
Abstract
We previously produced pigs with a latent oncogenic TP53 mutation. Humans with TP53 germline mutations are predisposed to a wide spectrum of early-onset cancers, predominantly breast, brain, adrenal gland cancer, soft tissue sarcomas and osteosarcomas. Loss of p53 function has been observed in >50% of human cancers. Here we demonstrate that porcine mesenchymal stem cells (MSCs) convert to a transformed phenotype after activation of latent oncogenic TP53R167H and KRASG12D, and overexpression of MYC promotes tumorigenesis. The process mimics key molecular aspects of human sarcomagenesis. Transformed porcine MSCs exhibit genomic instability, with complex karyotypes, and develop into sarcomas on transplantation into immune-deficient mice. In pigs, heterozygous knockout of TP53 was sufficient for spontaneous osteosarcoma development in older animals, whereas homozygous TP53 knockout resulted in multiple large osteosarcomas in 7–8-month-old animals. This is the first report that engineered mutation of an endogenous tumour-suppressor gene leads to invasive cancer in pigs. Unlike in Trp53 mutant mice, osteosarcoma developed in the long bones and skull, closely recapitulating the human disease. These animals thus promise a model for juvenile osteosarcoma, a relatively uncommon but devastating disease.
Similar content being viewed by others


Introduction
Animal models of human cancers are crucial for the development of urgently needed diagnostic and therapeutic techniques. Genetically modified mice are widely used, but their small size and short lifespan preclude some preclinical studies. It is, for example, difficult to scale down radiological, thermal or surgical treatment of tumours, or perform longitudinal studies of tumour progression and remission, or longer-term response to therapy.
Mouse and human cancer biology also differ. Murine cells are more easily transformed in vitro than human cells,1 and the set of genetic events required for tumorigenesis is different.2 Mouse models may therefore not always provide the best representation of human disease.
Pigs are increasingly important in biomedicine and offer valuable complementary resources for cancer research. The number of pigs genetically modified to replicate human diseases has increased dramatically.3 Valuable models such as cystic fibrosis and diabetes are established.4, 5 Work is also proceeding towards genetically defined porcine cancer models, such as inactivation of BRCA1 for breast cancer,6 mutation of TP537 and, as we have reported, knockout and conditional activation of mutant TP53,8 latent oncogenic KRAS mutation9 and truncating mutations of APC to model colorectal cancer.10 The relevance of pig cancer models depends on how closely they resemble human disease. Porcine cancer biology is a new field and many fundamental questions remain open. Not least, does replication of single or combined human oncogenic mutation(s) in a pig have an equivalent effect on cell transformation and tumorigenesis?
Sarcomas are a group of tumours originating from the mesenchyme. They fall into two categories: those with disease-specific chromosomal translocations, and those with complex unstable karyotypes. Sarcomas of the second type, for example, fibrosarcoma, liposarcoma, chondrosarcoma and osteosarcoma, often have TP53 and RB1 mutations.11, 12 Consistent with this, transformation of human mesenchymal stem cells (MSCs) requires disruption of the p53 and Rb tumour-suppressor pathways, stabilisation of MYC, activation of oncogenic RAS and telomere maintenance.13 We report that targeted mutation of endogenous porcine TP53 and KRAS in primary MSCs results in neoplastic transformation and tumorigenesis. Molecular analysis shows the process closely resembles human sarcomagenesis.
Osteosarcoma is a relatively rare solid tumour but the most common primary bone cancer. It predominantly affects young people and is highly malignant, requiring aggressive surgical resection and cytotoxic chemotherapy. Five-year survival for patients with metastatic osteosarcoma is only around 30%.14 We report that pigs with heterozygous and homozygous inactivation of TP53 consistently develop osteosarcomas, providing a new model of osteosarcoma at human scale to understand and treat this devastating disease.
Results
Oncogenic modification of porcine MSCs
Human sarcomas frequently originate from MSCs, often initiated by lesions affecting the p53 tumour-suppressor pathway. Other alterations, including oncogenic KRAS mutations or MYC amplification, may occur later during tumorigenesis. To show that similar molecular events lead to sarcomagenesis in a porcine model, we mutated the endogenous TP53 and KRAS genes in porcine MSCs.
The porcine TP53R167H mutation is orthologous to human TP53R175H. In a first round of gene targeting, we introduced a Cre-inducible latent mutant allele, TP53LSL-R167H, into porcine MSCs and derived one MSC cell clone homozygous for the mutant allele (TP53LSL-R167H/LSL-R167H), which in unrecombined form is a TP53 knockout.8 For brevity, this genotype is referred to as MSC-P. It was subjected to a second round of gene targeting to introduce a G to A substitution into KRAS codon 12 (G12D) and a floxed transcriptional terminator cassette (LSL) into the first intron, generating the Cre-inducible allele KRASLSL-G12D (Supplementary Figures S1a–d). The resulting genotype (TP53LSL-R167H/LSL-R167H, KRASLSL-G12D/+) is termed MSC-PK. One MSC-PK cell clone was transduced with Cre and subclones were isolated that had excised LSL cassettes from both mutant TP53 alleles and the mutated KRAS allele (Figure 1b), termed MSC-PKC.

Transformation of porcine MSCs. (a) Overview of stepwise transformation of genetically modified porcine MSCs. (b) Left: Cre-mediated excision of transcriptional termination cassette from the KRASLSL-G12D allele. Cell types as shown. PCR amplification products across the site of the LSL-BS cassette in KRAS intron 1. Predicted fragment sizes: wild-type KRAS 167 bp; non-recombined KRASLSL-G12D 1486 bp; Cre-excised KRASL-G12D 201 bp. Right: Cre-mediated excision of transcriptional termination cassette from both TP53LSL-R167H alleles. Cell types as shown. PCR amplification products across the site of LSL-NEO cassette in TP53 intron 1. Predicted fragment sizes: wild-type TP53 198 bp; non-recombined TP53LSL-R167H 1929 bp; Cre-excised TP53L-R167H 254 bp. (c) Ras activation assay and p53 western blotting analysis. Cre-recombined MSC-PKC and MSC-PKCM cells show increased levels of active GTP-bound Ras proteins (21 kDa) as well as abundant levels of mutant p53-R167H proteins (46 kDa). (d) EdU (5-ethynyl-2’-deoxyuridine) incorporation during S phase. Data are consistent with the enhanced proliferative capacity of genetically modified derivatives relative to wild-type MSCs. P-values: MSC-PK=0.0727; MSC-PKC=0.0042; MSC-PKCM=0.0019. (e) Upper row: Loss of contact inhibition. MSC-PKCM cells form multi-layered foci when cultured at higher densities. Scale bars indicate 400 μm. Lower row: Anchorage-independent growth in soft agar. Wild-type MSCs grow as single cells in semi-solid medium, whereas MSC-PK, MSC-PKC and MSC-PKCM cells form three-dimensional colonies. Scale bars indicate 400 μm. (f) Upper: H&E-stained section of paucicellular nodule formed by injected MSC-PKC cells. The nodule consists mainly of matrigel with fat islands, capillary sprouts and isolated cells with slightly irregular nuclei. Scale bar indicates 500 μm. The arrow indicates an area of higher cellularity. Lower: Mesenchymal cells with slightly irregular nuclei embedded in a myxoid stroma are evident at higher magnification. Scale bar indicates 100 μm. (g) H&E-stained sections of MSC-PKCM derived tumour. This tumour is mostly highly cellular, but a matrix-rich area is still present, for example, as indicated by an asterisk. In the cellular areas, large tumour cells with pleomorphic nuclei are evident. A chronic inflammatory reaction is also present. Scale bar indicates 200 μm.
The MYC gene is often amplified in human cancers15 and has a pivotal role in transformation of human primary cells.2, 16, 17 We added a porcine MYC expression vector to one MSC-PKC cell clone and derived a pool (MSC-PKCM) that showed an average 1.7-fold increase in MYC mRNA expression and a similar increase in MYC protein (Supplementary Figures S2a and b). These genetic modifications are summarised in Figure 1a.
Reverse transcriptase PCR (RT–PCR) and sequence analysis showed that MSC-PKC and MSC-PKCM cells expressed mutant p53-R167H, wild-type KRAS and mutant KRAS-G12D mRNAs and biochemical evidence demonstrated increased levels of activated GTP-bound Ras and accumulation of mutant p53-R167H protein (Figure 1c), similar to their human and murine counterparts.18, 19
The progressive genetic modifications also resulted in phenotypic changes, such as accelerated cellular proliferation (Figure 1d), loss of contact inhibition in MSC-PKCM cells (Figure 1e top) and anchorage-independent growth (Figure 1e bottom).
Genetically modified porcine MSCs form sarcomas in a xenotransplantation model
To analyse tumorigenic potential in vivo, porcine cells expressing mutant TP53R167H and KRASG12D with and without additional MYC (MSC-PKC and MSC-PKCM) were xenografted into two immunodeficient mice. After 45 days, MSC-PKC cells formed a small nodule in one of the four injection sites with few atypical cells (Figure 1f). MSC-PKCM cells gave rise to tumours in three of the four injection sites in the other animal, classified as low-grade fibroblastic sarcomas (Figure 1g). Two such tumours were explanted and cultured to derive a porcine sarcoma cell line (poSARCO).
Molecular analysis of transformed MSCs and sarcomas
Microarray analysis was carried out to identify transcriptional changes associated with the stages of porcine MSC transformation. Figure 2a and Supplementary Table S1 show expression profiles for six groups of genes associated with cellular transformation: cellular transformation, telomere maintenance, cell cycle control, p53 target genes, apoptosis, and chromosomal instability (CIN).

Expression profiles of cancer-related genes in transformed MSCs and sarcoma-derived tumour cells. (a) Differential gene expression heat maps relative to wild-type MSCs. Genes are grouped by functional categories, as indicated. The key at bottom right indicates fold-change values (log2 scale) represented as a colour gradient from blue (downregulation) to red (upregulation). See also Supplementary Table S2. (b) Sequence analysis of KRAS RT–PCR products amplified from exon 1 to exon 4. Cell types as shown. Codon 12 is indicated by a red box. RT–PCR products carrying the GAT sequence (G12D mutation) are the predominant species in poSARCO cells. (c) PoSARCO cells express higher levels of active GTP-bound Ras proteins (21 kDa) than the parental MSC-PKCM cells.
Cellular transformation
TP53 was not expressed in uninduced MSC-PK cells but upregulated after Cre activation. MYC expression increased in MSC-PKCM owing to exogenous expression. KRAS expression increased at each stage of cellular transformation and was the highest in poSARCO cells, likely owing to spontaneous gene amplification, as in many tumours.20 Quantitative PCR (Q-PCR) analysis revealed two KRAS copies in wild-type MSCs, MSC-PKC and MSC-PKCM, while poSARCO had six. Sequence analysis of KRAS mRNAs (exon 1 to exon 4) detected higher expression of KRAS-G12D than wild-type KRAS in poSARCO (Figure 2b). PoSARCO cells also exhibited increased levels of activated GTP-bound Ras protein (Figure 2c).
Primary human cells can escape cellular senescence through simultaneous inactivation of p53 and Rb.21 The CDKN2A gene products p16INK4α and p14ARF (p19ARF in mice) regulate the Rb and p53 pathways,22, 23, 24 but their roles differ between humans and mice25 and little is known about the pig. As shown in Figure 2a, RB1 expression remained low in the MSC derivatives, but p14ARF (ARF) expression varied with the stages of cellular transformation. The microarrays used did not include p16INK4α, so p16INK4α and p14ARF expression was determined by Q-RT–PCR. Figure 3a shows that p16INK4α expression was diminished in MSC-PK cells, almost absent in MSC-PKC cells and similarly low in MSC-PKCM cells. Investigation of 21 CpG dinucleotides in the 5′ region of p16INK4A revealed that none were methylated in wild-type MSCs and progressively greater methylation in uninduced MSC-PK, activated MSC-PKC cells and the MSC-PKCM pool (Figure 3b).

p16INK4, p14ARF and TERT expression. (a) Expression of porcine p16INK4α and p14ARF relative to wild-type MSCs. Cell types as shown. Values are normalised to GAPDH mRNA expression. Left: Downregulation of p16INK4α mRNA expression in stepwise modified MSC derivatives. Right: Increased p14ARF mRNA expression in modified porcine MSC derivatives. (b) CpG methylation analysis of the porcine P16INK4A locus (−10 to +256 bp relative to transcription start). Circles indicate methylation sites: filled circles represent methylated and open circles non-methylated sites. The proportion of total sites methylated is indicated at right. Cell types as shown. (c) Telomerase reverse transcriptase (TERT) expression is not activated in transformed porcine MSC derivatives. Porcine induced pluripotent stem cells (piPSC) are shown as a positive control for porcine TERT expression; a diagnostic 387-bp DNA fragment is evident. GAPDH was amplified (567 bp) as a control as indicated.
Telomere maintenance
No reactivated TERT expression was observed (Figures 2a and 3c) in transformed MSC derivatives, but there was evidence for activation of the ALT mechanism, as frequently observed in human MSC-derived sarcomas.26 BLM, FEN1, FANCD2 and, to a lesser extent, FANCA were upregulated. Genes involved in telomere elongation and capping, RAD51, RAD54B and BRCA2, were also upregulated, while shelterins TERF1 and TERF2 and POT1 remained low, possibly indicating telomere deprotection (Figure 2a, Supplementary Table S1).
Cell cycle control
Loss of Rb function results in deregulated G1 checkpoint control and enhanced cellular proliferation. Although RB1 expression remained at low baseline levels, several cyclin genes and their interacting cyclin-dependent kinases were upregulated in the genetically modified MSCs and poSARCO cells (Figure 2a, Supplementary Table S1).
p53 target genes and genes associated with apoptosis
Reduced expression of the key feedback regulator MDM2 indicates that mutant p53-R167H has impaired ability to transactivate its targets. This extends to genes important for cell cycle arrest, such as CDKN1A, CDKN1B and GADD45A, expression of which was lower than in wild-type MSCs and unaffected by induction of p53-R167H expression. Impaired capacity for apoptosis is indicated by lower expression of components of the death receptor pathway, for example, FAS, TNFSF10 (TRAIL) and TNFRSF10B (KILLER/DR5); and of the mitochondrial pathway, for example, BBC3 (PUMA), BAX and CASP6 (Figure 2a, Supplementary Table S1).
Chromosomal instability
Several CIN-associated genes were strongly upregulated, including the spindle assembly checkpoint machinery genes AURKA, AURKB, BUB1, BUB1B and MAD2L1;27, 28 regulators of sister chromatid segregation PTTG1 and ESPL1;29 and CDC20, a subunit of the anaphase-promoting complex/cyclosome, which is overexpressed in several human cancers and associated with poor prognosis30 (Figure 2a, Supplementary Table S1).
To investigate the dysfunction of mitotic checkpoints suggested by deregulated expression of cell cycle control genes, cells were treated with nocadazole to disrupt the mitotic spindle and induce mitotic arrest. The number of cells in G0/G1, S and G2/M phase determined by flow cytometry is shown in Figure 4a. Figure 4b shows the ratio between nocodazole treated and untreated cells in G1 phase. Human colon cancer cell lines known to have CIN and mitotic checkpoint deficiency (SW480, CaCo2) and chromosomally stable human cell lines of non-tumour origin (HEK293) or from colon cancer (HCT116, microsatellite unstable) provided comparisons. G1-peak ratios ranged from 0.05 in HEK293, which show the strongest blocking response to nocodazole, to 0.2 for CaCo2 cancer cells, which showed a weak response. Transformed porcine MSCs showed elevated ratios of cells in G1 phase after nocodazole treatment, similar to the CIN-positive cell lines SW480 and CaCo2, and poSARCO cells displayed an even higher ratio (Figure 4b).

Chromosomal instability. (a) The number of cells in G0/G1, S and G2/M phase determined by flow cytometry, after growth under normal conditions (control), and 24 h after treatment with nocodazole. Representative DNA content histograms are shown for cell types as indicated. Wild-type MSCs show a strong increase in the G2/M-phase peak in response to nocadazole. In contrast, p53-deficient cell types (MSC-P, MSC-PK) and cells that express mutant p53-R167H (MSC-PKC,MSC-PKCM poSARCO) show no such increase but feature polyploid DNA peaks after nocodazole treatment. (b) Mitotic block index. The ratio of G1 peak in nocodazole-treated cells compared with control conditions (mean±s.d. for n=4 assays) shown for human cell lines and porcine MSC derivatives as indicated. A low index value indicates an effective mitotic checkpoint arrest after nocodazole treatment, as shown for human HEK293 (non-tumour origin) and HCT116 cells (colon cancer cells, chromosomally stable but DNA microsatellite unstable). In contrast, human SW480 and CaCo2 cells have high index values, indicating failure to induce G2/M arrest. MSC-P cells have intermediate index values, whereas MSC-PKC and MSC-PKCM clones, and especially poSARCO cells, have highly elevated index values, indicative of mitotic checkpoint deficiency. The tumour-derived poSARCO cells feature significantly increased index numbers, compared with p53-deficient MSC-P (P=0.0055), whereas MSC-PKC and MSC-PKCM cells do not differ significantly from MSC-P.
In summary, these data suggest that porcine MSCs resemble human MSCs in requiring perturbation of the p53, Rb, KRAS and MYC signalling pathways combined with a spontaneous telomerase-independent immortalisation step to convert to a fully transformed phenotype.
TP53 inactivation in pigs is sufficient for development of osteosarcomas
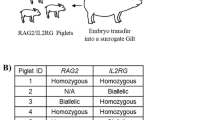
We determined whether pigs carrying the latent TP53LSL-R167H mutation8 in heterozygous or homozygous form would spontaneously develop tumours. Animals were regularly observed for alterations in health and wellbeing, and individuals were slaughtered periodically for necropsy examination.
To date, nine heterozygous knockout pigs aged up to 32 months have been examined by necropsy, as summarised in Table 1. There was no evidence of tumours or other abnormalities in animals <16 months. Of the five older animals, four had tumours (ID: 34, 36, 47, 60), and one animal (ID: 49) had disseminated calcifying, ossifying lesions but no identifiable primary tumour. Figure 5a shows gross morphology and histology of tumours from two heterozygous and one homozygous animal.

Osteosarcomas in TP53 knockout pigs. (a) Upper: Osteoblastic osteosarcoma at left tuber olecrani in heterozygous knockout animal ID: 36. H&E-stained section shows tumour cells (T) with eccentric, hyperchromatic nuclei that produce osteoid (O). Multinucleate giant cells (arrows) with features of osteoclasts are scattered throughout the neoplasia. Scale bar indicates 100 μm. Lower: Osteoblastic and chondroblastic osteosarcoma at the skull basis in heterozygous knockout animal ID: 47. H&E-stained section shows tumour cells (T) that produce osteoid (O) and a chondroid matrix (C). Scale bar indicates 100 μm. (b) Upper: Osteoblastic osteosarcoma of the skull infiltrating os sphenoidale and vomer in homozygous knockout animal ID: 242. Polygonal tumour cells (T) and islands of osteoid (O). Scale bar indicates 100 μm. Lower: osteoblastic osteosarcoma in the bone marrow of left femur in homozygous knockout animal ID: 242. Multinucleated giant cells (arrows) were scattered throughout the neoplasia. Scale bar indicates 100 μm.
Lesions from three pigs (animal ID: 34, 36, 47) were determined by histology as osteoblastic osteosarcomas. Two animals (animal ID: 34, 36) had osteosarcomas within long bones (metaphyseal region of the tibia and tuber olecrani) (Figure 5a top). The other animal (animal ID: 47) had a tumour in the nasal conchae, classified as an osteoblastic and chondroblastic osteosarcoma (Figure 5a bottom). Animal 60 showed a calcified fibrous tumour of the mandible.
We examined two F2 generation homozygous TP53LSL-R167H/LSL-R167H knockout pigs (animal ID: 242, 336). These both grew more slowly than wild-type and heterozygous siblings, weighing 85–90 kg at 8 months while normal weight for this age and breed is 120–130 kg. Other than reduced size, neither showed ill effects or signs of distress in early life, but at 8 months pig 242 quickly lost condition and exhibited laboured breathing, and pig 336 developed paralysis of the hind legs. Both were slaughtered. Necropsy of pig 242 revealed multifocal osteoblastic osteosarcoma in the skull, left femur, left tibia, left humerus and pelvic bone. Figure 5b shows gross morphology and histology of the skull and left femur. Necropsy of pig 336 revealed multifocal osteoblastic osteosarcoma in the vertebral column, right femur and right ulna. The eighth thoracic vertebra was fractured, probably causing the limb paralysis.
Over the past 6 years, similar necropsies have been performed on 60 wild-type pigs aged between 12 and 48 months at the Bavarian Animal Health Service, none of which displayed any tumours. Moreover, there have been very few reports of spontaneous osteosarcomas in wild-type pigs.31, 32
Radiation resistance
Several mutant p53 isoforms confer resistance to radiation.33 p53-deficient MSCs or MSCs expressing mutant p53 show increased resistance to 137Cs irradiation (Figure 6a). Similarly, cells derived from an osteosarcoma on the left tibia of homozygous TP53 knockout pig 242 showed increased radioresistance compared with control cells derived from a tumour-free bone of the same animal (Figure 6b). Tumour-derived and normal cell isolates were characterised as positive for osteocalcin, osteonectin, type 1 collagen, alkaline phosphatase and vimentin and negative for EpCam.

Transformed MSCs and osteosarcoma-derived cells show increased resistance to radiation. (a) Survival of cells after 137Cs irradiation after 2 weeks of culture. Left: Colonies stained with crystal violet; cell types and doses as indicated. Right: Mean survival±s.d., n=4. No significant differences were observed at 10 Gy irradiation. However, at 2 Gy transformed MSC showed increased viability compared with WT-MSCs: MSC-P (P=0.0184), MSCK-PKC (P=0.0500). MSC-PKCM cells did not attain significance (P=0.1558). Results were similar at the intermediate dose 6 Gy: MSC-P (P=0.0011); MSCK-PKC (P=0.0081); MSC-PKCM (P=0.0874). (b) Osteosarcoma cells show increased radioresistance. Cell survival in response to irradiation was assessed after 2 weeks of cell culture, as above. Cells were derived from a tumour on the left tibia of homozygous TP53 knockout pig (animal ID: 242) compared with cells from a tumour-free bone of the same animal. Tumour cells showed increased clonal growth after irradiation.
Multinucleation and CIN
TP53 mutations are associated with CIN and human osteosarcomas have genome-wide DNA instability.34 Osteosarcoma cells isolated from TP53 heterozygous pig 47 and TP53 homozygous knockout pig 242 were checked for mitotic abnormalities, as were the p53-deficient MSC clones (MSC-P, MSC-PK), cell clones with activated TP53-R167H mutation (MSC-PKC, MSC-PKCM) and tumour-derived poSARCO cells. Multinucleated cells with fragmented nuclei were common in all samples (Supplementary Figures S3a and b). Many osteosarcoma-derived cells showed nuclear abnormalities, such as giant nuclei, multinucleated cells, micronuclei, lagging chromosomes (anaphase bridges) and abnormal spindle apparatus (Supplementary Figures S3b and c).
Chromosome counting also revealed that only 34.7% of metaphase spreads (n=174) from osteosarcoma cells (homozygous TP53 knockout pig 242) were karyotypically normal, compared with 77% from wild-type MSCs (n=100).
Discussion
In vitro studies of sarcomagenesis with human and mouse MSCs revealed that murine MSCs can be transformed by abrogation of p53 function alone.35 Human MSCs are more refractory, requiring disruption of both the Rb and p53 pathways to bypass senescence in combination with ectopic expression of TERT, oncogenic HRASV12 and SV40 small T antigen.13 We report that porcine MSCs resemble human MSCs, requiring perturbation of p53, KRAS and MYC signalling pathways with spontaneous Rb pathway inactivation and telomerase-independent immortalisation steps to convert to a fully transformed phenotype. Immortalisation via the ALT mechanism rather than telomerase reactivation accords with findings from human mesenchymal malignancies.26
There have been two other reports of neoplastic transformation of porcine primary cells in vitro, based on overexpression of randomly integrated oncogenic transgenes,36, 37 but these are generally expressed at non-physiological levels. Our findings are based on targeted mutation of endogenous genes, more closely mimicking natural lesions that underlie cancer.
We demonstrate that null mutation of TP53 results in spontaneous osteosarcomas in both heterozygous (>20 months) and homozygous (7–8 months) form. Rapid disease onset is an important practical advantage for future use of this model and compares well with mice, where homozygous Trp53 knockout animals develop neoplasms around 6 months of age.38 The natural lifespan of a pig is approximately 10–15 years.
The effects of osteosarcoma and the necessary surgery can be devastating. There is an urgent need for animal models to improve methods of surgical management, develop new drugs and understand the molecular basis of disease initiation and progression. Most osteosarcomas are sporadic and of unknown cause but can be induced by radiation treatment. They frequently have TP53 and RB1 mutations34, 39 and alterations affecting cell cycle checkpoints, such as reduced p16INK4A expression.40 Patients with Li-Fraumeni syndrome and hereditary retinoblastoma are predisposed to develop osteosarcomas.41, 42 Disruption of p53 often leads to genomic instability, defective nocodazole-induced mitotic spindle checkpoints and resistance to radiation, all of which we observed. We showed that cells derived from porcine osteosarcomas are resistant to radiation, consistent with radioresistance of human osteosarcomas. Similar to human osteosarcoma cell lines and tumour samples from patients, porcine osteosarcoma cells displayed nuclear abnormalities and atypical mitotic figures (anaphase bridges, abnormal spindle apparatus). Moreover, metaphase spreads of porcine osteosarcoma-derived cells displayed highly variable chromosome numbers, again similar to human data.43 As up to 50% of human osteosarcomas have alterations in the TP53 gene44 and various cytogenetic abnormalities,45 we consider that our porcine osteosarcoma model provides a valuable platform for studying such genetic changes.
Genetically modified mice have so far been the natural focus for modelling osteosarcoma. Germline Trp53 inactivation in mice results in diverse cancers, with ~25% osteosarcomas in heterozygotes and ~4% osteosarcomas in homozygotes, which mainly develop lymphomas.46 The high incidence of other tumours is a problem that has motivated development of improved mouse osteosarcoma models with Cre-mediated conditional deletion of Trp53 in the osteogenic lineage, sometimes in combination with Rb1. These models show highly penetrant osteosarcoma formation but have been criticised because murine primary tumours predominantly affect the axial skeleton, while human osteosarcomas are most common in the long bones of the limbs.47
Work in rats is less advanced than in mice, but a similar mixed tumour spectrum has been reported, with approximately half of heterozygous Trp53 knockout rats developing osteosarcomas, while most homozygotes develop haemangiosarcoma.48
Yucatan pigs have been described that carry the TP53R167H mutation.7 Pigs heterozygous for this mutant allele were reported to be free of tumours up to 30 months, while homozygous pigs mainly developed lymphomas and some osteogenic tumours. These findings differ from ours, perhaps owing to the different breed, the p53 mutation or both.
Many different p53 mutations have been analysed in humans and mice. Human TP53R175H (orthologous to porcine TP53R167H) is thought to impart gain-of-function properties with dominant-negative effect. Mutant p53-R175H inhibits wild-type p53 interaction with promoter elements,49 advances angiogenesis,50 and promotes epithelial–mesenchymal transition.51 Genotype–phenotype analysis of Li-Fraumeni families revealed that patients carrying such a mutation in the core p53 DNA-binding domain show more highly penetrant cancer phenotypes with higher incidence and earlier onset than those with TP53 inactivation mutations.52, 53 These observations are also reflected in mouse studies. Mice heterozygous for Trp53R172H (orthologous to human TP53R175H and porcine TP53R167H) developed more tumours and a different tumour spectrum than heterozygous knockout mice.54
We previously showed that p53-deficient porcine cells develop a transformed phenotype with more rapid cell doubling, growth in semi-solid medium and resistance to the chemotherapeutic drug doxorubicin and that these characteristics were more pronounced in porcine cells that express mutant p53-R167H,8 suggesting that porcine p53-R167H is a gain-of-function mutation, as per murine and human data. An important advantage of our model is that the TP53 knockout allele can be activated to express mutant p53-R167H, so it will, in future, be possible to generate animals that express p53-R167H for direct comparison with p53 inactivation in the same breed.
To the best of our knowledge, ours is the first report of TP53 knockout pigs. The anatomical locations of osteosarcomas observed are mainly the long bones, skull and mandible. Analysis of homozygous p53 deficiency is so far restricted to two animals but indicates accelerated tumour development and, unlike rodents, no change in tumour spectrum relative to heterozygous knockout.
Our priority is now to further investigate how accurately the model represents the human disease. The initiation and progression of human osteosarcoma are not well understood and we hope the porcine model will help elucidate the molecular pathways and driver mutations involved. Imaging studies will enable non-invasive longitudinal investigation of individual animals to identify early-stage tumours, analyse disease progression and investigate metastasis. Several practical advantages of the model are already apparent, not least its simplicity, with no need for tissue-specific gene inactivation or multiple engineered mutations. It is also helpful that osteosarcomas form relatively late in heterozygotes, allowing normal breeding and expansion of the herd, and with short latency in homozygotes that can be produced as required.
Materials and methods
Reagents
Chemicals were obtained from Sigma-Aldrich Chemie GmbH (Munich, Germany) unless otherwise specified; cell culture media and supplements were obtained from ThermoFisher Scientific (Waltham, MA, USA) or Invitrogen (Carlsbad, CA, USA) unless otherwise specified.
Animals
Animal studies were approved by the Government of Upper Bavaria (permit number 55.2-1-54-2532-34-09) and performed according to the German Animal Welfare Act and European Union Normative for Care and Use of Experimental Animals and were approved by the Institutional Animal Care and Use Committees of Technische Universität München, and Regierung von Oberbayern.
Nine TP53LSL-R167H/+ and two TP5LSL-R167H/LSL-R167H Landrace pigs aged 7.5–32 months, male and female, were generated and raised in our own facilities with food and water provided ad libitum.
Two 7- and 10-week-old female NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Jackson Lab (Bar Harbor, ME, USA) and maintained in specific pathogen-free facilities with food and water provided ad libitum.
Necropsy examination and tumour analysis
Pigs were humanely killed and complete necropsy examination was carried out at the Tiergesundheitsdienst Bayern (Bavarian Animal Health Service). Specimens were fixed, embedded, sectioned and stained by standard methods. Bone specimens were first decalcified in Ossa Fixona (Waldeck GmbH, Münster, Germany).
Porcine primary cells and cell lines
Porcine primary cells and cell lines were derived in house and regularly screened for mycoplasma contamination. Cell lines of human origin (Hek293, HCT1116, SW480,CaCo2) were obtained from ATCC or DSMZ (Braunschweig, Germany), cultured in Dulbecco's Modified Eagle's Medium (DMEM) with foetal calf serum (7%), 1% Pen/Strep and 1% glutamine and tested every 2 months for mycoplasma infection. To avoid contamination and phenotype changes, cells were kept as frozen stocks and cultured for 4 weeks maximum.
Generation of TP53R167H, KRASG12D double gene targeted porcine MSC clones
Derivation and culture of porcine TP53 gene-targeted MSC clones has been described previously.8 The vector KRAS-BSR (Supplementary Figure S1a) is the same as vector KRAS-NEO, which has been described previously,9 but contains the blasticidin resistance gene (bsr) instead of neo.
KRAS-BSR targeted cell clones were identified by: 5′ and 3′ junction PCR, RT–PCR, and restriction fragment-length polymorphism to confirm the G to A mutation, as described elsewhere.9 Cre-mediated induction of TP53R167H and KRASG12D alleles was confirmed by PCR and RT–PCR analysis as described elsewhere.8, 9 All PCR primers used and diagnostic fragments amplified are shown in Supplementary Table S2.
Porcine MYC expression vector
This vector comprised the mouse phosphoglycerate kinase promoter directing expression of 1.361 kb porcine MYC cDNA linked to 98 bp MYC 5′ and 178 bp MYC 3′ untranslated regions (NCBI accession number NM_001005154.1: 494…2128). A bsr selectable marker linked by an internal ribosome entry sequence was placed 3′ of the MYC coding sequence.
Cre recombinase protein transduction
Cre protein was produced in vitro with the vector pTriEx-HTNC (Addgene plasmid 13763, Cambridge, MA, USA) as described55, 56 and transduced as described previously.8 After 96 h, cells were plated by limiting dilution into 96-well plates to derive single-cell clones.
Bisulphite sequencing
In all, 500 ng genomic DNA samples were converted using the EpiTect Fast Bisulphite Conversion Kit (Qiagen, Germantown, MD, USA) by standard methods. A 266-bp P16INK4A fragment (NCBI accession number AJ316064.1: 299…565) was amplified using primers BS-p16F and BS-p16R with GoTaq Polymerase (Promega, Madison, WI, USA). Thermal cycling conditions were: 30 min, 95 °C; then 35 cycles of: 30 s, 95 °C; 30 s, 54 °C; 30 s, 72 °C; followed by 5 min, 72 °C. Amplified PCR products were processed as described previously.57
Copy number variation analysis
Q-PCR was carried out using Fast SybrGreen PCR MasterMix and run on 7500 Fast Real-Time PCR System (both from Applied Biosystems, Foster City, CA, USA). Relative quantification of KRAS copy number was performed with primers CNV-KRASF and CNV-KRASR. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) was amplified using primers CNV-GAPDHF and CNV-GAPDHR. Relative quantification of copy number alterations was performed in 10 μl with 10 ng genomic DNA. Thermal cycling conditions were 10 min, 95 °C; then 40 cycles of: 15 s, 95 °C; 1 min, 60 °C. Samples were run in triplicate, data were normalised to GAPDH and copy numbers were calculated by the ΔΔCT method.
Quantitative real-time RT–PCR
Relative quantification of gene expression was carried out by two-step Q-RT–PCR using Fast SybrGreen PCR MasterMix. Primers used were: p16INK4α RT-p16F and RT-p16R; p14ARF RT-p14F and RT-p14R; MYC RT-MycF and RT-MycR; and GAPDH RT-GAPDHF and RT-GAPDHR. Reactions were performed in 10 μl. Thermal cycling parameters were as above. Samples were assayed in triplicate, relative expression was normalised to GAPDH expression and fold-differences were calculated by the ΔΔCT method.
Western blotting analysis
Protein lysates were prepared as described previously.8 Total protein (40 μg) was loaded in each lane and separated by 15% sodium dodecyl sulphate–polyacrylamide gel electrophoresis. Blotting and detection of p53 and GAPDH was as described previously.58 Porcine MYC was detected with anti-c-MYC (N-262, Santa Cruz Biotechnology, Heidelberg, Germany) diluted 1:250.
Ras activation assay
Ras activity was analysed using the Ras Activation Assay Kit (Merck, Millipore, Darmstadt, Germany). Total protein (1 mg) was incubated with 10 μg Raf-1 RBD agarose for 3 h at 4 °C with gentle agitation. Western blotting and detection were carried out according to the manufacturer's instructions.
Growth characteristics of porcine cells
Cycling and non-cycling cells in the G0/G1 peak were differentiated by 2-h pulse-labelling with 5-ethynyl-2'-deoxyuridine and then staining with the Click-iT EdU Flow Cytometry Assay Kit (ThermoFisher). For the focus formation assay, 1 × 105 cells were plated onto 10-cm cell culture dishes and cultured for 4 weeks, and the medium was changed every 2–3 days. Anchorage-independent growth assay of primary and transformed porcine cells was carried out as described.8
Tumour growth and isolation of porcine sarcoma-derived tumour cells
Groups of 1 × 107 cells suspended in chilled DMEM were mixed 1:1 with high concentration matrigel basement membrane matrix (BD Biosciences, Bedford, MA, USA) and implanted subcutaneously into NSG mice. Mice were humanely killed, tumours excised, fixed, sectioned and H&E (haemotoxylin and eosin) stained by standard methods. Cells were derived from a minced piece of tumour digested in DMEM supplemented with 200 U/ml collagenase type IV (Worthington, Lakewood, NJ, USA) at 37 °C for 24 h, then centrifuged at 100 r.c.f. for 5 min. Cells were resuspended in standard porcine MSC medium,8 cultured at 37 °C, 5% CO2 and passaged at regular intervals.
Establishment of cell cultures from porcine osteosarcoma
Cell cultures were established from osteosarcomas as described above for tumour xenografts, cultured and passaged by standard methods.
Gene expression profiling and data analysis
cDNA was prepared from total RNA by standard methods. Gene expression experiments were conducted on custom Sus Scrofa 12-plex microarrays containing 142 073 probes targeting 17 261 genes. Probe selection and array design is described elsewhere.59 In all, 200 ng double-stranded cDNA was Cy3-labelled using the 'One colour DNA Labelling Kit' (Roche NimbleGen, Madison, WI, USA). A minimum of three technical replicates of each sample were loaded, and raw data intensities were collected using a NimbleGen MS200 scanner. Background corrected, quantile normalised and RMA (Robust Multi-Array Analysis) normalised probe intensities were generated using the NimbleScan v2.6 software (Roche NimbleGen). Average signal intensities were calculated for each probe set and log2 transformed. Data were analysed using the Partek Genomics Suite software, v6.6 (Partek, St Louis, MO, USA). Significant differentially expressed genes were identified for a false-discovery rate P-value of 0.05 and twofold change.
Indirect immunofluorescence staining
Immunostaining and fluorescence micrography were performed as described previously.60 Antibodies and reagents used were: anti-α-tubulin (MABT205, Calbiochem, San Diego, CA, USA), 4′,6-diamidino-2-phenyl indole (DAPI) and TRITC-Phalloidin (P1951, Sigma-Aldrich), secondary antibodies coupled to fluorophores (Jackson Immunoresearch, West Grove, PA, USA).
Cell cycle analysis
Mitotic arrest was induced with 100 ng/ml nocadazole for 20 h. Cells were harvested, washed, resuspended in ice-cold Ca2+/Mg2+-free phosphate-buffered saline with 1 mg/ml glucose, fixed in 70% ice-cold ethanol overnight at 4 °C and incubated 1 h at room temperature in the dark with 50 μg/ml propidium iodide and 0.4 mg/ml RNaseA (Qiagen). Cells were washed, resuspended in phosphate-buffered saline with 0,5% (v/v) bovine serum albumin and 0.01% (w/v) NaN3 and strained before fluorescence-activated cell sorting analysis. Cell cycle profiles were measured using a FACSCalibur device (BD Biosciences, San Jose, CA, USA) with the CellQuest Pro software (BD Biosciences). Doublets, dead cells and debris were excluded, and single-parameter DNA histograms were analysed with the FlowJo software (Tree Star, Ashland, OR, USA) to identify G1 and G2/M peaks and the S-phase populations from univariate distribution curves.
Cell irradiation clonogenic assay
Aliquots of 5 × 105 cells were irradiated with 2, 6 and 10 Gy from a 137Cs source (GE Healthcare Buchler, Braunschweig, Germany), seeded in six-well plates and cultured under standard conditions overnight. A total of 1 × 102 cells were seeded on 10-cm dishes and cultivated under standard conditions for 14 days. Plates were fixed and stained with 0.1% crystal violet by standard methods, and colonies were counted by two observers.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 5 and GraphPad InStat3 (GraphPad Software, La Jolla, CA, USA). Data are presented as mean±s.d. Comparison between two data sets was carried out using Student’s unpaired t-test, after prior testing for Gaussian distribution. Non-normally distributed sample sets were tested with Mann–Whitney U-tests. All statistical comparisons were carried out at a 0.05 threshold level of significance.
Karyotype analysis
Primary porcine cells were treated with 0.8 μg/ml colcemid for 20 h, dissociated with 10 × trypsin/EDTA, treated with 0.8% Na-citrate/75 mm KCl/H2O (1:1:1) and fixed with ice-cold methanol/acetic acid (3:1). Metaphase spreads were treated with 0.03 mg/ml pepsin in 0.01m HCl, 45 s at 37 °C, DAPI stained and mounted with antifade.
Accession codes
References
Rangarajan A, Hong SJ, Gifford A, Weinberg RA . Species- and cell type-specific requirements for cellular transformation. Cancer Cell 2004; 6: 171–183.
Kendall SD, Linardic CM, Adam SJ, Counter CM . A network of genetic events sufficient to convert normal human cells to a tumorigenic state. Cancer Res 2005; 65: 9824–9828.
Flisikowska T, Kind A, Schnieke A . Genetically modified pigs to model human diseases. J Appl Genet 2013; 55: 53–64.
Rogers CS, Stoltz DA, Meyerholz DK, Ostedgaard LS, Rokhlina T, Taft PJ et al. Disruption of the CFTR gene produces a model of cystic fibrosis in newborn pigs. Science 2008; 321: 1837–1841.
Renner S, Fehlings C, Herbach N, Hofmann A, von Waldthausen DC, Kessler B et al. Glucose intolerance and reduced proliferation of pancreatic beta-cells in transgenic pigs with impaired glucose-dependent insulinotropic polypeptide function. Diabetes 2010; 59: 1228–1238.
Luo Y, Li J, Liu Y, Lin L, Du Y, Li S et al. High efficiency of BRCA1 knockout using rAAV-mediated gene targeting: developing a pig model for breast cancer. Transgenic Res 2011; 20: 975–988.
Sieren JC, Meyerholz DK, Wang XJ, Davis BT, Newell JD Jr, Hammond E et al. Development and translational imaging of a TP53 porcine tumorigenesis model. J Clin Invest 2014; 124: 4052–4066.
Leuchs S, Saalfrank A, Merkl C, Flisikowska T, Edlinger M, Durkovic M et al. Inactivation and inducible oncogenic mutation of p53 in gene targeted pigs. PLoS ONE 2012; 7: e43323.
Li S, Edlinger M, Saalfrank A, Flisikowski K, Tschukes A, Kurome M et al. Viable pigs with a conditionally-activated oncogenic KRAS mutation. Transgenic Res 2015; 24: 509–517.
Flisikowska T, Merkl C, Landmann M, Eser S, Rezaei N, Cui X et al. A porcine model of familial adenomatous polyposis. Gastroenterology 2012; 143: 1173–1175, e1171-1177.
Helman LJ, Meltzer P . Mechanisms of sarcoma development. Nat Rev Cancer 2003; 3: 685–694.
Mohseny AB, Hogendoorn PC . Concise review: mesenchymal tumors: when stem cells go mad. Stem Cells 2011; 29: 397–403.
Funes JM, Quintero M, Henderson S, Martinez D, Qureshi U, Westwood C et al. Transformation of human mesenchymal stem cells increases their dependency on oxidative phosphorylation for energy production. Proc Natl Acad Sci USA 2007; 104: 6223–6228.
Mirabello L, Troisi RJ, Savage SA . Osteosarcoma incidence and survival rates from 1973 to 2004: data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009; 115: 1531–1543.
Beroukhim R, Mermel CH, Porter D, Wei G, Raychaudhuri S, Donovan J et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010; 463: 899–905.
Drayton S, Rowe J, Jones R, Vatcheva R, Cuthbert-Heavens D, Marshall J et al. Tumor suppressor p16INK4a determines sensitivity of human cells to transformation by cooperating cellular oncogenes. Cancer Cell 2003; 4: 301–310.
Boehm JS, Hession MT, Bulmer SE, Hahn WC . Transformation of human and murine fibroblasts without viral oncoproteins. Mol Cell Biol 2005; 25: 6464–6474.
Midgley CA, Lane DP . p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene 1997; 15: 1179–1189.
Scheffzek K, Ahmadian MR, Kabsch W, Wiesmuller L, Lautwein A, Schmitz F et al. The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 1997; 277: 333–338.
Singh A, Greninger P, Rhodes D, Koopman L, Violette S, Bardeesy N et al. A gene expression signature associated with "K-Ras addiction" reveals regulators of EMT and tumor cell survival. Cancer Cell 2009; 15: 489–500.
Smogorzewska A, de Lange T . Different telomere damage signaling pathways in human and mouse cells. EMBO J 2002; 21: 4338–4348.
Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW . Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88: 593–602.
Wei W, Hemmer RM, Sedivy JM . Role of p14(ARF) in replicative and induced senescence of human fibroblasts. Mol Cell Biol 2001; 21: 6748–6757.
Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ et al. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev 1998; 12: 2424–2433.
Rangarajan A, Weinberg RA . Opinion: comparative biology of mouse versus human cells: modelling human cancer in mice. Nat Rev Cancer 2003; 3: 952–959.
Lafferty-Whyte K, Cairney CJ, Will MB, Serakinci N, Daidone MG, Zaffaroni N et al. A gene expression signature classifying telomerase and ALT immortalization reveals an hTERT regulatory network and suggests a mesenchymal stem cell origin for ALT. Oncogene 2009; 28: 3765–3774.
Gollin SM . Mechanisms leading to chromosomal instability. Semin Cancer Biol 2005; 15: 33–42.
Rao CV, Yamada HY, Yao Y, Dai W . Enhanced genomic instabilities caused by deregulated microtubule dynamics and chromosome segregation: a perspective from genetic studies in mice. Carcinogenesis 2009; 30: 1469–1474.
Holland AJ, Cleveland DW . Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 2009; 10: 478–487.
Karra H, Repo H, Ahonen I, Loyttyniemi E, Pitkanen R, Lintunen M et al. Cdc20 and securin overexpression predict short-term breast cancer survival. Br J Cancer 2014; 110: 2905–2913.
Seva J, Pallares FJ, Gomez MA, Bernabe A . Osteoblastic osteosarcoma in a fattening pig. Vet Rec 2001; 148: 147–148.
Williamson MM, Byrne A . Mandibular osteosarcoma in a pig. Aust Vet J 2006; 84: 202–203.
Okaichi K, Ide-Kanematsu M, Izumi N, Morita N, Okumura Y, Ihara M . Variations in sensitivity to ionizing radiation in relation to p53 mutation point. Anticancer Res 2008; 28: 2687–2690.
Overholtzer M, Rao PH, Favis R, Lu XY, Elowitz MB, Barany F et al. The presence of p53 mutations in human osteosarcomas correlates with high levels of genomic instability. Proc Natl Acad Sci USA 2003; 100: 11547–11552.
Rubio R, Garcia-Castro J, Gutierrez-Aranda I, Paramio J, Santos M, Catalina P et al. Deficiency in p53 but not retinoblastoma induces the transformation of mesenchymal stem cells in vitro and initiates leiomyosarcoma in vivo. Cancer Res 2010; 70: 4185–4194.
Adam SJ, Rund LA, Kuzmuk KN, Zachary JF, Schook LB, Counter CM . Genetic induction of tumorigenesis in swine. Oncogene 2007; 26: 1038–1045.
Schook LB, Collares TV, Hu W, Liang Y, Rodrigues FM, Rund LA et al. A genetic porcine model of cancer. PLoS ONE 2015; 10: e0128864.
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr, Butel JS et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356: 215–221.
Wadayama B, Toguchida J, Shimizu T, Ishizaki K, Sasaki MS, Kotoura Y et al. Mutation spectrum of the retinoblastoma gene in osteosarcomas. Cancer Res 1994; 54: 3042–3048.
Maitra A, Roberts H, Weinberg AG, Geradts J . Loss of p16(INK4a) expression correlates with decreased survival in pediatric osteosarcomas. Int J Cancer 2001; 95: 34–38.
Ognjanovic S, Olivier M, Bergemann TL, Hainaut P . Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer 2012; 118: 1387–1396.
Abramson DH, Ellsworth RM, Kitchin FD, Tung G . Second nonocular tumors in retinoblastoma survivors. Are they radiation-induced? Ophthalmology 1984; 91: 1351–1355.
Al-Romaih K, Bayani J, Vorobyova J, Karaskova J, Park PC, Zielenska M et al. Chromosomal instability in osteosarcoma and its association with centrosome abnormalities. Cancer Genet Cytogenet 2003; 144: 91–99.
Tsuchiya T, Sekine K, Hinohara S, Namiki T, Nobori T, Kaneko Y . Analysis of the p16INK4, p14ARF, p15, TP53, and MDM2 genes and their prognostic implications in osteosarcoma and Ewing sarcoma. Cancer Genet Cytogenet 2000; 120: 91–98.
Boehm AK, Neff JR, Squire JA, Bayani J, Nelson M, Bridge JA . Cytogenetic findings in 36 osteosarcoma specimens and a review of the literature. Pediatr Pathol Mol Med 2000; 19: 359–376.
Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994; 4: 1–7.
Guijarro MV, Ghivizzani SC, Gibbs CP . Animal models in osteosarcoma. Front Oncol 2014; 4: 189.
van Boxtel R, Kuiper RV, Toonen PW, van Heesch S, Hermsen R, de Bruin A et al. Homozygous and heterozygous p53 knockout rats develop metastasizing sarcomas with high frequency. Am J Pathol 2011; 179: 1616–1622.
Willis A, Jung EJ, Wakefield T, Chen X . Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004; 23: 2330–2338.
Khromova NV, Kopnin PB, Stepanova EV, Agapova LS, Kopnin BP . p53 hot-spot mutants increase tumor vascularization via ROS-mediated activation of the HIF1/VEGF-A pathway. Cancer Lett 2009; 276: 143–151.
Kogan-Sakin I, Tabach Y, Buganim Y, Molchadsky A, Solomon H, Madar S et al2011. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell Death Differ 2011; 18: 271–281.
Birch JM, Blair V, Kelsey AM, Evans DG, Harris M, Tricker KJ et al. Cancer phenotype correlates with constitutional TP53 genotype in families with the Li-Fraumeni syndrome. Oncogene 1998; 17: 1061–1068.
Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P et al. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res 2003; 63: 6643–6650.
Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 2004; 119: 847–860.
Peitz M, Pfannkuche K, Rajewsky K, Edenhofer F . Ability of the hydrophobic FGF and basic TAT peptides to promote cellular uptake of recombinant Cre recombinase: a tool for efficient genetic engineering of mammalian genomes. Proc Natl Acad Sci USA 2002; 99: 4489–4494.
Munst B, Patsch C, Edenhofer F . Engineering cell-permeable protein. J Vis Exp 2009; 28: pii 1627.
Venhoranta H, Li S, Salamon S, Flisikowska T, Andersson M, Switonski M et al. Non-CpG hypermethylation in placenta of mutation-induced intrauterine growth restricted bovine foetuses. Biochem Biophys Res Commun 2014; 444: 391–394.
Merkl C, Leuchs S, Saalfrank A, Kind A, Schnieke A . RNA interference in pigs: comparison of RNAi test systems and expression vectors. Mol Biotech 2011; 48: 38–48.
Heckel T, Schmucki R, Berrera M, Ringshandl S, Badi L, Steiner G et al. Functional analysis and transcriptional output of the Göttingen minipig genome. BMC Genomics 2015; 16: 932.
Martini M, Gnann A, Scheikl D, Holzmann B, Janssen KP . The candidate tumor suppressor SASH1 interacts with the actin cytoskeleton and stimulates cell-matrix adhesion. Int J Biochem Cell Biol 2011; 43: 1630–1640.
Acknowledgements
We thank Susanne Christan, Vanessa Klein, Lena Glashauser, Melanie Honz, Friedrich Fassler, Thomas Wagner, Astrid Nagel and Birgit Isele-Rüegg for excellent technical assistance and Steffen and Viola Loebnitz and Josef Reim for devoted animal husbandry. This work was supported by the German Research Foundation (DFG) (grant number SCHN 971/3-2) and the German Federal Ministry of Education and Research (BMBF) (grant number 16EX1024D). We are members of COST Action BM1308 'Sharing Advances on Large Animal Models' (SALAAM).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogenesis website
Supplementary information
Rights and permissions
Oncogenesis is an open-access journal published by Nature Publishing Group. This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Saalfrank, A., Janssen, KP., Ravon, M. et al. A porcine model of osteosarcoma. Oncogenesis 5, e210 (2016). https://doi.org/10.1038/oncsis.2016.19
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2016.19
This article is cited by
-
Elevated circulating Hsp70 levels are correlative for malignancies in different mammalian species
Cell Stress and Chaperones (2023)
-
Swine models for translational oncological research: an evolving landscape and regulatory considerations
Mammalian Genome (2022)
-
A novel method for isolation and culture of primary swine gastric epithelial cells
BMC Molecular and Cell Biology (2021)
-
Porcine model elucidates function of p53 isoform in carcinogenesis and reveals novel circTP53 RNA
Oncogene (2021)
-
Porcine pancreatic ductal epithelial cells transformed with KRASG12D and SV40T are tumorigenic
Scientific Reports (2021)
