removal of the pyridinium salt by filtration followed by evaporation, extraction from CHCl3/saturated aqueous NH4Cl and purification by silica gel column chromatography yielded 2-(3-nitrophenyl) 3-methyl bicyclo[2.2.1]hept-5-ene-2,3-dicarboxylate as a colorless crystal. Yield: 75%. MS (m/z): calcd. for M+ 317.29; found 317.72. 1H NMR (CDCl3, p.p.m.): δ 8.05 (1H), 7.97 (1H), 7.49 (2H), 6.41 (1H), 6.19 (1H), 3.63 (3H), 3.49 (2H), 3.27 (2H), 1.55 (1H), 1.42 (1H). 13C NMR (CDCl3, p.p.m.): δ 172.7, 170.8, 151.1, 148.6, 135.7, 134.4, 129.9, 128.4, 120.6, 117.5, 52.0, 48.8, 48.6, 47.9, 47.0, 46.2. IR (cm−1): 2984 (νC-H), 1736 (νC=O), 1528 (νN-O), 1206 (νC-O). Anal. Calcd. for C16H15NO6: C, 60.57; H, 4.77; N, 4.41%. Found: C, 60.47; H, 4.67; N, 4.41%. The product (0.50 g, 1.58 mmol) was reduced with SnCl2·2H2O (1.07 g, 4.72 mmol) in ethanol at 80 °C under N2 for 15 h. Extraction with ethyl acetate/saturated aqueous NaCl and purification by alumina column chromatography using ethyl acetate as an eluent yielded 2 as pale yellow crystals. Yield: 57%. MS (m/z): calcd. for M+ 287.31; found 287.10. 1H NMR (CDCl3, p.p.m.): δ 6.97 (1H), 6.39 (1H), 6.21 (2H), 6.17 (1H), 6.13 (1H), 5.22 (2H), 3.58 (1H), 3.52 (3H), 3.46 (1H), 3.18 (1H), 3.12 (1H), 1.43 (1H), 1.33 (1H). 13C NMR (CDCl3, p.p.m.): δ 172.1, 170.3, 151.4, 149.8, 135.0, 134.6, 129.1, 111.0, 108.3, 106.7, 51.2, 48.0, 47.5, 47.4, 45.9, 45.8. IR (cm−1): 3376 (νN-H), 2982 (νC-H), 1756 (νC=O), 1248 (νC-O). Anal. Calcd. for C16H17NO4: C, 66.89; H, 5.96; N, 4.88%. Found: C, 66.88; H, 5.91; N, 4.65%.
Polymerization
The copolymer 4 (Scheme 2) was prepared by ring-opening metathesis polymerization (ROMP), according to the procedure as previously reported in our literature for the TEMPO-substituted norbornene,37 with slight modifications as follows. To a mixture of 1 and 2 at various compositions (0.1 M in total) in dehydrated THF was added the Grubb’s second generation catalyst (5 mol%) under nitrogen. After stirring at 25 °C for several hours, the mixture was poured into hexane/diethyl ether (1/1 in v/v) to precipitate the product 4, which was collected by filtration and washed repeatedly with hexane to give a pale purple powder (Table 1). The product with a lower composition of 2 than 50% (that is, entries 3 and 4 in Table 1) was soluble in CHCl3, CH2Cl2, THF and CH3CN. The monomer composition in the copolymer was determined by elemental analysis as shown in Table 1. It may be noted that NMR was unavailable for characterization of these copolymers because of the paramagnetic nature of the radical pendant moieties. Unpaired electron density of the polymers was determined from magnetic susceptibilities obtained by SQUID measurements.37 The densities were 95% (entry 3) and 94% (entry 4) per 1 unit in the copolymer, which were roughly in agreement with the composition of the copolymer based on the elemental analysis.
Crosslinking
The copolymer 4 (20 mg) with a composition of m/n=7/3 (entry 4 in Table 1) was dissolved in CH3CN (10 ml) containing 0.1 M TBAClO4. Then, it was electropolymerized onto an ITO/glass electrode by scanning the electrode potential in the range of 0–0.85 V vs Ag/AgCl at 50 mV s−1 to give a homogeneous purple layer of 5 (Scheme 3), which was rinsed with CH3CN repeatedly.
Polyaniline-enriched layer of 5 was prepared by the electropolymerization of 4 in the presence of polyaniline as follows. To an aqueous solution of aniline/HCl was added a slight excess of ammonium persulfate, which was then stirred at 0 °C for several hours. The product was collected by filtration, washed with water and aqueous HCl, and dried to give a dark green powder of polyaniline in 58% with Mn=1.3 × 104, which was soluble in NMP and THF. A solution of the copolymer 4 with a composition of m/n=7/3 (entry 4 in Table 1) in ethyl lactate was mixed with a solution of polyaniline in NMP with a weight ratio of 4/polyaniline=9/1, which was coated on an ITO/glass electrode with a surface area of A=1 cm2 using a bar coater. The resulting layer was 1.5 μm in thickness. The electrode was then immersed in CH3CN containing 0.1 M TBAClO4, which was maintained at a potential of 1.0 V vs Ag/AgCl to undergo crosslinking by the electrolysis of the aniline unit in 4 and polyaniline in the layer. After the electrolysis with an electricity of 0.026 C, the electrode was rinsed with CH3CN repeatedly to give the polyaniline-enriched layer of 5.
The crosslinked polymer 6, a control material without the conducting path, was prepared by the procedure described in our previous literature,37 with slight modifications as follows. The monomers 1 and 3 with a molar ratio of 9/1 (0.1 M in total) was dissolved in CH2Cl2 to which was added the Grubb’s second generation catalyst. The resulting solution was deaerated and stirred at 40 °C. The copolymer was isolated by pouring the reaction mixture into hexane to precipitate the product, which was collected by filtration and washed with hexane repeatedly. The copolymer was obtained as a red powder in 81% yield with Mn=1.0 × 104 (Mw/Mn=1.8) and a monomer composition of 8.5/1.5 based on the elemental analysis, which was soluble in CH2Cl2, CHCl3 and CH3CN. Crosslinking of the copolymer spin-coated on an electrode was accomplished by photodimerization of the coumarin unit, which proceeded under the irradication of light at 8.1 J cm−2 using a low-pressure Hg lamp. The progress of the crosslinking was monitored by the gradual decrease in intensity of the absorption band at λmax=320 nm for the coumarin unit, which disappeared after 20 min on the completion of the dimerization in the polymer layer. Survival of the TEMPO radical during the course of the irradiation was supported by the unpaired electron density determined by the SQUID measurements, which amounted to more than 0.9 per TEMPO unit.
Preparation of a Poly(1-oxy-2,2,6,6-tetramethylpiperidinyl vinyl ether)/polyaniline Composite Layer
Poly(1-oxy-2,2,6,6-tetramethylpiperidinyl vinyl ether) (PTVE) was prepared according to the method described in the previous literature,15 with slight modification as follows. A mixture of 4-hydroxy-2,2,6,6-tetramethylpiperidin-1-oxyl, vinyl acetate, an iridium catalyst [IrCl(cod)]2 and sodium carbonate in dehydrated toluene was stirred at 90 °C for 5 h. After concentrating the mixture and adding hexane, the resulting mixture was further stirred for 10 min. Purification of the product by silica gel column chromatography with hexane/ethyl acetate (5/1 in v/v) as an eluent gave the monomer 1-oxy-2,2,6,6-tetramethylpiperidinyl vinyl ether as a red powder. The monomer and a catalyst, boron trifluoride diethyl etherate, was dissolved in dehydrated 1,2-dichloroethane maintained at −78 °C, which was then heated to −25 °C and kept stirring for 20 h. After quenching the polymerization by the addition of methanol and pouring the mixture into hexane, PTVE was obtained as a red powder (Mn=1.2 × 104). The oxidized product of the polymer, PTVE+ in which all of the nitroxide radicals were converted to oxoammonium hexafluorophosphate, was prepared by slowly adding 60% hexafluorophosphoric acid to an aqueous solution of PTVE and stirring the resulting solution for 1 h followed by addition of an aqueous solution of sodium hypochlorite to precipitate the crude product, which was dissolved in CH3CN and purified by precipitation from H2O to yield a pale green powder of PTVE+ as the hexafluorophosphate salt. The PTVE+/polyaniline composite layer was prepared either by spin-coating a mixture of a CH3CN solution of PTVE+ and a dispersion of polyaniline in methanol (PTVE+/polyaniline=9/1 in w/w) on an electrode, or by electropolymerizing aniline at an electrode modified with a preformed layer of PTVE+, which was accomplished by maintaining the PTVE+-modified electrode at 1.0 V vs Ag/AgCl in aqueous HCl solution of aniline.
Methods
Electrochemical analyses were carried out in a conventional cell under argon. The auxiliary electrode was a coiled platinum wire. The reference electrode was a commercial Ag/AgCl immersed in a solution of 0.1 M TBAClO4 in CH3CN. The formal potential of the ferrocene/ferrocenium couple was 0.45 V vs this Ag/AgCl electrode. An ALS 660B Electrochemical Analyzer (ALS Co., Tokyo, Japan) was employed to obtain the voltammograms.
1H and 13C NMR spectra were recorded on a JEOL JNM-LA500 (JEOL Co., Tokyo, Japan) or Bruker AVANCE 600 spectrometer (Bruker Japan Co., Tokyo, Japan) with chemical shifts downfield from tetramethylsilane as the internal standard. Infrared spectra were obtained using a Jasco FT-IR 410 spectrometer (Jasco Co., Tokyo, Japan) with potassium bromide pellets. Molecular weight measurements were done by gel permeation chromatography using a TOSOH HLC8220 instrument (TOSOH Co., Tokyo, Japan) with CHCl3 or THF as the eluent. Calibration was done with polystyrene standards. Elemental analyses were performed using a Perkin-Elmer PE-2400 II (Perkin-Elmer Japan Co., Tokyo, Japan) and a Metrohm 645 multi-DOSIMAT (Metrohm Japan Co., Tokyo, Japan). Two parallel analyses were performed for each sample. Mass spectra were obtained using a JMS-SX102A (JEOL Co.) or Shimadzu GCMS-QP5050 spectrometer (Shimadzu Co., Tokyo, Japan). The magnetization and the magnetic susceptibility of the powdery polymer samples were measured by a Quantum Design MPMS-7 SQUID magnetometer (Quantum Design Inc., San Diego, CA, USA). The magnetic susceptibility was measured from 1.95 to 270 K in a 0.5 T field.
Results and Discussion
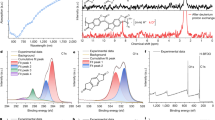
An excellent functional group tolerance established for ROMP of norbornene has allowed the synthesis of a number of polynorbornene derivatives bearing various functional groups. A polynorbornene-polythiophene hybrid has been obtained by preparing polynorbornene bearing 2,2′:5′,2″-terthiophen-3′-yl pendant group, followed by in-situ electropolymerization of the terthiophene unit within the polymer.69 Similar approach has been employed to integrate various conducting polymers such as PEDOT into polynorbornene.70, 71 However, one could state that the polythiophene-based conducting path might be incompatible with the radical polymers because of the dedoping process anticipated for the p-doped thienylene chain to parallel with the redox mediation process of the radical polymers equilibrated near 0.8 V vs Ag/AgCl. Reasoning that conducting polymers with more negative doping potentials should be compatible with the radical polymers, we integrated polyaniline into the TEMPO-substituted polynorbornene. The copolymer 4 with a composition of m/n=7/3, prepared by the ROMP of the corresponding monomers 1 and 2 (Table 1), was subjected to electropolymerization of the aniline pendants by the electrolysis of the solution of 4 with an ITO/glass electrode. Cyclic voltammetry revealed a gradual increase in the current for the TEMPO/TEMPO+ redox couple during the repeated potential scanning (Figure 1b), which resulted in the formation of 5 on the ITO/glass as a pale purple layer. With the progress of the elecropolymerization, new absorption bands ascribed to the quinoid imine unit in the polyaniline chain emerged at λmax=1107 cm−1 and 650 nm in the infrared and visible spectra, respectively. The lack of diffusion tail in the cyclic voltammogram of the layer of 5 at E1/2=0.78 V vs Ag/AgCl (Figure 1a) and the amounts of charge integrated for the charging and discharging processes that were both almost coincided with the amount of the TEMPO units in the layer demonstrated a reversible charge storage throughout the polymer layer. The much broader electrochemical response from the polyaniline chain near 0.7 V was hidden behind the wave for the TEMPO unit. An intriguing aspect of the electropolymerization was the capability of tuning the layer thickness according to the amount of charge passed during the potentiostatic electrolysis of 4 (Figure 1c). The thickness increased almost linearly with the amount of the electricity up to more than 1 μm, maintaining the apparent homogeneity and transparency of the layer. However, the electropolymerized layer of 5 was not obtained when hexafluorophosphates or tetrafluoroborates were employed as the electrolyte anion in place of the perchlorate, which indicated that the property of the layer depended on the nature of the dopant anion in the polyaniline chain.

(a) Cyclic voltammogram (5 cycles) for a thin layer of 5 on an ITO/glass electrode obtained by electrolytic polymerization of 4 (m/n=7/3) followed by thorough washing with CH3CN to remove unreacted 4 in the layer. The electrolyte contained 0.1 M TBAClO4 in CH3CN. Scan rate=5 mV s−1. (b) Cyclic voltammogram recorded during the electropolymerization of 4 (m/n=7/3) on the ITO per glass. The electrolyte was a solution of 4 (20 mg) in CH3CN (10 ml) containing 0.1 M TBAClO4. Scan rate=50 mV s−1. (c) Plots of a layer thickness of 5 versus the amount of charge consumed during the electropolymerization of 4 (m/n=7/3) by potentiostatic electrolysis at 0.9 V vs Ag/AgCl. The electrolyte conditions were as in (b).
The effect of polyaniline as the conducting path was highlightened in the cyclic voltammogram obtained for thick layers (Figure 2). While a broad wave was recorded for the 1.5-μm thick layer of 6 as a result of the increase in resistance with the thickness, a quasi-reversible response with a smaller peak-to-peak separation was obtained for the layer of 5, which suggested the decrease in the substantial diffusional length.

Cyclic voltammograms for thick layers of 5 (red curve) and 6 (black curve). The conditions for the electropolymerization of 4 and the electrolyte were as in Figure 1a. The thickness was 1.5 μm for both 5 and 6.
The reduced resistance of the polymer layer by the integration of the polyaniline chain reflected in excellent power-rate capability. Galvanostatic charging/discharging curves revealed the higher rate performance for the layer of 5 that maintained more than 50% of the formula weight-based theoretical charge-storage density even at a high rate of 120 C (that is, within 30 s) than that for 6 (Figure 3), where 1 C represented the current density required to complete full charging or discharging in 1 h. The reduced resistance of the layer also led to the less significant overvoltage required for the charging and discharging processes, which was evidenced by the smaller shift in plateau potentials at high C rates.

Galvanostatic chronopotentiomery for charging and discharging of 5 (a) and 6 (b) at the rate of 10, 20, 40, 80 and 120 C with the descending order of capacity. For discharging at various C rates, the layers were charged in advance at 10 C. The layers of 5 and 6 were as in Figure 2. The electrolyte conditions were as in Figure 1a.
Attempt to increase the polyaniline composition in 5 was examined with a view to further improve the rate performance for the charging/discharging process. However, electrolopolymerization of the aniline-enriched 4 with a composition of m/n=5/5 (entry 3 in Table 1) resulted only in the formation of very thin layers with a thickness of less than 300 nm. The copolymers 4 with higher aniline composition were all insoluble, which limited the use as the monomer for the electropolymerization. In contrast, the polyaniline-enriched layer of 5 was successfully prepared by an in-situ electropolymerization of the preformed layer of a 4/polyaniline composite with a composition of 9/1 (in w/w) on the ITO/glass electrode. Interestingly, dissolution of 4 during the electropolymerization was suppressed by the presence of polyaniline as the conducting additive, which suggested that polyaniline also acted as a binder in the composite layer. Electrolysis of the layer affected the crosslinking of the aniline unit in 4 with polyaniline to provide the layer of polyaniline-enriched 5, which was characterized by an excellent robustness of the charging/discharging cycles without dissolution of the polymer into the electrolyte or decrease in the charge-storage capacity. Indeed, the robustness of the charging/discharging cyclability was a good indication of the crosslinking. Layers of 5 with higher polyaniline contents, prepared from the 4/polyaniline composites with 7/3 and 6/4 (in w/w), both resulted in the loss of unreacted or excess polyaniline that dissolved into the electrolyte. The crosslinking was also evidenced by a control experiment, in which a composite layer of a copolymer of 1 and 3 (that is, the precursor of 6) and polyaniline was electrolyzed to give a layer of the unreacted copolymer that readily dissolved into the electrolyte.
Galvanostatic charging/discharging experiments for the polyaniline-enriched layer of 5, prepared from the 4/polyaniline composite with a composition of 9/1 (in w/w), revealed an exceptionally high rate capability that retained almost 50% of the theoretical charge-storage density even at a very fast charging/discharging at 360 C (Figure 4a). The rate performance significantly improved from that of the layer of 5 prepared by the electropolymerization of 4 (Figure 4b), suggesting the effect of integrated polyaniline as the conducting path. It may be added that the process of preparing the polyaniline-enriched 5 would be advantageous over the electropolymerization of dissolved 4, because all of the 4 molecules in the preformed layer is retained in the resulting composite layer.

Chronopotentiomeric responses for (a) the polyaniline-enriched layer of 5 prepared by the electropolymerization of 4 in the presence of polyaniline (9/1 in w/w) and (b) a repeat of Figure 3a, at very high rates. The C rates were 10, 40, 80, 120, 240 and 360 C with the descending order of capacity. For discharging at various C rates, the layers were charged in advance at 10 C. The electrolyte conditions were as in Figure 1a.
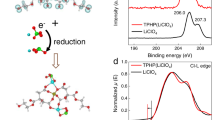
Nyquist plots obtained from the impedance spectroscopy of the polyaniline-enriched layer of 5, equilibrated at each electrode potential (Figure 5a), revealed a smaller semicircle and thus a less resistive charge-transfer process than those obtained for the photo-crosslinked layer of 6 (Figure 5b), which was in agreement with the higher rate performance obtained with 5. The radius of the semicircle, or the charge-transfer resistance RCT, was minimized when the layers were equilibrated near the redox potential of E1/2=0.8 V vs Ag/AgCl (curves C), based on the mechanism of the redox mediation through the electron self-exchange reaction between the neighboring sites.37 The decrease in RCT for 5, compared with 6, was the most significant when the electrode potential was maintained at 1.0 V vs Ag/AgCl, which was indicative of the enhanced effect of polyaniline as the conducting path in the fully doped state of the emeraldine base.

Nyquist plots for impedance analysis of (a) the polyaniline-enriched layer of 5 and (b) 6. The electrode potentials were 0.6 (A), 0.7 (B), 0.8 (C), 0.9 (D) and 1.0 V vs Ag/AgCl (E). The electrolyte conditions were as in Figure 1a.
Attempts to prepare the hybrid of the radical polymer and polyaniline without the chemical bond between the two components was examined by an exhaustive mechanical grinding method. For this purpose, an aqueous solution of a hydrophilic radical polymer, PTVE+, was employed with the dispersion of polyaniline in methanol. However, the resulting composite layer was more resistive than the polyaniline-enriched layer of 5. Incorporation of polyaniline into the preformed layer of PTVE+ by the electropolymerization of aniline at the PTVE+-coated anode also gave resistive layers with low-rate performances. These results demonstrated that the chemical bond formation by the crosslinking of the radical polymer with polyaniline most likely affected the integration of the two dissimilar polymers to yield the molecular hybrid. Incorporation of the self-doped polyaniline chain into the radical polymers with a view to further reduce the charge-transfer resistance, together with the exploration of radical-bearing aliphatic polymers with higher affinity to the π-conjugated chain, is the topic of our continuous investigation.
Conclusion
A hybrid of the electroconducting π-conjugated polymer and the non-conjugated redox-mediating polymer at a molecular level was prepared by integrating polyaniline into polynorbornene populated with redox-active TEMPO sites. The synthetic approach was based on the in-situ electropolymerization of the aniline pendants incorporated in the copolymer, which took advantage of the functional group tolerance of the ROMP of norbornene. The obtained layer was characterized by the significant power-rate capability as a result of the presence of the conducting path to reduce the charge-transfer resistance, giving rise to very fast charging and discharging of the layer within several seconds and yet maintaining the substantial capacity and plateau voltage. The present approach is viewed as a practical strategy to improve the performance of various redox mediators with diffusion-limited mass transfer processes.

Norbornenedicarboxylates with TEMPO (1), aniline (2) and coumarin (3) functional groups.

Synthesis of TEMPO- and aniline-substituted polynorbornene (4) by ROMP.

Poly(TEMPO-substituted norbornene) crosslinked with electropolymerized polyaniline (5) and photodimerized coumarin37 (6).
References
Murray, R. W . Molecular Design of Electrode Surfaces (Wiley-Interscience, New York, NY, USA, 1992).
Nishide, H . & Oyaizu, K . Toward flexible batteries. Science 319, 737–738 (2008).
Oyaizu, K . & Nishide, H . Radical polymers for organic electronics: a radical departure from conjugated polymers? Adv. Mater. 21, 2339–2344 (2009).
Blauch, D. N . & Saveant, J. M . Dynamics of electron hopping in assemblies of redox centers. Percolation and diffusion. J. Am. Chem. Soc. 114, 3323–3332 (1992).
Nishihara, H ., Kanaizuka, K ., Nishimori, Y . & Yamanoi, Y . Construction of redox- and photo-functional molecular systems on electrode surface for application to molecular devices. Coord. Chem. Rev. 251, 2674–2687 (2007).
Kato, F ., Kikuchi, A ., Okuyama, T ., Oyaizu, K . & Nishide, H . Nitroxide radical molecules as highly reactive redox mediators in dye-sensitized solar cells. Angew. Chem. Int. Ed. 124, 10324–10327 (2012).
Suga, T ., Pu, Y.-J ., Oyaizu, K . & Nishide, H . Electron-transfer kinetics of nitroxide radicals as an electrode-active material. Bull. Chem. Soc. Jpn 77, 2203–2204 (2004).
Yonekuta, Y ., Oyaizu, K . & Nishide, H . Structural implication of oxoammonium cations for reversible organic one-electron redox reaction to nitroxide radicals. Chem. Lett. 36, 866–867 (2007).
Suga, T ., Takeuchi, S ., Ozaki, T ., Sakata, M ., Oyaizu, K . & Nishide, H . Synthesis of poly(oxoammonium salt)s and their electrical properties in the organic thin film device. Chem. Lett. 38, 1160–1161 (2009).
Choi, W ., Harada, D ., Oyaizu, K . & Nishide, H . Aqueous electrochemistry of poly(vinylanthraquinone) for anode-active materials in high-density and rechargeable polymer/air batteries. J. Am. Chem. Soc. 133, 19839–19843 (2011).
Chae, I. -S ., Koyano, M ., Sukegawa, T ., Oyaizu, K . & Nishide, H . Redox equilibrium of a zwitterionic radical polymer in a non-aqueous electrolyte for novel Li+ host material in a Li-ion battery. J. Mater. Chem. A 1, 9608–9611 (2013).
Chae, I. -S ., Koyano, M ., Oyaizu, K . & Nishide, H . Self-doping inspired zwitterionic pendant design of radical polymers toward a rocking-chair-type organic cathode-active material. J. Mater. Chem. A 1, 1326–1333 (2013).
Sukegawa, T ., Omata, H ., Masuko, I ., Oyaizu, K . & Nishide, H . Anionic polymerization of 4-methacryloyloxy-TEMPO using an MMA-capped initiator. ACS Macro Lett. 3, 240–243 (2014).
Bugnon, L ., Morton, C. J. H ., Novak, P ., Vetter, J . & Nesvadba, P . Synthesis of poly(4-methacryloyloxy-TEMPO) via group-transfer polymerization and its evaluation in organic radical battery. Chem. Mater. 19, 2910–2914 (2007).
Koshika, K ., Sano, N ., Oyaizu, K . & Nishide, H . An ultrafast chargeable polymer electrode based on the combination of nitroxide radical and aqueous electrolyte. Chem. Commun. 836–838 (2009).
Koshika, K ., Chikushi, N ., Sano, N ., Oyaizu, K . & Nishide, H . A TEMPO-substituted polyacrylamide as a new cathode material: an organic rechargeable device composed of polymer electrodes and aqueous electrolyte. Green Chem. 12, 1573–1575 (2010).
Hyakutake, T ., Park, J. Y ., Yonekuta, Y ., Oyaizu, K ., Nishide, H . & Advincula, R . Nanolithographic patterning via electrochemical oxidation of stable poly(nitroxide radical)s to poly(oxoammonium salt)s. J. Mater. Chem. 20, 9616–9618 (2010).
Zhuang, X ., Xiao, C ., Oyaizu, K ., Chikushi, N ., Chen, X . & Nishide, H . Synthesis of amphiphilic block copolymers bearing stable nitroxyl radicals. J. Polym. Sci. A 48, 5404–5410 (2010).
Zhuang, X ., Zhang, H ., Chikushi, N ., Zhao, C ., Oyaizu, K ., Chen, X . & Nishide, H . Biodegradable and electroactive tempo-substituted acrylamide/lactide copolymer. Macromol. Biosci. 10, 1203–1209 (2010).
Nishide, H ., Koshika, K . & Oyaizu, K . An environmentally benign battery based on organic radical polymers. Pure Appl. Chem. 81, 1961–1970 (2009).
Koshika, K ., Kitajima, M ., Oyaizu, K . & Nishide, H . A rechargeable battery based on hydrophilic radical polymer-electrode and its green assessment. Green Chem. Lett. Rev. 2, 169–174 (2009).
Koshika, K ., Sano, N ., Oyaizu, K . & Nishide, H . An aqueous electrolyte-type rechargeable device utilizing a hydrophilic radical polymer-cathode. Macromol. Chem. Phys. 210, 1989–1995 (2009).
Behrends, F ., Wagner, H ., Studer, A ., Niehaus, O ., Pottgen, R . & Eckert, H . Polynitroxides from alkoxyamine monomers: structural and kinetic investigations by solid state NMR. Macromolecules 46, 2553–2561 (2013).
Sukegawa, T ., Kai, A ., Oyaizu, K . & Nishide, H . Synthesis of pendant nitronyl nitroxide radical-containing poly(norbornene)s as ambipolar electrode-active materials. Macromolecules 46, 1361–1367 (2013).
Oyaizu, K ., Sukegawa, T . & Nishide, H . Dual dopable poly(phenylacetylene) with nitronyl nitroxide pendants for reversible ambipolar charging and discharging. Chem. Lett. 40, 184–185 (2011).
Nesvadba, P ., Bugnon, L ., Maire, P . & Novák, P . Synthesis of a novel spirobisnitroxide polymer and its evaluation in an organic radical battery. Chem. Mater. 22, 783–788 (2009).
Suga, T ., Sugita, S ., Ohshiro, H ., Oyaizu, K . & Nishide, H . P- and n-type bipolar redox-active radical polymer: toward totally organic polymer-based rechargeable devices with variable configuration. Adv. Mater. 23, 751–754 (2011).
Suga, T ., Ohshiro, H ., Sugita, S ., Oyaizu, K . & Nishide, H . Emerging n-type redox active radical polymer for a totally organic polymer-based rechargeable battery. Adv. Mater. 21, 1627–1630 (2009).
Oyaizu, K ., Kawamoto, T ., Suga, T . & Nishide, H . Synthesis and charge transport properties of redox-active nitroxide polyethers with large site density. Macromolecules 43, 10382–10389 (2010).
Nguyen, M. T ., Diaz, A. F ., Dementev, V. V . & Pannell, K. H . Electrochemical and electrochromic properties of poly(dialkylsilyleneferrocenylene) films. Chem. Mater. 6, 952–954 (1994).
Grumelli, D. E ., Garay, F ., Barbero, C. A . & Calvo, E. J . Dynamics of ion exchange between self-assembled redox polyelectrolyte multilayer modified electrode and liquid electrolyte. J. Phys. Chem. B 110, 15345–15352 (2006).
Barbero, C ., Calvo, E. J ., Etchenique, R ., Morales, G. M . & Otero, M . An EQCM electroacoustic study of poly(vinylferrocene) modified electrodes in different aqueous electrolytes. Electrochim. Acta 45, 3895–3906 (2000).
Inzelt, G . & Bacskai, J . Electrochemical quartz crystal microbalance study of the swelling of poly(vinylferrocene) films. Electrochim. Acta 37, 647–654 (1992).
Yonekuta, Y ., Susuki, K ., Oyaizu, K ., Honda, K . & Nishide, H . Battery-inspired non-volatile and rewritable memory architectures: a radical polymer-based organic device. J. Am. Chem. Soc. 129, 14128–14129 (2007).
Oyaizu, K ., Suga, T ., Yoshimura, K . & Nishide, H . Synthesis and characterization of radical-bearing polyethers as an electrode-active material for organic secondary batteries. Macromolecules 41, 6646–6652 (2008).
Bard, A. J . & Faulkner, L. R . Electrochemical Methods, Fundamentals and Applications 2nd edn (Wiley, New York, NY, USA, 2001).
Oyaizu, K ., Ando, Y ., Konishi, H . & Nishide, H . Nernstian adsorbate-like bulk layer of organic radical polymers for high-density charge storage purposes. J. Am. Chem. Soc. 130, 14459–14461 (2008).
Nakahara, K ., Oyaizu, K . & Nishide, H . Electrolyte anion-assisted charge transportation in poly(oxoammonium cation/nitroxyl radical) redox gels. J. Mater. Chem. 22, 13669–13673 (2012).
Tokue, H ., Oyaizu, K ., Sukegawa, T . & Nishide, H . TEMPO/viologen electrochemical heterojunction for diffusion controlled redox mediation: a highly rectifying bilayer-sandwiched device based on cross reaction at interface between dissimilar redox polymers. ACS Appl. Mater. Interfaces 6, 4043–4049 (2014).
Nakahara, K ., Oyaizu, K . & Nishide, H . Organic radical battery approaching practical use. Chem. Lett. 40, 222–227 (2011).
Janoschka, T ., Hager, M. D . & Schubert, U. S . Powering up the future: radical polymers for battery applications. Adv. Mater. 24, 6397–6409 (2012).
Gracia, R . & Mecerreyes, D . Polymers with redox properties: materials for batteries, biosensors and more. Polym. Chem. 4, 2206–2214 (2013).
Novak, P ., Muller, K ., Santhanam, K. S. V . & Haas, O . Electrochemically active polymers for rechargeable batteries. Chem. Rev. 97, 207–281 (1997).
Oyaizu, K . & Nishide, H . Macromolecular complexes leading to high performance energy devices. Macromol. Symp. 317–318, 248–258 (2012).
Oyaizu, K ., Niibori, Y ., Takahashi, A . & Nishide, H . BODIPY-Sensitized photocharging of anthraquinone-populated polymer layers for organic photorechargeable air battery. J. Inorg. Organomet. Polym. 23, 243–250 (2013).
Sano, N ., Tomita, W ., Hara, S ., Min, C.-H ., Lee, J. -S ., Oyaizu, K . & Nishide, H . Polyviologen hydrogel with high-rate capability for anodes toward an aqueous electrolyte-type and organic-based rechargeable device. ACS Appl. Mater. Interfaces 5, 1355–1361 (2013).
Takahashi, Y ., Hayashi, N ., Oyaizu, K ., Honda, K . & Nishide, H . Totally organic polymer-based electrochromic cell using tempo-substituted polynorbornene as a counter electrode-active material. Polym. J. 40, 763–767 (2008).
Takahashi, Y ., Oyaizu, K ., Honda, K . & Nishide, H . Low-energy driven electrochromic devices using radical polymer as transparent counter electroactive material. J. Photopolym. Sci. Technol. 20, 29–34 (2007).
Beaujuge, P. M ., Amb, C. M . & Reynolds, J. R . Spectral engineering in π-conjugated polymers with intramolecular donor-acceptor interactions. Acc. Chem. Res. 43, 1396–1407 (2010).
Oyaizu, K ., Ikeda, H ., Hayo, N ., Kato, F . & Nishide, H . Ionic liquid-inspired redox shuttles: properties of a ferrocenylimidazolium salt as an efficient mediator for dye-sensitized solar cell. Chem. Lett. 43, 1134–1136 (2014).
Kato, R ., Kato, F ., Oyaizu, K . & Nishide, H . Redox-active hydroxy-TEMPO radical immobilized in Nafion layer for an aqueous electrolyte-based and dye-sensitized solar cell. Chem. Lett. 43, 480–482 (2014).
Oyaizu, K ., Hayo, N ., Sasada, Y ., Kato, F . & Nishide, H . Enhanced bimolecular exchange reaction through programmed coordination of a five-coordinate oxovanadium complex for efficient redox mediation in dye-sensitized solar cells. Dalton Trans. 42, 16090–16095 (2013).
Kato, F ., Hayashi, N ., Murakami, T ., Okumura, C ., Oyaizu, K . & Nishide, H . Nitroxide radicals for highly efficient redox mediation in dye-sensitized solar cells. Chem. Lett. 39, 464–465 (2010).
Murakami, T ., Kato, F ., Oyaizu, K . & Nishide, H . Porphyrin-dye sensitized solar cell utilizing nitroxide radical mediator. J. Photopolym. Sci. Technol. 23, 353–355 (2010).
Guo, W ., Su, J ., Li, Y. H ., Wan, L. J . & Guo, Y. G . Nitroxide radical polymer/graphene nanocomposite as an improved cathode material for rechargeable lithium batteries. Electrochim. Acta 72, 81–86 (2012).
Kim, Y ., Jo, C ., Lee, J ., Lee, C. W . & Yoon, S . An ordered nanocomposite of organic radical polymer and mesocellular carbon foam as cathode material in lithium ion batteries. J. Mater. Chem. 22, 1453–1458 (2012).
Oyaizu, K ., Hatemata, A ., Choi, W . & Nishide, H . Redox-active polyimide/carbon nanocomposite electrodes for reversible charge storage at negative potentials: expanding the functional horizon of polyimides. J. Mater. Chem. 20, 5404–5410 (2010).
Oyaizu, K ., Choi, W . & Nishide, H . Functionalization of poly(4-chloromethylstyrene) with anthraquinone pendants for organic anode-active materials. Polym. Adv. Technol. 22, 1242–1247 (2011).
Choi, W ., Ohtani, S ., Oyaizu, K ., Nishide, H . & Geckeler, K. E . Radical polymer-wrapped SWNTs at a molecular level: high-rate redox mediation through a percolation network for a transparent charge-storage material. Adv. Mater. 23, 4440–4443 (2011).
Choi, W ., Endo, S ., Oyaizu, K ., Nishide, H . & Geckeler, K. E . Robust and efficient charge storage by uniform grafting of TEMPO radical polymer around multi-walled carbon nanotubes. J. Mater. Chem. A 1, 2999–3003 (2013).
Lin, C. H ., Chou, W. J . & Lee, J. T . Three-dimensionally ordered macroporous nitroxide polymer brush electrodes prepared by surface-initiated atom transfer polymerization for organic radical batteries. Macromol. Rapid Commun. 33, 107–113 (2013).
Lin, C. H ., Chau, C. M . & Lee, J. T . Synthesis and characterization of polythiophene grafted with a nitroxide radical polymer via atom transfer radical polymerization. Polym. Chem. 3, 1467–1474 (2012).
Hung, M. K ., Wang, Y. H ., Lin, C. H ., Lin, H. C . & Lee, J. T . Synthesis and electrochemical behaviour of nitroxide polymer brush thin-film electrodes for organic radical batteries. J. Mater. Chem. 22, 1570–1577 (2012).
Ibe, T ., Kaiho, S ., Oyaizu, K . & Nishide, H . Electronic communication in the formation of a quartet molecule 2,6,10-tris [bis(p-methoxyphenyl)aminium]triphenylene. Chem. Lett. 39, 356–357 (2010).
Nishide, H ., Doi, R ., Oyaizu, K . & Tsuchida, E . Electrochemical and ferromagnetic couplings in 4,4’,4’’-(1,3,5-benzenetryl)tris(phenoxyl) radical formation. J. Org. Chem. 66, 1680–1685 (2001).
Oyaizu, K ., Yamamoto, K ., Ishii, Y . & Tsuchida, E . Synthesis and characterization of nickel dithiocarbamate complexes bearing ferrocenyl subunits. Chem. Eur. J. 5, 3193–3201 (1999).
Yoshihara, S ., Isozumi, H ., Kasai, M ., Yonehara, H ., Ando, Y ., Oyaizu, K . & Nishide, H . Improving charge/discharge properties of radical polymer electrodes influenced strongly by current collector/carbon fiber interface. J. Phys. Chem. B 114, 8335–8340 (2010).
Yoshihara, S ., Katsuta, H ., Isozumi, H ., Kasai, M ., Oyaizu, K . & Nishide, H . Designing current collector/composite electrode interfacial structure of organic radical battery. J. Power Sources 196, 7806–7811 (2011).
Kumar, A ., Jang, S. -Y ., Padilla, J ., Otero, T. F . & Sotzing, G. A . Photopatterned electrochromic conjugated polymer films via precursor approach. Polymer 49, 3686–3692 (2008).
Kang, H. A ., Bronstein, H. E . & Swager, T. M . Conductive block copolymers integrated into polynorbornene-derived scaffolds. Macromolecules 41, 5540–5547 (2008).
Yan, J ., Ye, Q ., Han, X . & Zhou, F . Step-by-step build-up of ordered p–n heterojunctions at nanoscale for efficient light harvesting. RSC Advances 3, 166–171 (2013).
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (No. 25107733) on the Innovative Areas, ‘Fusion Materials: Creative Development of Materials and Exploration of Their Function through Molecular Control’ from MEXT, Japan. This work was partially supported by Grants-in-Aid for Scientific Research (Nos. 24225003, 25288056 and 26620108) from MEXT, Japan. We thank Dr Takeo Suga for helpful discussion on the polymer synthesis.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Oyaizu, K., Tatsuhira, H. & Nishide, H. Facile charge transport and storage by a TEMPO-populated redox mediating polymer integrated with polyaniline as electrical conducting path. Polym J 47, 212–219 (2015). https://doi.org/10.1038/pj.2014.124
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2014.124
This article is cited by
-
Reversible and high-density energy storage with polymers populated with bistable redox sites
Polymer Journal (2023)
