
Abstract
In this study, the use of triblock (A–B–A) oligomers of ɛ-caprolactone (ɛ-CL) (A) and PEG400 (B) as stabilizers (SB) for the copolymerization of L-lactide (LLA) and ɛ-CL in supercritical carbon dioxide (scCO2) was investigated. To determine the effect of CO2-philic and polymer-philic segments on copolymerization, oligomers with three different average molecular weights (Mw=2000–6000 Da) were synthesized by changing the PEG400/ɛ-CL ratio. Copolymerizations were confirmed by 1H-nuclear magnetic resonance (NMR), 13C-NMR and differential scanning calorimeter data. It was possible to copolymerize LLA and ɛ-CL in scCO2 without any SB; however, the polymerization yields and average molecular weights were low, and significant aggregate formations were detected. Recipes featuring only 5% SB were successfully applied to reach high polymerization yields of ∼85% and polymers with average molecular weights greater than 20 kDa. When the polymer-philic segment of the SB increased, both the yield and molecular weight of the copolymer also increased significantly, resulting in white powdery products.
Similar content being viewed by others
Introduction
The disadvantages of traditional solvents, such as chloroform and toluene, have compelled scientists to introduce new solvents in polymerization protocols. In the past two decades, supercritical fluids have emerged as attractive alternative solvents for polymer synthesis and processing.1 Supercritical carbon dioxide (scCO2) is the most preferred solvent because of its remarkable properties, such as an easily accessible critical temperature (Tc=31.1 °C) and pressure (Pc=73.8 bar) values, no toxic solvent residues, ease of product recovery and being non-flammable, inert and inexpensive. Under supercritical conditions, carbon dioxide exhibits gas-like diffusivity and liquid-like density. These unique properties can be tuned easily by changing the temperature and pressure.2 There are a number of studies on the synthesis, processing, modification and purification of synthetic and natural polymers in scCO2.3, 4, 5, 6, 7, 8
Polymers obtained at the end of polymerization have low solubilities in scCO2. This issue has brought about many heterogeneous polymerization processes, such as precipitation, emulsion, suspension and dispersion polymerizations.8 One of the major areas of study in the heterogeneous polymerization in scCO2 is radical polymerization.9, 10 Different types of heterogeneous radical polymerizations such as nitroxide-mediated polymerization,11, 12, 13 atom transfer radical polymerization14, 15, 16 and reversible addition fragmentation chain transfer17, 18 polymerization have been investigated in scCO2.
The ring-opening homo- and copolymerization of L-lactide (LLA), ɛ-caprolactone (ɛ-CL) and glycolide in scCO2 has attracted much attention throughout the past decade. All of these linear aliphatic polyesters are biodegradable and widely used in a variety of medical applications, such as controlled drug release, absorbable sutures and scaffolds.8, 19 There are many routes for the polymerization of these monomers. The ring-opening polymerization of these lactones is the most popular polymerization protocol.20, 21, 22, 23 One of the main (target) issues is to reach high molecular weights and polymerization yields without any aggregation in the end product, which could be achieved by using proper stabilizers (SBs) and initiator systems. Ganapathy et al.8 reported the use of a poly(dimethyl siloxane) (PDMS)-based structure as a SB in the polymerization of LLA. Although PDMS-b-poly(methacrylic acid) and PDMS-b-poly(acrylic acid) formed discrete (PLLA) microparticles, acetylated PDMS-based copolymers could not stabilize the polymerization and produced hard agglomerates. Stassin and Jerome24 reported the homopolymerization of LLA and copolymerization with ɛ-CL in the presence of dibutyltin dimethoxide as an initiator. Random copolymers were synthesized, and the reactivity of monomers in scCO2 was compared with that of monomers in toluene. Bergeot et al.25 described the polymerization of ɛ-CL in scCO2 with different metal alkoxides as initiators. They demonstrated the effect of different types of metal alkoxides on the reactivity of the monomer. Stassin et al.26, 27, 28 also reported the synthesis of poly(ɛ-caprolactone) (PCL) in scCO2 using different metal initiators. They also determined the effect of temperature and pressure on polymerization. The emulsion and precipitation syntheses of poly(D,L-lactide-co-glycolide) using a CO2 soluble SB, poly(perfluoroocytyl), were reported by Hile and Pishko.29, 30 Bratton et al.31, 32 used blocks of PCL with KrytoxFSL and PCL-perfluoropolyether-PCL as SBs in PLLA synthesis in scCO2. A series of fluorinated diblock and triblock copolymers of PCL and poly(heptadecafluorodecylacrylate) were synthesized as SBs by the ring-opening polymerization of ɛ-CL and atom transfer radical polymerization of the acrylate by Grignard et al.33 They used these copolymers in the dispersion ring-opening polymerization of ɛ-CL in scCO2. They determined the effect of the composition and structure of the polymeric SBs on the stabilization of PCL microspheres. Mazarro et al.34 reported the ring-opening copolymerization of D,L-lactide and glycolide in scCO2 using zinc(II) 2-ethylhexanoate (Zn(Oct)2) as a catalyst instead of stannous octoate (Sn(Oct)2). Experiments were performed at various reaction times, pressures and stirring rates, and the results obtained were similar to those for (Sn(Oct)2). Apart from carbon dioxide, chlorodifluoromethane (R22) was also used as a supercritical fluid in the polymerization of ɛ-CL and LLA. Prabowo et al.35 investigated the copolymerization of ɛ-CL and LLA initiated by Sn(Oct)2 in supercritical R22 at various reaction times, temperatures and amounts of monomer and catalyst under a pressure of 250 bar. A reaction time of 10 h and a temperature of 130 °C were determined as the optimum conditions for obtaining an 86-kDa copolymer. Sarbu et al.36 used propylene and CO2 to synthesize a series of poly(ether-carbonate) copolymers. They demonstrated that these copolymers could be readily dissolved in CO2 at low pressures. Lee et al.37 synthesized a 50:50 random copolymer of reversible addition fragmentation chain transfer-terminated vinyl acetate and vinyl butyrate. They reported high solubility values for these copolymers, which were successfully used to perform dispersion polymerization in scCO2.
To the best of our knowledge, these studies used only nonfluorine- and nonsiloxane-based SBs. There are many limitations to using fluorine- and siloxane-based SBs, such as their high cost and the potential toxic side effects of the nondegradable residual materials in medical applications. Our groups are interested in using these biodegradable polyesters to prepare tissue engineering scaffolds.38, 39, 40 Therefore, we have sought SBs that are degradable and safe for medical use to polymerize LLA and ɛ-CL in scCO2. In this study, we synthesized triblock copolymers of PEG, which is one of the safest water-soluble polymers used in various medical applications, and ɛ-CL, which is one of the co-monomers used in the final polymer product. We expected that the polymer-philic segment would interact with the polymer and anchor the growing particles, whereas the solvent-philic (CO2-philic) segment would prevent the aggregation of the growing polymer particles.31, 32 In our previous study, we investigated the use of triblock oligomers of ɛ-CL and PEG400 as biodegradable/biocompatible SBs in PLLA synthesis in scCO2. A significant improvement was achieved in the yield, molecular weight and morphology of the resulting polymer.41 In this article, we report the copolymerization of LLA and ɛ-CL in the presence of triblock oligomers as SBs.
Experimental procedure
Materials
L-lactide (Aldrich, Steinheim, Germany, 98%) was recrystallized in toluene and dried under reduced pressure to remove solvent overnight and stored at 4 °C. Polyethylene glycol with a molecular weight of 400 (PEG400, Fluka, Steinheim, Germany), ɛ-caprolactone (ɛ-CL, >99%, Fluka), tin (II) ethyl hexanoate (Sn(Oct)2, 95%, Sigma, Steinheim, Germany), n-butanol (Aldrich), methanol (Merck, Darmstadt, Germany), toluene (Merck), triphenylphosphine (Sigma-Aldrich, Steinheim, Germany) and tetrahydrofuran (Merck) were used as received. Choloroform (Fluka, HPLC grade) was filtered and then sonicated for 15 min before use. Carbon dioxide with high purity (>99.5) was obtained from a local company (OKSAN, Ankara, Turkey).
Synthesis of triblock stabilizer
Sn(Oct)2 was used as a catalyst in the bulk polymerization of triblock (A–B–A) oligomers of PCL-PEG400-PCL. The polymerization protocol and solubility tests performed are detailed in our previous publication.38 To determine the effect of the lengths of hydrophilic/hydrophobic segment on copolymerizaton in scCO2, the bulk polymerization of the oligomers was conducted by using three different ratios of hydrophilic oligomer (PEG 400) to hydrophobic co-monomer (ɛ-CL) in the initial media: 1/10, 1/25 and 1/50 (mol/mol). For simplification, these SBs are denoted as SB1, SB2 and SB3, respectively, in the text below.
Solubility tests on the triblock copolymers were performed in a 100-ml high-pressure stainless reactor (THAR R100, Thar Process, Inc., Pittsburgh, PA, USA) with two sapphire windows on both sides by cloud point experiments using 5% w/w (0.1875 g, equivalent to a loading of 5% w/w (SB/monomer)) for all of the SBs in a temperature range of 60–100 °C.
Synthesis of poly(LLA/ɛ-CL) in scCO2
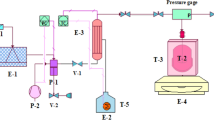
A 100-ml high-pressure stainless reactor system (THAR R100) with two sapphire windows on both sides was used for copolymerization in scCO2. This system consists of a high-pressure pump (THAR, P series) used to pressurize CO2, an automated back-pressure regulator (THAR, ABPR200), a temperature controller (THAR, CN6), a mechanical mixer (Minarik Corporation, South Beloit, IL, USA; MM23101C) with four blades and a computer to control the entire system. Sn(Oct)2 and n-butanol were used as the initiator and catalyst, respectively, for all of the copolymerizations. The proper amount of triphenyl phosphine was added to the copolymerizaton medium to eliminate/reduce transesterification side reactions.42
We were very careful to dry the whole system and remove any traces of water. The reactor was purged with carbon dioxide at room temperature and low pressure (65 bar) for 15 min. Afterwards, the reactor was loaded with monomer and other ingredients. The ɛ-CL to LLA ratio was selected to be 80/20 w/w by considering how these copolymers would ultimately be used. In a typical experiment, the reactor was charged with 3.75 g total monomer (3.75% w/v to CO2), the desired amount of the triblock copolymer SB (0–20% w/w to total monomer), 0.0375 g of triphenyl phosphine (1% w/w to total monomer), 15 μl n-butanol and 30 μl Sn(Oct)2. The reactor was closed and purged with CO2 (10 g min−1) for 15 min at 40 °C and 65 bar to remove the air; heated to the polymerization temperature of 80 °C; pressurized to the polymerization pressure of 100 bar; and then mixed at a constant stirring rate of 700 r.p.m. for 48 h. At the end of the polymerization, the temperature control was switched off and the reactor was cooled to ambient temperature. Then, the mixer was stopped. Dragged polymerization products were collected to cold methanol via venting. Because the degassing procedure may affect the morphology of copolymer products in scCO2, careful attention was paid in applying the same degassing procedure to all of the runs in the same manner. We were very careful to collect the products left in the reactor system. To remove the polymerization products along the wall of the reactor, the reactor was washed with tetrahydrofuran, pressurized to 65 bar at room temperature, and vented using cold methanol. This procedure was repeated until it was assured that no residue remained in the reactor system. The product in methanol was filtered and dried under reduced pressure at room temperature. It is important to note that to determine the reproducibility of the copolymerizations, some runs were repeated a minimum of three times under exactly the same conditions. The results are given as the average and standard deviation of the results of these repeated experiments (see Table 1).
Characterization
The yield values were determined gravimetrically. In the copolymerization experiment, because triblock SB remained in the product, the yields were calculated by subtracting the amount of triblock copolymer SB initially loaded. The molecular weights of the SBs and copolymerization products were determined by gel permeation chromatography. Polystyrene samples were used, with the Mark–Houwink equation as a correction factor, as standards to obtain a calibration curve. Gel permeation chromatography analysis was conducted with a refractive index detector (Shimadzu LC 10AD, Shimadzu, Kyoto, Japan) and chloroform as the solvent and eluent at a flow rate of 1 ml min−1. 1H-nuclear magnetic resonance (NMR) and 13C-NMR spectra were recorded at 25 °C in chloroform on a Bruker DPX-400 (Bruker, Billerica, MA, USA). Differential scanning calorimeter (DSC) analysis was performed with a Shimadzu DSC 60 (Shimadzu). In a typical DSC analysis, the sample was first cooled to −80 °C and then heated to 200 °C. To determine the morphology of the copolymerization products, samples were analyzed by scanning electron microscopy (HITACHI TM1000, Hitachi, Schaumburg, IL, USA) under vacuum.
Results and Discussion
Three different triblock SBs (SB1, SB2 and SB3) were synthesized using different amounts of PEG and ɛ-CL in the recipes, in which the PEG/ɛ-CL ratios were 1/10, 1/25 and 1/50, respectively. The properties of these SBs are provided in our previous publication in which the SBs were used in the homopolymerization of L-lactide in scCO2 media. These SBs, the average molecular weights (Mn) and heterogeneity indices (HI, in parentheses) of which were calculated from the gel permeation chromatography data to be 1380 (1.62), 2140 (1.80) and 2670 (2.26), respectively, were also used in the copolymerization of L-lactide with ɛ-CL.41, 42 It is important to note that under the polymerization conditions (80 °C, 100 bar), all of the SBs were partially soluble (they form cloudy mixtures in the reactor) and their solubilities were inversely proportional to their molecular weight.41 Because of their triblock structure, the SBs are expected to adopt a U shape in scCO2.
The chemical structure of both the SB and poly(LLA/CL) with corresponding peaks and also the representative 1H-NMR spectra of the copolymerization products, which indicates copolymer formation, are shown in Figure 1. The characteristic peaks of the SB are also apparent in these spectra at around 3.5–4.3 p.p.m. This means that SB remained in the product. This is expected because the ɛ-CL part is very compatible with the copolymer carrying ɛ-CL segments; therefore, it would be difficult to remove. Note that both ɛ-CL and PEG are highly biocompatible and that ɛ-CL is degradable; thus, there will be no limitations for the use of this product in medical applications.43 Thus far, we have not attempted to remove the SB, which is the main advantage of this materials and the point to be stressed in this article.

Representative 1H-nuclear magnetic resonance (NMR) spectra of the copolymerization products: (a) with stabilizer (5% SB2) and (b) without stabilizer. Chemical structure of poly(LLA/ɛ-CL) and triblock stabilizers and corresponding peaks for 1H-NMR peaks are shown above the spectra. ɛ-CL, ɛ-caprolactone; LLA, L-lactide.
Table 1 summarizes the results obtained in this study. It is notable that even without using any SB, very high polymerization yields and molecular weights were achieved. However, unfortunately, the products were in aggregated form. This was one of the most important points we considered when planning this study and was also mentioned in similar studies reported in the related literature. It should be noted that because the triblock SBs (SBs) remain in the product, we have subtracted the mass of SB from the product mass when calculating the yield values (Table 1). In almost all of the cases in which we used triblock oligomers as SBs, the polymerization yield increased noticeably, which was one of the aims of this study. In the related literature, similar results were obtained. Ganapathy et al.8 investigated the polymerization of LLA with a PDMS-based SB in scCO2. Without the SB, they reported that an aggregated morphology for PLLA was produced with 78% yield at the end of the polymerization process (80 °C, 207 bar, 47 h). In the presence of the SB (10% wt), the polymerization resulted in a fine powder with an 85% yield and a number average molecular weight (Mn) value of 12 kDa under the same polymerization conditions. Bratton et al.31 reported the use of a block of PCL–perfluoropolyether–PCL as a SB in PLLA synthesis. In the absence of the SB, the product adopted an aggregated form that was obtained at an 85% yield with a Mn value of 12 kDa (polymerization conditions: 80 °C, 241 bar, 48 h and 300 r.p.m.). A remarkable improvement was reported both in yield and molecular weight values for polymerizations featuring 10% SB.
The type of SB used here produced significant effects on the average molecular weight and polymerization yield. It should be noted that SB1 has the lowest ɛ-CL content, whereas SB3 has the highest. The co-monomer during copolymerization is also ɛ-CL. It seems that SBs with higher ɛ-CL contents interact with the copolymer being formed and better stabilize the system, which in turn results in copolymers with higher yields and much higher molecular weights. In the case of SB1, because the ɛ-CL content is low, stabilization was not very good, and the average molecular weight was quite low compared with that of the other samples. The product formed a gel because of the low molecular weight of the copolymer. Both the yield and, especially, the molecular weight increased with the increase in the ɛ-CL content (for SB2 and SB3), and products in white powder form (no aggregation) were obtained.
As shown in Table 1, increasing amounts of the SB (from 5 to 10%) first increased both the polymer yield and molecular weight very significantly; however, a further increase in the SB content during copolymerization produced the opposite effect. It seems that there is a certain (optimum) SB concentration for the copolymerization of LLA with ɛ-CL in scCO2 using PEG/ɛ-CL SBs. It is likely that this polymerization is heterogeneous to different extents, depending on the initial recipe and reaction time; however, this behavior cannot be easily explained, and more detailed experimental analysis is required.
We repeated experiments at two different temperatures (80 and 100 °C) for polymerizations with and without stabilizer (SB2; see Table 1). Increasing the temperature had a positive effect when ‘no stabilizer’ was used. Meanwhile, the effects ‘with stabilizer’ were negative: the average molecular weight decreased and the molecular weight distribution increased, both significantly.
Table 1 also shows heterogeneity indices (HI), which were determined from the gel permeation chromatography data. Notably, the lowest HI value was observed in the case where there was no SB in the recipe. Using a SB increased the HI values (a wider molecular weight distribution). It is noteworthy that the oligomeric SBs remain in the polymer product taken from the reactor, which widens the distribution as expected. Also, a second small peak is observed corresponding to the SBs, as shown in Figure 2.

Representative gel permeation choromatograms of copolymerization products obtained without using stabilizer (a) and with three different amounts (5% (b), 10% (c) and 20% (d)) of SB2 at 80 °C.
Representative DSC thermograms of the copolymerization products in scCO2 for different types of SBs are shown in Figure 3. As indicated, all of the thermograms resemble each other, and two peaks around 30 and 130 °C correspond to the glass transition and melting points of the copolymer, respectively. The presence of the glass transition point around 30 °C may be attributed to block PLLA segments. Owing to low loading content of SBs, no thermal peaks for the SB were observed. Because the HI values of the copolymerization products were quite high, the corresponding melting point ranges were broad.

Representative differential scanning calorimeter thermograms of copolymerization products obtained without using stabilizer (a) and with three different stabilizers (SB1 (b), SB2 (c) and SB3 (d)). The stabilizer concentration in the recipe and the polymerization temperature were 5% and 80 °C, respectively.
To gain more information about the copolymer structure, the 13C-NMR spectra of the selected copolymerization products were obtained. The carbonyl (−C=O) region (168–175 p.p.m.) of the two copolymer products obtained with (SB2 5%) and without (no SB) any SB are shown in Figure 4. Detailed information regarding the product synthesized with SB is also given in Table 2. The underlined groups indicate the spectrum peaks that correspond to the carbonyl groups. From the sequences of monomers in the product, it is clear that LLA and ɛ-CL were polymerized in random and block segments. DSC analysis also indicates the presence of block PLLA segments. Considering the 13C-NMR and DSC analysis, we have concluded that the copolymer products synthesized assumed a block and random structure.

Representative 13C-nuclear magnetic resonance spectra of copolymerization products obtained without using stabilizer (a) and with stabilizers (b) (SB2). The stabilizer concentration in the recipe and the polymerization temperature were 5% and 80 °C, respectively.
Using the data provided by 13C-NMR analysis, the weight percentages of LLA and ɛ-CL in copolymer structure were calculated from the average length of LLA (LLA) and ɛ-CL (LCL) (Table 2) using equations (1 and 2) given below. Here, ‘I’ is the respective peak intensity in the 13C-NMR spectrum.44 By calculating these values and multiplying the ratio of the molecular weight of the monomers, a 33/67 weight ratio of ɛ-CL/LLA was determined for the product synthesized using SB2 (5%). Note that the initial ratio of ɛ-CL/LLA was 20/80. It is clear that ɛ-CL was incorporated into the copolymer structure to a greater extent than LLA because ɛ-CL is more soluble than LLA in scCO2. Similar results were obtained for all runs of this study.


To determine the effect of the SB loading on the particle size, scanning electron microscopy analysis of the copolymer products was performed. Representative scanning electron microscopy images of copolymer products obtained without SB and with 5 and 20% SBs are given in Figures 5a–c, respectively. In the absence of the SB, the product formed an aggregated solid phase (Figure 5a). When the SB was used, the copolymer product formed random porous shapes (Figures 5b and c). This may be attributed to the partial solubility of the SBs in scCO2. The SB molecules were at the interface between the scCO2 and the polymer, stabilizing the polymer particles during polymerization.

Representative scanning electron microscopy images of copolymer products: (a) without any stabilizer (SB); (b) with SB2 (5%) and (c) with SB2 (20%) at 80 °C.
Conclusions
Copolymers of LLA and ɛ-CL in the form of random porous shapes were successfully synthesized by ring-opening polymerization in scCO2 (80 °C, 100 bar) in the presence of triblock (A–B–A–) copolymers, with ɛ-CL and PEG as SBs. In our previous study, the proper chemical structure and partial solubility of the SBs were confirmed. The use of even moderate (5%) SB contents prevented aggregation and led to random copolymer products (SB and copolymer mixture). By considering the polymerization yield, the average molecular weight and heterogeneity indices, it can be concluded that the most suitable recipe contains SB2 with an initial concentration of 10%. In this case, these three parameters correspond to values of 86.5%, 28.1 kDa and 1.97, respectively.
References
Betts, D. E., McClain, J. B. & DeSimone, J. M. The importance of surfactants for polymerizations in carbon dioxide, high pressure chemical engineering. Proc. Tech. Proceed. 12, 23–30 (1996).
Kendall, J. L., Canelas, D. A., Young, J. L. & DeSimone, J. M. Polymerizations in supercritical carbon dioxide. Chem. Rev. 99, 543–563 (1999).
Kıran, E. Polymerization and polymer modification reactions in near and supercritical fluids, supercritical fluids: fundamentals and applications. NATO Sci. Ser. E: Appl. Ser. 366, 253–276 (1998).
Tai, H., Popov, V. K., Shakeskeff, K. M. & Howdle, S. M. Putting the fizz into chemistry: applications of supercritical carbon dioxide in tissue engineering, drug delivery and synthesis of novel block copolymers. Bioche. Soc. Trans. 35, 516–521 (2007).
Tsivintzelis, I., Pavlidou, E. & Panayiotou, C. Biodegradable polymer foams prepared with supercritical co2–ethanol mixtures as blowing agents. J. Sup. Flu. 42, 265–272 (2007).
Teng, X., Ren, J. & Gu, S. Preparation and characterization of porous PDLLA/HA composite foams by supercritical carbon dioxide technology. J. Biomed. Mat. Res. Part B Appl. Biomat. 81, 185–193 (2006).
Tai, H., Mather, M. L., Howard, D., Wang, W., White, L. J., Crowe, J. A., Morgan, S. P., Chandra, A., Williams, D. J., Howdle, S. M. & Shakeskeff, K. M. Control of pore size and structure of tissue engineering scaffolds produced by supercritical fluid processing. Eur. Cells Mat. 14, 64–77 (2007).
Ganapathy, H. S., Hwang, H. S., Jeong, Y. T., Lee, W. K. & Lim, K. T. Ring-opening polymerization of L-Lactide in supercritical carbon dioxide using PDMS based stabilizers. Eur. Polym. J. 43, 119–126 (2007).
Zetterlund, PerB., Aldabbagh, F. & Okubo, M. Controlled/living heterogeneous radical polymerization in supercritical carbon dioxide. J. Polym. Sci. Part A Polym. Chem. 47, 3711–3728 (2009).
Thurecht, K. J. & Howdle, S. M. Controlled dispersion polymerization in supercritical carbon dioxide. Aust. J. Chem. 62, 3711–3956 (2009).
McHale, R., Aldabbagh, F., Zetterlund, P. B. & Okubo, M. Nitroxide-mediated radical precipitation polymerization of styrene in supercritical carbon dioxide. M. Macromol. Chem. Phys. 208, 1813–1822 (2007).
McHale, R., Aldabbagh, F., Zetterlund, P. B., Minami, H. & Okubo, M. Nitroxide-mediated radical dispersion polymerization of styrene in supercritical carbon dioxide using a poly(dimethylsiloxane-b-methylmethacrylate) stabilizer. Macromolecules 39, 6853–6860 (2006).
Aldabbagh, F., Zetterlund, P. B. & Okubo, M. Improved control in nitroxide-mediated radical polymerization using supercritical carbon dioxide. Macromolecules 41, 2732–2734 (2008).
Xia, J., Johnson, T. S., Gaynor, G., Matyjaszewski, K. & DeSimone, J. Atom transfer radical polymerization in supercritical carbon dioxide. Macromolecules 32, 4802–4805 (1999).
Grignard, B., Jerome, C., Calberg, C., Jerome, R., Wang, W., Howdle, S. M. & Detrembleur, C. Dispersion atom transfer radical polymerization of vinyl monomersin supercritical carbon dioxide. Macromolecules 41, 8575–8583 (2008).
Grignard, B., Jerome, C., Calberg, C., Jerome, R., Wang, W., Howdle, S. M. & Detrembleur, C. Copper bromide complexed by fluorinated macroligands: towards microspheres by ATRP of vinyl monomers in scCO2 . Chem. Commun. 3, 314–316 (2008).
Gregory, A. M., Thurecht, K. J. & Howdle, S. M. Controlled dispersion polymerization of methyl methacrylate in supercritical carbon dioxide via RAFT. Macromolecules 41, 1215–1222 (2008).
Thurecht, K. J., Gregory, A. M., Wang, W. & Howdle, S. M. ‘Living’ polymer beads in supercritical CO2 . Macromolecules 40, 2965–2967 (2007).
Domb, A. J., Kieman, N., Sheskin, T., Benzolila, A., Sloger, J. & Teomim, D. in Polymeric Biomaterials (ed. Dumitriu, S.) Part I Ch. 4, 91–122 (Marcel Dekker, Inc, 1993).
Thurecht, K. J., Heise, A., deGeus, M., Villarroya, S., Zhou, J., Wyatt, M. F. & Howdle, S. M. Kinetics of enzymatic ring-opening polymerization of 〈epsilon〉-caprolactone in supercritical carbon dioxide. Macromolecules 39, 7967–7972 (2006).
Zhou, J., Villarroya, S., Wang, W., Wyatt, M. F., Duxbury, C. J., Thurecht, K. J. & Howdle, S. M. One-step chemoenzymatic synthesis of poly(〈epsilon〉-caprolactone-block-methyl methacrylate) in supercritical CO2 . Macromolecules 39, 5352–5358 (2006).
Zhou, J., Villarroya, S., Wang, W., Wyatt, M. F., Duxbury, C. J., Thurecht, K. J. & Howdle, S. M. One-step chemoenzymatic synthesis of poly(ɛ-caprolactone-block- methyl methacrylate) in supercritical CO2 . Macromolecules 40, 2276 (2007).
Villarroya, S., Zhou, J., Duxbury, C. J., Heise, A. & Howdle, S. M. Synthesis of semifluorinated block copolymers containing poly(〈epsilon〉-caprolactone) by the combination of ATRP and enzymatic ROP in scCO2 . Macromolecules 39, 633–640 (2006).
Stassin, F. & Jerome, R. Polymerization of (L,L)-lactide and copolymerization with B-caprolactone Initiated by dibutyltin dimethoxide in supercritical carbon dioxide. J. Polym. Sci. Part A Polym. Chem. 43, 2777–2789 (2005).
Bergeot, V., Tassaing, T., Besnard, M., Cansel, F. & Mingotaud, A. F. Anionic ring-opening polymerization of ɛ-caprolactone in supercritical carbon dioxide: parameters influencing the reactivity. J. Sup. Flu. 28, 249–261 (2004).
Stassin, F., Halleux, O. & Jerome, R. Ring-opening polymerization of epsilon-caprolactone in supercritical carbon dioxide. Macromolecules 34, 775–781 (2001).
Stassin, F. & Jerome, R. Effect of pressure and temperature upon tin alkoxide-promoted ring-opening polymerization of ɛ-caprolactone in supercritical carbon dioxide. Chem. Commun. 2, 232–233 (2003).
Stassin, F. & Jerome, R. Contribution of supercritical CO2 to the preparation of aliphatic polyesters and materials thereof. Macromol. Symp. 217, 135–146 (2004).
Hile, D. D. & Pishko, M. V. Ring-opening precipitation polymerization of poly(D,L-lactide-co-glycolide) in supercritical carbon dioxide. Macromol. Rapid Commun. 20, 511–514 (1999).
Hile, D. D. & Pishko, M. V. Emulsion copolymerization of D,L-lactide and glycolide in supercritical carbon ddioxide. J. Polym. Sci. Part A Polym. Chem. 39, 562–570 (2001).
Bratton, D., Brown, M. & Howdle, S. M. Suspension polymerization of L-lactide in supercritical carbon dioxide in the presence of a triblock copolymer stabilizer. Macromolecules 36, 5908–5911 (2003).
Bratton, D., Brown, M. & Howdle, S. M. Novel fluorinated stabilizers for ring-opening polymerization in supercritical carbon dioxide. J. Polym. Sci. Part A Polym. Chem. 43, 6573–6585 (2005).
Grignard, B., Stassin, F., Calberg, C., Jerome, R. & Jerome, C. Synthesis of biodegradable poly-ɛ-caprolactone microspheres by dispersion ring-opening polymerization in supercritical carbon dioxide. Biomacromolecules 9, 3141–3149 (2008).
Mazarro, R., de Lucas, A., Gracia, I. & Rodriguez, J. F. Copolymerization of D,L-lactide and glycolide in supercritical carbon dioxide with zinc octoate as catalyst. J. Biomed. Mat. Res. Part B Appl. Biomat. 85B, 196–203 (2008).
Prabowo, B., Choi, D. H. & Kim, S. H. Copolymerization of L-lactide and ɛ-caprolactone in supercritical fluid. Macromol. Res. 17, 575–579 (2009).
Sarbu, T., Styranec, T. & Beckman, E. J. Non-porous polymers with very high solubility in supercritical CO2 down to low pressures. Nature 405, 165–168 (2000).
Lee, H., Terry, E., Zong, M., Arrowsmith, N., Perrier, S., Thurecht, K J. & Howdle, S. M. Successful dispersion polymerization in supercritical CO2 using polyvinylalkylate hydrocarbon surfactants synthesized and anchored via RAFT. J. Am. Chem. Soc. 130, 12242–12243 (2008).
Piskin, E., Isoglu, I. A., Bolgen, N., Vargel, I., Griffiths, S., Cavusoglu, T., Korkusuz, P., Guzel, E. & Cartmell, S. In vivo performance of simvastatin-loaded electrospun spiral-wound polycaprolactone scaffolds in reconstruction of cranial bone defects in the rat model. J. Biomed. Mater. Res. A 90A, 1137–1151 (2009).
Aydin, H. M., El Haj, A. J., Piskin, E. & Yang, Y. Improving pore interconnectivity in polymeric scaffolds for tissue engineering. J. Tissue Eng. Regen. Med. 3, 470–476 (2009).
Aydin, H. M., Yang, Y., Kohler, T., El Haj, A., Muller, R. & Piskin, E. Interaction of osteoblasts with macroporous scaffolds made of PLLA/PCL blends modified with collagen and hydroxyapatite. Adv. Eng. Mater. 11, B83–B88 (2009).
Yılmaz, M., Eğri, S., Yıldız, N., Çalımlı, A. & Pişkin, E. Dispersion polymerization of L-lactide in supercritical carbon dioxide. J. Polym. Res. doi: 10.1007/s10965-010-9497-y (in press).
Zhu, W., Xie, W., Tong, X. & Shen, Z. Amphiphilic biodegradable poly(CL-b-PEG-b-CL) triblock copolymers prepared by novel rare earth complex: synthesis and crystallization properties. Eu. Polym. J. 43, 3522–3530 (2007).
Zhou, S., Deng, X. & Yang, H. Biodegradable poly(ɛ-caprolactone)-poly(ethylene glycol) block copolymers: characterization and their use as drug carriers for a controlled delivery system. Biomaterials 24, 3563–3570 (2003).
Shen, Y., Zhu, K. J., Shen, Z. & Yao, K. Synthesis and characterization of highly random copolymer of ɛ-caprolactone and D,L-lactide using rare earth catalyst. J. Polym. Sci. Part A Polym. Chem. 34, 1799–1805 (1996).
Acknowledgements
This study was supported by the Turkish Scientific and Technological Research Council of Turkey (TUBITAK Projects No: 108M155 and 105T509), Turkish Ministry of Industry (San-Tez Project No: 00020.STV.2006-1) and EU-FP6-NoE:Expertissues. Erhan Pişkin was supported by the Turkish Academy of Sciences as a full member. The support of Biyomedtek and the Hacettepe University, Center for Bioengineering is also kindly acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Yılmaz, M., Eğri, S., Yıldız, N. et al. Ring-opening copolymerization of L-lactide and ɛ-caprolactone in supercritical carbon dioxide using triblock oligomers of caprolactone and PEG as stabilizers. Polym J 43, 785–791 (2011). https://doi.org/10.1038/pj.2011.70
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pj.2011.70
