
Abstract
Transcription factors of the RUNX family (RUNXs), which play pivotal roles in normal development and neoplasia, are regulated by various post-translational modifications. To understand the molecular mechanisms underlying the regulation of RUNXs, we performed a large-scale functional genetic screen of a fly mutant library. The screen identified dPias (the fly ortholog of mammalian PIASs), an E3 ligase for the SUMO (small ubiquitin-like modifier) modification, as a novel genetic modifier of lz (the fly ortholog of mammalian RUNX3). Molecular biological analysis revealed that lz/RUNXs are sumoylated by dPias/PIAS1 at an evolutionarily conserved lysine residue (K372 of lz, K144 of RUNX1, K181 of RUNX2 and K148 of RUNX3). PIAS1-mediated sumoylation inhibited RUNX3 transactivation activity, and this modification was promoted by the AKT1 kinase. Importantly, PIAS1 failed to sumoylate some RUNX1 mutants associated with breast cancer. In nude mice, tumorigenicity was promoted by RUNX3 bearing a mutation in the sumoylation site, but suppressed by wild-type RUNX3. Our results suggest that RUNXs are sumoylated by PIAS1, and that this modification could play a critical role in the regulation of the tumor-suppressive activity of these proteins.
Similar content being viewed by others


Introduction
In mammals, the RUNX family of genes consists of RUNX1/AML1, RUNX2 and RUNX3.1 The Drosophila orthologs of mammalian RUNX family genes are Runt and lozenge (lz) that respectively control body segmentation and eye development.2 All RUNX family members share the central Runt domain, through which the proteins interact with CBFβ and bind to a specific DNA sequence. The RUNX family members play pivotal roles in normal development and neoplasia. RUNX1, which is required for hematopoiesis, is the most frequent target of chromosomal translocations associated with human leukemia.3, 4 RUNX1 is also involved in other types of cancer: for example, mutations in both RUNX1 and CBFB (which encodes CBFβ) are of clinical significance in breast cancer.5, 6 Deletions of RUNX1 in esophageal cancer have also been identified.7 RUNX2 is essential for osteogenesis,8 and Runx2-knockout mice display complete bone loss because of arrested osteoblast maturation.9, 10 RUNX3 is involved in neurogenesis11 and thymopoiesis,12 and also functions as a tumor suppressor. Targeted deletion of Runx3 in mice induces hyperplasia of the gastric epithelium, and loss of RUNX3 expression because of hypermethylation of the RUNX3 promoter region is associated with human gastric cancer.13 Subsequent studies have revealed that inactivation of RUNX3 is associated not only with gastric cancer but also with cancers of the lung, bladder, colon and other organs.13, 14, 15, 16, 17, 18 Paradoxically, expression of RUNX3 is increased in some cancers, including skin cancer,19 head and neck squamous cell carcinoma20 and ovarian cancer.21 RUNXs can be regulated by a multitude of covalent post-translational modifications, including phosphorylation, ubiquitination and acetylation.22 For example, RUNX3 is phosphorylated by various kinases,22 acetylated by p30023 and ubiquitinated by Mdm2 E3 ubiquitin ligase.24, 25
The small ubiquitin-like modifier (SUMO) is covalently linked to a variety of proteins and deconjugated by SUMO-specific proteases.26 In mammals, three SUMO proteins are expressed: SUMO1 (also known as PIC1, UBL1, Sentrin, GMP1 and SMT3C), SUMO2 (also known as SMT3A) and SUMO3 (also known as SMT3B). The sumoylation cycle is remarkably similar to that of ubiquitination. Mature SUMO is activated by the E1 enzyme, conjugated by the E2 enzyme and ligated to its substrate by the E3 ligase. Upon completion of the process, SUMO can be dissociated from the substrate by a deconjugation enzyme and recycled. Drosophila has only one form of E3, dPias (also called Su(var)2-10 or Zimp), that is required for normal blood cell and eye development.27 The PIAS family was originally identified by screening for proteins that interact with signal transducer and activator of transcription.28 Mammals have four genes encoding E3 ligases: PIAS1, PIAS2 (also called PIASxα and β spliced forms), PIAS3 and PIAS4 (also called PIASy). Members of the PIAS family can either activate or repress transactivation activity of target protein, depending on the target gene and interactions with transcriptional regulators.28, 29
Several lines of evidence point to a role for the SUMO modification pathway in tumorigenesis. Sumoylation can regulate the activities of important tumor-suppressor proteins, including p53, retinoblastoma protein (pRB), p63, p73 and murine double minute 2 (Mdm2).30, 31 For example, p53 is modified by SUMO1 at a single site, K386,32 and the sumoylation of p53 promotes apoptosis.33 Consistent with this, PIAS1 is frequently downregulated in multiple epithelial tumor types.34
In this study, we performed a large-scale functional genetic screen of a Drosophila mutant library and identified dPias as a novel lz/RUNX modifier. We also show that dPias/PIAS sumoylates lz/RUNXs at an evolutionarily conserved single lysine residue, and that this modification can regulate the tumor-suppressor activity of RUNXs.
Results
A large-scale genetic-modifier screen identified dPias as a regulator of lz/RUNXs
The fly ortholog of mammalian RUNXs, lz, regulates eye development.35, 36, 37 To identify genetic modifiers for lz/RUNXs, we performed a large-scale functional genetic screen38 using the GenExel fly mutant library (Taejon, Korea).39 Among the candidate mutant lines, G2410 had a P-element insertion in the promoter region of dPias that encodes SUMO E3 ligase. We further confirmed the genetic interaction between dPias and lz using UAS-dPias and UAS-lz. Glass multimer reporter (GMR)-Gal4-driven eye-specific expression of either UAS-lz or UAS-dPias led to a weak rough-eye phenotype (Supplementary Figure S1A; UAS-lz, UAS-dPias). When both lz and dPias were expressed by the same driver, the rough-eye phenotype was markedly exacerbated (Supplementary Figure S1A; UAS-lz+UAS-dPias). However, overexpression of a dPias mutant defective in SUMO E3 ligase activity (UAS-Pias-C388A) did not affect the eye phenotype of lz-overexpressing flies (Supplementary Figure S1B).
Next, we obtained two independent inducible RNA interference (RNAi) alleles of dPias (UAS-dPias-RNAi) that knocked down dPias in the eye when induced by Gal4 drivers (Supplementary Figure S1C). The GMR-driven RNAi-mediated knockdown of dPias led to a severe rough-eye phenotype (Supplementary Figure S1C, left). Notably, GMR-driven overexpression of lz in fly eyes dramatically reduced the severity of the dPias-knockdown phenotype (Supplementary Figure S1C, right). These results suggest that dPias contributes to fly eye development by regulating lz, and that E3 enzyme activity of dPias is essential for this regulation. Reverse transcriptase–PCR analysis confirmed overexpression of dPias and dPias-C388A and knockdown of dPias1 by the heat-shock40–Gal4 driver (Supplementary Figure S1D).
Mammalian PIAS1 sumoylates RUNX family members
We next investigated whether RUNX3 interacts with one or more mammalian PIASs. To this end, we coexpressed Myc-tagged RUNX3 (Myc-RUNX3) with hemagglutinin (HA)-tagged PIAS1, PIAS2α, PIAS2β, PIAS3 or PIAS4 (HA-PIASs) in HEK293 cells and monitored the interactions of these proteins by co-immunoprecipitation (co-IP)35 and immunoblotting (IB). RUNX3 interacted most strongly with PIAS1, but also bound PIAS3 and PIAS4 (Figure 1a).

Mammalian PIAS1 sumoylates RUNX family members. (a) HA-tagged human PIAS1, PIAS2α, PIAS12β, PIAS3 or PIAS4 were coexpressed with Myc-tagged RUNX3 in HEK293 cells. The RUNX3-PIAS interaction was measured by immunoprecipitation35 and IB. (b) HA-PIAS1 or HA-PIAS1-C351A (defective in SUMO E3 ligase activity) was coexpressed with FLAG-SUMO1 and Myc-RUNX3, and RUNX3 sumoylation and the RUNX3-PIAS1 interaction were analyzed by IP and IB. (c) HA-PIAS1 and FLAG-SUMO1 were expressed in HEK293 cells. Sumoylation of endogenous RUNX3 was detected by IP with anti-RUNX3 antibody (5G4) and IB with anti-FLAG antibody. (d) HEK293 cell were transfected with FLAG-SUMO1 and treated with si-PIAS1 (100 nM). Two independent si-PIAS1 RNAs #1 and #2 were used. Sumoylation of endogenous RUNX3 was detected by IP with anti-RUNX3 antibody (5G4) and IB with anti-FLAG antibody. (e) Myc-tagged RUNX1, RUNX2 or RUNX3 were coexpressed with HA-PIAS1 and FLAG-SUMO1 (wild-type (G) or G>A mutant (A)), and sumoylation of the RUNX family members was analyzed by IB. (f) FLAG-tagged SUMO1, SUMO2 and SUMO3 were coexpressed with HA-PIAS1 and Myc-RUNX3. Lysates were analyzed by IB using the indicated antibodies.
To identify the region of RUNX3 essential for the interaction with PIAS1, we coexpressed serial deletion constructs of Myc-RUNX3 with HA-PIAS1 and analyzed their interactions by IP and IB. PIAS1 co-IP with all RUNX3 deletion mutants with the exception of RUNX3-ΔRunt that lacks the Runt domain (Supplementary Figure S2). Thus, RUNX3 interacts with PIAS1 through the Runt domain.
To examine the sumoylation of RUNX3 by PIAS1, we coexpressed FLAG-SUMO1 and Myc-RUNX3 with HA-PIAS1. Coexpression of FLAG-SUMO1 with Myc-RUNX3 resulted in a shift of a small amount of RUNX3 by ∼17 kDa (Figure 1b, indicated by the arrowhead), similar to other sumoylated proteins. The intensity of the faint shifted band was markedly increased by coexpression of HA-PIAS1 (Figure 1b, indicated by arrow). However, HA-PIAS1-C351A, an E3 activity-defective mutant,41 failed to sumoylate RUNX3, even though the mutant PIAS1 could interact with RUNX3 (Figure 1b, RX3/PIAS1). IP with anti-Myc (RUNX3) antibody followed by IB with anti-FLAG (SUMO1) antibody detected sumoylated RUNX3 (Figure 1b).
To determine whether endogenous RUNX3 is sumoylated by PIAS1, we coexpressed HA-PIAS1 and FLAG-SUMO1 in HEK293 cells and measured sumoylated RUNX3 by IP and IB. Endogenous RUNX3 was also sumoylated weakly by expression of SUMO1 alone, and this sumoylation was increased by coexpression of PIAS1 (Figure 1c). Consistent with this, small interfering RNA-mediated knockdown of PIAS1 markedly reduced sumoylation of endogenous RUNX3 (Figure 1d). These results indicate that RUNX3 is post-translationally sumoylated by PIAS1.
We next examined whether other RUNX family members could also be sumoylated by PIAS1. Coexpression of Myc-RUNXs with HA-PIAS1 and FLAG-SUMO1 resulted in band shifts of RUNX1 and RUNX2 as well as RUNX3, suggesting that all three RUNXs are target of PIAS1-mediated sumoylation (Figure 1e).
The sumoylation reaction requires the presence of a C-terminal diglycine18 sequence on the SUMO molecule that is necessary for covalent attachment of SUMO to the target lysine residue. We mutated these C-terminal GG residues to alanines (SUMO1-G>SUMO1-A) and measured the efficiency of sumoylation of RUNXs by the mutant protein. PIAS1 failed to conjugate SUMO1-A to RUNXs, suggesting that SUMO modification of RUNX3 is because of the formation of a covalent bond involving diglycine (Figure 1e).
As noted above, the SUMO family contains three functional members. To gain insight into the specificity of sumoylation of RUNX3, we measured the efficiency of sumoylation of RUNX3 by SUMO1, SUMO2 and SUMO3. All three SUMOs could be conjugated with RUNX3 by PIAS1, and the reaction was most efficient with SUMO1 (Figure 1f).
RUNX3 is sumoylated on lysine 148
To identify the sumoylation site of RUNX3, we tested the PIAS1-mediated sumoylation of truncated RUNX3 derivatives. The results revealed that the sumoylation site resides within amino acids18 1–187 of RUNX3 that contains seven lysine residues (Figure 2a).

RUNX3 is sumoylated on lysine 148 that is evolutionarily conserved. (a) Schematic representation of human RUNX3. Lysine residues around the Runt domain are indicated. To identify the region required for sumoylation, Myc-RUNX3 deletion mutants were coexpressed with HA-PIAS1 and FLAG-SUMO1 in HEK293 cells. RUNX3 sumoylation was analyzed by IB. (b) Each lysine (K) residue of RUNX3 was individually mutated to arginine (R), and each Myc-RUNX3-KR mutant was coexpressed with HA-PIAS1 and FLAG-SUMO1 in HEK293 cells. RUNX3 sumoylation was analyzed by IB. (c) Amino acid sequence around the RUNX3 sumoylation site (K148) was compared among RUNX3 family members. c, chicken; d, Drosophila melanogaster; h, human; m, mouse; r, rat; x, Xenopus laevis; z, zebra fish. The conserved lysine residues used for RUNX3 sumoylation are indicated by shadow. GenBank accession numbers for each protein: human RUNX1, NP_001001890.1; human RUNX2, NP_001265407; human RUNX3, NP_004341.1; mouse, NP_062706.2; rat, NP_569109.1; chicken, XP_001232978, Xenopus: NP_001182313.1; zebrafish, NP_571679.2; Drosophila, AAC47196. (d) Myc-RUNX1-WT and Myc-RUNX1-K144R were coexpressed with HA-PIAS1 and FLAG-SUMO1 in HEK293 cells. RUNX1 sumoylation was analyzed by IB. (e) Myc-RUNX2-WT and Myc-RUNX2-K187R were coexpressed with HA-PIAS1 and FLAG-SUMO1 in HEK293 cells. RUNX2 sumoylation was analyzed by IB. (f) Myc-lz-WT and Myc-lz-372 were coexpressed with HA-dPias and FLAG-dSumo in HEK293 cells. The lz sumoylation was analyzed by IB.
SUMO conjugation often occurs on lysine (K) residues within the general minimal consensus sequence ΨKxD/E (Ψ is a large hydrophobic residue).42 Because this consensus sequence is absent from the amino acid sequence of RUNX3, we individually replaced each of the seven lysine residues in the Runt domain with arginine (R). To test the effect of each mutation on RUNX3 sumoylation, lysates of cells coexpressing each Myc-tagged lysine mutant of RUNX3 with SUMO1 and PIAS1 were subjected to IB. RUNX3 was not sumoylated when K148 was replaced by R, whereas sumoylation of RUNX3 was unaffected by replacement of other lysine residues (Figure 2b), indicating that K148 is a major site for RUNX3 sumoylation.
Sumoylation of RUNX family members by dPias/PIAS is evolutionarily conserved
Sequence comparisons show that the K148 residue and the surrounding amino acid sequence of RUNX3 are evolutionally conserved from flies to humans (Figure 2c). To determine whether other RUNX family members are also modified by sumoylation through this conserved K residue, we mutated corresponding residues K144 of RUNX1 (RUNX1-K144R) and K187 of RUNX2 (RUNX2-K187R). Analysis of the effect on PIAS-mediated sumoylation revealed that these mutations abolished PIAS1-mediated sumoylation of RUNX1 (Figure 2d) and RUNX2 (Figure 2e), suggesting that all three mammalian RUNX family members can be modified by PIAS1-mediated sumoylation at the conserved K residue.
Similarly, Drosophila lz was also sumoylated by dPias (Figure 2f). K372 of lz corresponds to K148 of RUNX3; when this residue was replaced by R (lz-K372R), lz was not sumoylated (Figure 2f). Thus, sumoylation of lz/RUNX by dPias/PIAS at a specific lysine residue is evolutionarily conserved.
PIAS1-mediated sumoylation inhibits transactivation activity of RUNX3
We then asked whether the PIAS1-mediated sumoylation regulates the transcriptional activity of RUNX3. To this end, we transfected HEK293 cells with the reporter plasmid 6xOSE-luciferase that contains six RUNX binding sites and responds to the expression of RUNX family members.43 Expression of RUNX3 increased the reporter activity up to 4.3-fold (Figure 3a). Notably, coexpression of FLAG-SUMO1 or HA-PIAS1 inhibited RUNX3-mediated reporter activity, and coexpression of both FLAG-SUMO1 and HA-PIAS1 nearly abolished activity (Figure 3a). Expression of RUNX3 with a mutation in the sumoylation site (RUNX3-K148R) also increased reporter activity as strongly as wild-type RUNX3, but this increase was not inhibited by the expression of FLAG-SUMO1 and HA-PIAS1 (Figure 3b).

PIAS1-mediated sumoylation inhibits transactivation activity of RUNX3. (a) Myc-RUNX3, FLAG-SUMO1 and HA-PIAS1 were transfected into HEK293 cells as indicated. The reporter activity of 6xOSE promoter luciferase was measured with a luminometer and normalized to that of pCMV-β-galactosidase, which was included as an internal control. *P<0.05 by Student’s t-test. (b) Myc-RUNX3-WT or Myc-RUNX3-K148R were co-transfected with FLAG-SUMO1 and HA-PIAS1 into HEK293 cells as indicated. The reporter activity of 6xOSE promoter luciferase was measured and normalized to that of pCMV-β-galactosidase. *P<0.05. (c) Myc-RUNX3 was co-transfected with FLAG-SUMO1 and HA-PIAS1 into HEK293 cells as indicated. The reporter activity of p21-promoter-luciferase (Chi et al.25) was measured and normalized to that of pCMV-β-galactosidase. *P<0.05. (d) Myc-RUNX3 (WT) or Myc-RUNX3-K148R (KR) was co-transfected with FLAG-SUMO1 and HA-PIAS1 into HEK293 cells as indicated. The levels of the expressed proteins and endogenous p21 were measured by IB. (e) Myc-RUNX3 (red) or Myc-RUNX3-K148R (red) was expressed with or without HA-PIAS1 (green) in HeLa cells, and the subcellular localization of each protein was visualized by immunostaining. Nuclei were stained with DAPI. (f) Myc-RUNX3-WT and Myc-RUNX3-K148R were coexpressed with increasing amounts of FLAG-CBFβ2 in HEK293 cells. The RUNX3-CBFβ2 interaction was analyzed by IP and IB. (g) Fixed amounts of Myc-RUNX3-WT, HA-PIAS1 and FLAG-SUMO1 were coexpressed with increasing amounts of HA-CBFβ2 in HEK293 cells. RUNX3 sumoylation was analyzed by IP and IB.
We have shown that the on/off switch governing p21Waf/Cip (p21) expression is tightly regulated and that RUNX3 is involved in both turning on and turning off p21 expression.17 To determine the effect of PIAS1-mediated RUNX3 sumoylation on p21 expression, we transfected HEK293 cells with a reporter plasmid expressing p21-promoter-luciferase.25 Expression of RUNX3 increased the reporter activity by up to fourfold, and coexpression of FLAG-SUMO1 and HA-PIAS1 inhibited RUNX3-mediated reporter activity (Figure 3c).
Next, we expressed Myc-RUNX3 or Myc-RUNX3-K148R with FLAG-SUMO1 and HA-PIAS1 in HEK293 cells and measured the expression levels of endogenous p21. Expression of RUNX3 with FLAG-SUMO1 and HA-PIAS1 decreased the p21 level, but Myc-RUNX3-K148R did not (Figure 3d). Together, these results suggest that PIAS1-mediated sumoylation inhibits the transactivation activity of RUNX3.
PIAS1-mediated sumoylation is not involved in nuclear localization of RUNX3
Sumoylation often contributes to the subcellular localization of transcriptional regulators. For example, the transcriptional corepressor CtBP is normally nuclear, but mutation of its single SUMO modification site results in cytoplasmic localization.44 To investigate whether SUMO modification contributes to the regulation of subcellular localization of RUNX3, we transfected HeLa cells with Myc-RUNX3, HA-PIAS1 or both constructs. Immunofluorescence analysis revealed that both RUNX3 and PIAS1 were localized to the nucleus (Figure 3e, top panels), and the nuclear localization of RUNX3 was not altered by coexpression of PIAS1 (Figure 3e, middle panels). RUNX3 with a mutation in the sumoylation site (Myc-RUNX3-K148R) was also localized to the nucleus (Figure 3e, bottom panels). These results suggest that PIAS1-mediated sumoylation is not involved in nuclear localization of RUNX3.
CBFβ2 does not interfere with PIAS1-mediated RUNX3 sumoylation
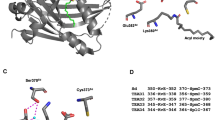
Because the sumoylation site of RUNX3 is located within the Runt domain through which RUNXs heterodimerize with CBFβ, we asked whether the K148 residue is involved in this interaction. The crystal structure of Runt domain/CBFβ/DNA45 shows that the K148 residue is exposed on the surface, suggesting that it is not involved in the RUNX3-CBFβ heterodimerization (Supplementary Figure S3A). To test this idea, we expressed Myc-RUNX3-WT or Myc-RUNX3-K148R with or without CBFβ2. IP and IB analysis revealed that RUNX3-K148R could effectively interact with CBFβ2 (Figure 3f).
We also analyzed whether CBFβ2 interferes with RUNX3 sumoylation. Expression of CBFβ2 increased the RUNX3 level and PIAS1-mediated RUNX3 sumoylation (Figure 3g). The level of sumoylated RUNX3 was proportional to the level of RUNX3 (Figure 3g). Our result suggests that CBFβ2 does not interfere with PIAS1-mediated RUNX3 sumoylation.
AKT1 stimulates RUNX3 sumoylation
To investigate how PIAS1-mediated RUNX3 sumoylation is regulated, we transfected HEK293 with Myc-RUNX3, HA-PIAS1 and SUMO1. The cells were synchronized by serum starvation, and then treated with serum, epidermal growth factor, fibroblast growth factor or transforming growth factor-β1 for 3 h. IB analysis revealed that serum, epidermal growth factor or fibroblast growth factor increased RUNX3 sumoylation, whereas transforming growth factor-β1 did not, suggesting that mitogenic signals stimulate RUNX3 sumoylation (Figure 4a).

AKT1 stimulates RUNX3 sumoylation. (a) HEK293 cell were transfected with Myc-RUNX3, HA-PIAS1 and FLAG-SUMO1, and then serum starved for 24 h. Cells were then treated with serum, epidermal growth factor (EGF; 1 μg/ml), fibroblast growth factor (FGF; 30 ng/ml) or transforming growth factor-β1 (TGFβ1; 5 nM) for 3 h. RUNX3 sumoylation was analyzed by IB. (b) Serially increasing levels of FLAG-AKT1 were coexpressed with Myc-RUNX3, HA-PIAS1 and FLAG-SUMO1 in HEK293 cells, and RUNX3 sumoylation was analyzed by IB. (c) HEK293 cells were transfected with HA-PIAS1 and FLAG-SUMO1 and treated with si-AKT1 (100 nM). Two independent si-AKT1 RNAs (#1 and #2) were used. Sumoylation of endogenous RUNX3 was detected by IP with anti-RUNX3 antibody (5G4) and IB with anti-FLAG antibody. (d) Amino-acid sequence around the RUNX3 sumoylation site (K148) was compared with that of RUNX2. The AKT1 phosphorylation sites of RUNX249 are indicated by arrows. The phosphorylation sites were mutated in RUNX3 to obtain RUNX3-T151,153A and RUNX3-T151,153E. RUNX3-WT, RUNX3-T151,153A or RUNX3-T151,153E was coexpressed with HA-PIAS1 and FLAG-SUMO1 in HEK293 cells, and RUNX3 sumoylation was analyzed by IB. (e) RUNX3-WT, RUNX3-T151,153A or RUNX3-T151,153E was coexpressed with FLAG-CBFβ2 in HEK293 cells, and the RUNX3-CBFβ2 interaction was analyzed by IP and IB.
We then examined the effect of ERK1/2 and AKT1 on RUNX3 sumoylation. Expression of ERK1 decreased RUNX3 sumoylation, whereas expression of ERK2 had no effect (Supplementary Figure S4A). In contrast, expression of FLAG-AKT1 increased RUNX3 sumoylation in a dose-dependent manner (Figure 4b). To understand the involvement of kinase activity of ATK1 in the PIAS1-mediated RUNX3 sumoylation, we expressed wild-type AKT1 (FLAG-AKT1-WT) or a kinase-defective AKT1 mutant (FLAG-AKT1-KD) with HA-PIAS1, FLAG-SUMO1 and RUNX3 and measured RUNX3 sumoylation. The result revealed that the kinase activity of AKT1 is required for the increase in RUNX3 sumoylation (Supplementary Figure S4B). Similarly, RNAi-mediated knockdown of AKT1 decreased PIAS1-mediated sumoylation of endogenous RUNX3 (Figure 4c). These results suggest that the AKT1 pathway stimulates RUNX3 sumoylation.
Because RUNX2 is phosphorylated by AKT1 at Ser/Thr residues46 that are evolutionarily conserved among the RUNX family members and are close to the sumoylation site (Figure 4d), we investigated whether AKT1 physically interacts with RUNX3 and stimulates PIAS1-mediated RUNX3 sumoylation through phosphorylation. Coexpression of AKT1 and serially deleted RUNX3, followed by IP and IB analysis, revealed that AKT1 interacts with RUNX3 via the Runt domain (Supplementary Figure S4C). We then mutated the putative AKT1 phosphorylation sites (Thr151 and Thr153) of RUNX3 to Ala or Glu and examined whether the mutated proteins could heterodimerize with CBFβ2 and become sumoylated. Mutations of Thr151 and Thr153 (to either Ala or Glu) markedly reduced both heterodimerization with CBFβ2 and PIAS1-mediated sumoylation relative to that of wild-type RUNX3 (Figures 4d and e). Unexpectedly, phosphomimicking mutations (Thr151 and Thr153 to Glu) failed to enhance PIAS1-mediated sumoylation. It is unclear why the phosphomimicking mutant failed to enhance sumoylation. We speculate that AKT1 may phosphorylate additional Ser/Thr residues and phosphomimicking mutations on Thr151 and Thr153 are not enough for enhancing sumoylation. Take together, these results suggest that AKT1 stimulates PIAS1-mediated RUNX3 sumoylation by phosphorylating RUNX3.
Sumoylation could be associated with the regulation of tumor-suppressor activity of RUNX family members
RUNX1 plays important roles in hematopoiesis, and its functional dysregulation leads to leukemia. Specific mutations of RUNX1 (R139Q, G141E and R142K) are frequently found in breast cancer.5, 6 Because these mutation sites are near the sumoylation site of RUNX1 (K144), we asked whether these mutations affect RUNX1 sumoylation. The R142K mutation did not affect RUNX1 sumoylation; however, the R139Q and G141E mutations markedly reduced RUNX1 sumoylation (Figure 5a). The R142K mutation, which does not affect RUNX1 sumoylation (Figure 5a), is known to abrogate heterodimerization with CBFβ.47 Therefore, the three mutants might have lost their tumor-suppressor activity for different reasons: namely, impaired SUMOylation (RUNX1-R139Q and RUNX1-G141E) or impaired heterodimerization with CBFβ (RUNX1-R142K).

Sumoylation regulates tumor-suppressor activity of RUNX family members. (a) Amino acid sequence around the RUNX3 sumoylation site (K148) was compared with that of RUNX1. The sites of RUNX1 mutations identified in breast cancers (R139Q, G141E and R142K) are indicated by arrows. Each of the mutated RUNX1 cDNAs was coexpressed with HA-PIAS1 and FLAG-SUMO1, and RUNX1 sumoylation was analyzed by IB. (b) Myc-RUNX3-WT and Myc-RUNX3-K148R were stably transfected into MKN28 gastric cancer cells that do not express RUNX3. Expression of the transfected genes was detected by IB with anti-RUNX3 antibody (5G4). (c) Tumorigenicity of MKN28 cells containing vector only (vector) or expressing RUNX3 or RUNX3-K148R were examined in nude mice. Cells were suspended in 0.85% phosphate-buffered saline (5 × 107 cells/ml) and 0.3 ml of cell suspension was injected subcutaneously. Tumor volumes were measured at the indicated times after injection, and average volumes and s.d. are indicated; n=5 for each group. (d) Tumors obtained at the end of the experiment shown in (c). One mouse injected with RUNX3-WT did not bear detectable tumors. (e) The weights of the tumors were measured at the end of the experiment, and the average weights are shown in the bar diagram. The P-values were calculated by Student’s t-test.
To understand the role of sumoylation in the tumor-suppressor activity of RUNX3, we established derivatives of the MKN28 gastric cancer cell line stably expressing wild-type RUNX3 (RUNX3-WT) or RUNX3-K148R (Figure 5b). MKN28 cells overexpressing RUNX3-WT grew more slowly than the control MKN28 cells (that is, MKN28 cells stably transfected with empty vector). However, expression of RUNX3-K148R did not significantly affect the growth of the cells (data not shown). We previously showed that MKN28 cells form rapidly growing tumors, and that tumor growth can be reduced by stable expression of exogenous RUNX3-WT.13 Therefore, we subjected cells expressing RUNX3-K148R to a tumorigenesis assay in nude mice. When inoculated into nude mice, RUNX3-WT-expressing MKN28 tumors grew more slowly than tumors derived from control MKN28 cells. In contrast, RUNX3-K148R-expressing MKN28 tumors grew faster than those of control cells (Figure 5c). At 25 days after injection, the tumors were removed from the mice and individually weighed. Whereas RUNX3-WT tumors weighed ∼75% as much as control tumors (25% reduction), tumors originating from RUNX3-K148R cells weighed 436% as much as the vector controls (Figures 5d and e). Thus, tumor growth was markedly promoted by RUNX3-K148R, but suppressed by RUNX3-WT.
Discussion
In this study, we identified dPias/PIAS1 as a novel lz/RUNX-modifying enzyme in a fly mutant library screen. Furthermore, we showed that dPias/PIAS1 sumoylates lz/RUNXs at K144 of RUNX1, K187 of RUNX2, K148 of RUNX3 and K372 of lz. Sumoylation of all the tested RUNX family members and the sequence conservation around the sumoylation sites suggest that dPias/PIAS1-mediated RUNX sumoylation is evolutionarily conserved from flies to humans.
SUMO modification of transcription factors can lead to transcriptional activation but is more often associated with transcriptional repression.48 We found that SUMO1 modification inhibits transactivation activity of RUNX3: overexpression of SUMO1 and PIAS1 led to decreased RUNX3-dependent transcriptional activity. This decrease was dependent on SUMO1 modification of RUNX3, as SUMO1/PIAS1 overexpression did not influence the transcriptional activity of RUNX3-K148R. Two general models have been proposed concerning the mechanisms underlying the functional alteration of proteins by SUMO. The first and most obvious idea is that a SUMO-protein conjugate could acquire altered affinity for a particular ligand.49 In the second model, SUMO ligation to a substrate blocks attachment of ubiquitin (or another ubiquitin-like protein) to the same substrate, possibly by competing for the same lysine residue.40 Notably in this regard, K148 of RUNX3 can also be modified by MDM2-mediated ubiquitination.25 Because the physiological significance of protein sumoylation is not fully understood, further study will be required to reveal how SUMO-1 modification inhibits the transcriptional activity of RUNX3.
Whereas dPias and lz positively cooperated in inducing a rough-eye phenotype in Drosophila, the transactivation activity of RUNX3 was inhibited by PIAS1 in mammalian cells. The results from Drosophila and mammalian cells are discordant in this respect. However, genetic experiments in Drosophila revealed that runt domain transcription factors function both to activate and to repress transcription of different downstream target genes.50 In addition, an appropriate level of lz activity is important for the development of an appropriate type of sense organ; for instance, both an increased level and a decreased level of lz activity can lead to a rough-eye phenotype (Gupta et al.51). Therefore, the results of the fly genetic analysis that we used in this study may suggest that overexpression of dPias positively cooperates in inducing a rough-eye phenotype by enhancing the repressor activity of lz.
In most situations, the proportion of a particular transcription factor that is SUMO modified at steady state is small. Similarly, only a small fraction of RUNX3 is sumoylated.26 Nevertheless, RUNX3 was maximally repressed by SUMO-1 and PIAS1, and mutation of the sumoylation site fully relieved this repression, as with other sumoylated transcriptions factors. To explain this phenomenon, it has been proposed that a transcription factor can be sumoylated and incorporated into a repression complex, after which the SUMO is deconjugated but the transcription factor remains within the repression complex.42 We speculate that the transactivation activity of RUNX3 might be inhibited through the same mechanism.
Protein functions are often regulated by interplay between different modifications. For example, some proteins are subject to phosphorylation-dependent sumoylation, such as heat-shock factors, GATA-1 and myocyte enhancer factor 2 (MEF2), all of which have the ΨKxExxSP motif.52 Within this motif, serine phosphorylation may contribute to sumoylation of lysine. Although RUNX3 lacks the ΨKxE motif, it has multiple Ser/Thr residues followed by sumoylation that are phosphorylated by AKT1 (Figure 4d). We found that growth factor (epidermal growth factor and fibroblast growth factor) stimulation or overexpression of AKT1 increased RUNX3 sumoylation, whereas mutations of the AKT1 phosphorylation site of RUNX3 decreased RUNX3 sumoylation. These results suggest that growth factor-stimulated AKT1 promotes RUNX3 sumoylation.
Inactivation of RUNX family members has been observed frequently in multiple tumor types. For example, RUNX1 is frequently mutated in leukemia and breast cancer, and RUNX3 expression is frequently inactivated by DNA hypermethylation in various cancers including gastric cancer.22, 53, 54 Furthermore, some RUNX1 mutants found in breast cancer are not sumoylated by PIAS1, implying that sumoylation is associated with tumor-suppressor activity. Notably in this regard, our tumorigenesis assay revealed that a sumoylation-defective mutant of RUNX3 (RUNX3-K148R) lacks tumor-suppressor activity: ectopic expression of RUNX3-K148R in the MKN28 gastric cancer cell line promoted tumor growth in nude mice. This result suggests that SUMO1 conjugation may play a role in regulation of the tumor-suppressor activity of RUNXs.
Unexpectedly, the RUNX3-K148R mutant did not just lose its tumor-suppressor activity, but it also gained very strong oncogenic activity. Although we do not understand the reason, we have previously reported a similar phenomenon with a RUNX3-R122C mutant identified in a gastric cancer patient; overexpression of the RUNX3-R122C mutant markedly increased tumorigenecity of the MKN28 gastric cancer cell line (Li et al.13). We speculate that RUNX3 may suppress some, as yet unidentified, survival factors and induce pro-apoptotic factors. Further analysis is required to identify the survival factors repressed by sumoylated RUNX3.
Although the functions of RUNXs in tumorigenesis have been extensively studied,55 regulation of their tumor-suppressor activities is not yet fully understood. Our results suggest that sumoylation might be one of the mechanisms involved in the regulation of RUNX tumor-suppressor activity. This interpretation is supported by the observations that epidermal growth factor receptor signaling is controlled by negative feedback regulation and that failure of this negative regulation is associated with diverse tumor types.34 In this context, it is worth mentioning that both RUNX3 and PIAS1 are involved in this negative regulation.34 Therefore, it is likely that epidermal growth factor/AKT1 pathway-triggered RUNX3 sumoylation, which leads to inhibition of RUNX3 transcriptional activity, might be associated with negative feedback regulation. Although our results suggest that sumoylation at K148 could regulate the tumor-suppressor activity of RUNX3, K148 is also modified by acetylation and ubiquitination,23 and hence it is possible that other types of modification at the site could also be involved in regulation.
In this study, we showed that RUNX family members are sumoylated by PIAS1 isoforms, and demonstrated that this modification could control the tumor-suppressor activity of RUNXs. Our results reveal a previously unknown RUNX modification that is critical for the regulation of RUNX activities.
Materials and methods
Fly strains and crosses
The pUAST-RUNX3 plasmid was constructed by subcloning the entire coding region of RUNX3 (Q13761-1) into pUAST, and then used to generate RUNX3-overexpressing transgenic flies (UAS-RX3). The lz3 fly line, various Gal4 lines such as heat-shock40- and GMR-Gal4 and all other UAS lines were obtained from Bloomington Stock Center (Bloomington, IN, USA). The fly lines v29448, v31625 and v32956 were obtained from Vienna Drosophila RNAi Center (Vienna, Austria). The EP GenExel library used in the screening was obtained from the BioMedical Research Center of the Korea Advanced Institute of Science and Technology (Taejon, Korea). UAS-lz fly was kindly provided by U Banerjee (University of California, Los Angeles, CA, USA). Drosophila stocks were maintained and cultured using standard cornmeal-yeast-agar medium at 25 °C.
A fly mutant library screening
Using eye-specific GMR-driven UAS-RUNX3 transgenic flies (GMR>RUNX3) that exhibit a rough-eye phenotype, ∼20 000 independent fly lines from the GenExel fly mutant library were screened,39 each of which possesses an EP element randomly inserted into its genome via mobilization of a transposable element. Double-mutant flies were obtained by crossbreeding each of the mutant flies with GMR>RUNX3 flies, and the eye phenotypes of the progeny flies were analyzed. Fly lines exhibiting increased or reduced expression of the rough-eye phenotype were chosen and crossed again with GMR>UAS-lz flies, and ultimately candidate EP lines were selected.
PCR primers, small interfering RNAs and antibodies
Primers used in Reverse transcriptase–PCR were as follows: dPias: 5′-ATGGTGCAGATGCTTCGAGTGGTC-3′ and 5′-CAGGCAAAAGCGCAGTTGAACCTG-3′; rp49: 5′-CACCAGTCGGATCGATATGC-3′ and 5′-CACGTTGTGCACCAGGAACT-3′. AKT1-specific or PIAS1-specific small interfering RNAs were purchased from Bioneer Co. (Daejeon, Korea): AKT1 #1 sense, 5′-GACAACCGCCAUCCAGACU-3′; AKT1 #1 antisense, 5′-AGUCUGGAUGGCGGUUGU C-3′; AKT1 #2 sense, 5′-CCUUUUCGACGCUUAACCU-3′; AKT1 #2 antisense, 5′-AGGUUAAGCGUCGAAAAGG-3′; PIAS1 #1 sense, 5′-AAGGUCAUUCUAGAGCUUUA-3′; PIAS1 #1, antisense 5′-UAAAGCUCUAGAAUGACCUU-3′; PIAS1 #2 sense, 5′-CGAAUGAACUUGGCAGAAA-3′; PIAS1 #2 antisense, 5′-UUUCUGCCCAAGUUCAUUCG-3′). Anti-Myc polyclonal (A-14), anti-Myc (9E10) monoclonal and anti-RUNX3 polyclonal (H-50) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-RUNX3 monoclonal (5G4) antibody was purchased from Abcam (Cambridge, UK). The anti-FLAG (M2), anti-AKT1, anti-PIAS1 and anti-tubulin antibodies were purchased from Sigma (St Louis, MO, USA). The Alexa Fluor 488 anti-mouse and Alexa Fluor 594 anti-rabbit antibodies were purchased from Molecular Probes (Invitrogen, Carlsbad, CA, USA). DAPI (4′,6-diamidino-2-phenylindole) was purchased from Sigma.
Plasmid constructs
The vectors expressing full-length RUNX3 (Myc-RUNX3) and RUNX deletion mutants fused to the Myc-epitope tag were constructed using the cytomegalovirus promoter-derived mammalian expression plasmid pCS4–3Myc. The cDNAs encoding human PIAS and SUMO isoforms were generated by PCR and then subcloned into the pCS4 vector to generate constructs for expression of HA-tagged PIAS1, PIAS2a, PIAS2b, PIAS3, PIAS4 and FLAG-tagged SUMO1, SUMO2 and SUMO3. Drosophila Pias (NM_001201959) and Drosophila Sumo (NM_058063.4) cDNAs were generated by PCR and then subcloned into the pCS4 vector to generate HA-dPias and FLAG-dSumo. Plasmids for expression of point mutants of lz, RUNX family members, dPias and PIAS1 were made by site-directed mutagenesis, and mutations were confirmed by DNA sequencing.
Cell culture and DNA transfection
HEK293 and HeLa cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA), and MKN28 cells were maintained in RPMI-1640 medium with the same supplements. Transient transfections were performed using Lipofectamine Plus reagent (Invitrogen) according to the manufacturer’s instructions.
Co-immunoprecipitations
Cells were lysed in 20 mM Tris-HCl (pH 7.4), 5 mM EDTA, 10 mM Na2P2O7, 10 mM NaF, 2 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 1% Nonidet P-40 and 1 × protease inhibitor mixture (Roche Applied Science, Basel, Switzerland). Cell lysates were incubated with the appropriate primary antibody for 2 h at 4 °C, and then with protein A/G-Sepharose for the next hour. Bound proteins were eluted by boiling, and then subjected to SDS–polyacrylamide gel electrophoresis followed by immunoblot analysis.
Immunoblot analysis
HEK293 cells were transiently transfected with the indicated expression plasmids, and cell extracts were prepared 24 h post transfection. Extracts were separated by SDS–polyacrylamide gel electrophoresis, and proteins were transferred electrophoretically to nitrocellulose membranes. Membranes were probed with the appropriate polyclonal or monoclonal primary antibody, followed by horseradish peroxidase-conjugated anti-rabbit, anti-mouse or anti-goat immunoglobulin G. Immunoreactive proteins were visualized by chemiluminescence according to the manufacturer’s instructions (Pierce, Rockford, IL, USA). Images of the immunoblots were acquired using an LAS-3000 mini imaging system (Fujifilm, Tokyo, Japan).
Immunofluorescence staining
HeLa cells were grown on 22 mm cover slips (Fisher, Waltham, MA, USA) and then transfected with the indicated expression vectors. Cells were washed with phosphate-buffered saline, and then fixed in a solution of 4% formaldehyde for 15 min at room temperature. Cells were incubated for 45 min in a solution of 10% fetal bovine serum in phosphate-buffered saline containing 0.1% Triton X-100, followed by incubation overnight at 25 °C with the indicated primary antibody, and then for 1 h at 25 °C with Alexa Fluor 488 anti-mouse or Alexa 594 anti-rabbit antibody. The cells were then stained with DAPI for 7 min. Cells were examined with a Leica TCS-NT laser scanning confocal microscope (Leica, Wetzlar, Germany) using an oil-immersion objective lens and the appropriate emission filters, settings and controls to exclude bleed-through effects.
Nude mouse xenograft model
Four- to five-week-old male athymic nude (nu/nu) mice were maintained at Chungbuk National University (Cheongju, South Korea) in accordance with the guidelines of the Chungbuk Animal Resource Center. MKN28 stable cell clones were suspended in sterile phosphate-buffered saline at a concentration of 2 × 106/200 μl and injected subcutaneously into the flanks of individual nude mice. After transplantation, tumor size was measured using calipers, and tumor volume was estimated according to the following formula: tumor volume (mm3)=L × W2/2, where L is the length and W is the width.
References
van Wijnen AJ, Stein GS, Gergen JP, Groner Y, Hiebert SW, Ito Y et al. Nomenclature for Runt-related (RUNX) proteins. Oncogene 2004; 23: 4209–4210.
Canon J, Banerjee U . Runt and Lozenge function in Drosophila development. Semin Cell Dev Biol 2000; 11: 327–336.
Look AT . E2A-HLF chimeric transcription factors in pro-B cell acute lymphoblastic leukemia. Curr Top Microbiol Immunol 1997; 220: 45–53.
Speck NA, Gilliland DG . Core-binding factors in haematopoiesis and leukaemia. Nat Rev Cancer 2002; 2: 502–513.
Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012; 486: 405–409.
Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 2012; 486: 353–360.
Dulak AM, Schumacher SE, van Lieshout J, Imamura Y, Fox C, Shim B et al. Gastrointestinal adenocarcinomas of the esophagus, stomach, and colon exhibit distinct patterns of genome instability and oncogenesis. Cancer Res 2012; 72: 4383–4393.
Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G . Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell 1997; 89: 747–754.
Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997; 89: 755–764.
Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 1997; 89: 765–771.
Inoue K, Ozaki S, Shiga T, Ito K, Masuda T, Okado N et al. Runx3 controls the axonal projection of proprioceptive dorsal root ganglion neurons. Nat Neurosci 2002; 5: 946–954.
Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T et al. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell 2002; 111: 621–633.
Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 2002; 109: 113–124.
Ito K, Lim AC, Salto-Tellez M, Motoda L, Osato M, Chuang LS et al. RUNX3 attenuates beta-catenin/T cell factors in intestinal tumorigenesis. Cancer Cell 2008; 14: 226–237.
Kim WJ, Kim EJ, Jeong P, Quan C, Kim J, Li QL et al. RUNX3 inactivation by point mutations and aberrant DNA methylation in bladder tumors. Cancer Res 2005; 65: 9347–9354.
Lee KS, Lee YS, Lee JM, Ito K, Cinghu S, Kim JH et al. Runx3 is required for the differentiation of lung epithelial cells and suppression of lung cancer. Oncogene 2010; 29: 3349–3361.
Lee YS, Lee JW, Jang JW, Chi XZ, Kim JH, Li YH et al. Runx3 inactivation is a crucial early event in the development of lung adenocarcinoma. Cancer Cell 2013; 24: 603–616.
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 2006; 38: 787–793.
Salto-Tellez M, Peh BK, Ito K, Tan SH, Chong PY, Han HC et al. RUNX3 protein is overexpressed in human basal cell carcinomas. Oncogene 2006; 25: 7646–7649.
Tsunematsu T, Kudo Y, Iizuka S, Ogawa I, Fujita T, Kurihara H et al. RUNX3 has an oncogenic role in head and neck cancer. PLoS ONE 2009; 4: e5892.
Lee CW, Chuang LS, Kimura S, Lai SK, Ong CW, Yan B et al. RUNX3 functions as an oncogene in ovarian cancer. Gynecol Oncol 2011; 122: 410–417.
Bae SC, Lee YH . Phosphorylation, acetylation and ubiquitination: the molecular basis of RUNX regulation. Gene 2006; 366: 58–66.
Jin YH, Jeon EJ, Li QL, Lee YH, Choi JK, Kim WJ et al. Transforming growth factor-beta stimulates p300-dependent RUNX3 acetylation, which inhibits ubiquitination-mediated degradation. J Biol Chem 2004; 279: 29409–29417.
Kim JH, Choi JK, Cinghu S, Jang JW, Lee YS, Li YH et al. Jab1/CSN5 induces the cytoplasmic localization and degradation of RUNX3. J Cell Biochem 2009; 107: 557–565.
Chi XZ, Kim J, Lee YH, Lee JW, Lee KS, Wee H et al. Runt-related transcription factor RUNX3 is a target of MDM2-mediated ubiquitination. Cancer Res 2009; 69: 8111–8119.
Hay RT . SUMO: a history of modification. Mol Cell 2005; 18: 1–12.
Betz A, Lampen N, Martinek S, Young MW, Darnell JE Jr . A Drosophila PIAS homologue negatively regulates stat92E. Proc Natl Acad Sci USA 2001; 98: 9563–9568.
Shuai K, Liu B . Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat Rev Immunol 2005; 5: 593–605.
Schmidt D, Muller S . PIAS/SUMO: new partners in transcriptional regulation. Cell Mol Life Sci 2003; 60: 2561–2574.
Seeler JS, Bischof O, Nacerddine K, Dejean A . SUMO the three Rs and cancer. Curr Top Microbiol Immunol 2007; 313: 49–71.
Kim KI, Baek SH . SUMOylation code in cancer development and metastasis. Mol Cell 2006; 22: 247–253.
Rodriguez MS, Desterro JM, Lain S, Midgley CA, Lane DP, Hay RT . SUMO-1 modification activates the transcriptional response of p53. EMBO J 1999; 18: 6455–6461.
Muller S, Berger M, Lehembre F, Seeler JS, Haupt Y, Dejean A . c-Jun and p53 activity is modulated by SUMO-1 modification. J Biol Chem 2000; 275: 13321–13329.
Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F et al. A module of negative feedback regulators defines growth factor signaling. Nat Genet 2007; 39: 503–512.
Crew JR, Batterham P, Pollock JA . Developing compound eye in lozenge mutants of Drosophila: lozenge expression in the R7 equivalence group. Dev Gene Evol 1997; 206: 481–493.
Daga A, Karlovich CA, Dumstrei K, Banerjee U . Patterning of cells in the Drosophila eye by Lozenge, which shares homologous domains with AML1. Genes Dev 1996; 10: 1194–1205.
Flores GV, Daga A, Kalhor HR, Banerjee U . Lozenge is expressed in pluripotent precursor cells and patterns multiple cell types in the Drosophila eye through the control of cell-specific transcription factors. Development 1998; 125: 3681–3687.
St Johnston D . The art and design of genetic screens: Drosophila melanogaster. Nat Rev Genet 2002; 3: 176–188.
Lee JH, Koh H, Kim M, Kim Y, Lee SY, Karess RE et al. Energy-dependent regulation of cell structure by AMP-activated protein kinase. Nature 2007; 447: 1017–1020.
Buschmann T, Fuchs SY, Lee CG, Pan ZQ, Ronai Z . SUMO-1 modification of Mdm2 prevents its self-ubiquitination and increases Mdm2 ability to ubiquitinate p53. Cell 2000; 101: 753–762.
Kahyo T, Nishida T, Yasuda H . Involvement of PIAS1 in the sumoylation of tumor suppressor p53. Mol Cell 2001; 8: 713–718.
Johnson ES . Protein modification by SUMO. Annu Rev Biochem 2004; 73: 355–382.
Ducy P, Karsenty G . Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol 1995; 15: 1858–1869.
Lin X, Sun B, Liang M, Liang YY, Gast A, Hildebrand J et al. Opposed regulation of corepressor CtBP by SUMOylation and PDZ binding. Mol Cell 2003; 11: 1389–1396.
Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K et al. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFbeta. Cell 2001; 104: 755–767.
Fujita T, Azuma Y, Fukuyama R, Hattori Y, Yoshida C, Koida M et al. Runx2 induces osteoblast and chondrocyte differentiation and enhances their migration by coupling with PI3K-Akt signaling. J Cell Biol 2004; 166: 85–95.
Osato M . Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene 2004; 23: 4284–4296.
Gill G . SUMO and ubiquitin in the nucleus: different functions, similar mechanisms? Genes Dev 2004; 18: 2046–2059.
Melchior F . SUMO—nonclassical ubiquitin. Annu Rev Cell Dev Biol 2000; 16: 591–626.
Wheeler JC, Shigesada K, Gergen JP, Ito Y . Mechanisms of transcriptional regulation by Runt domain proteins. Semin Cell Dev Biol 2000; 11: 369–375.
Gupta BP, Flores GV, Banerjee U, Rodrigues V . Patterning an epidermal field: Drosophila lozenge, a member of the AML-1/Runt family of transcription factors, specifies olfactory sense organ type in a dose-dependent manner. Dev Biol 1998; 203: 400–411.
Hietakangas V, Anckar J, Blomster HA, Fujimoto M, Palvimo JJ, Nakai A et al. PDSM, a motif for phosphorylation-dependent SUMO modification. Proc Natl Acad Sci USA 2006; 103: 45–50.
Ito Y . Oncogenic potential of the RUNX gene family: ‘overview’. Oncogene 2004; 23: 4198–4208.
Chuang LS, Ito Y . RUNX3 is multifunctional in carcinogenesis of multiple solid tumors. Oncogene 2010; 29: 2605–2615.
Blyth K, Cameron ER, Neil JC . The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer 2005; 5: 376–387.
Acknowledgements
S-C Bae was supported by the research grant of Chungbuk National University in 2012. IIY Park was supported by the Engineering Foundation and Cooperative Research Program for Agriculture Science & Technology Development of Korea (PJ009588).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogenesis website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 3.0 Unported License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Kim, JH., Jang, JW., Lee, YS. et al. RUNX family members are covalently modified and regulated by PIAS1-mediated sumoylation. Oncogenesis 3, e101 (2014). https://doi.org/10.1038/oncsis.2014.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2014.15
Keywords
This article is cited by
-
PIAS1 impedes vascular endothelial injury and atherosclerotic plaque formation in diabetes by blocking the RUNX3/TSP-1 axis
Human Cell (2023)
-
Hepatoprotective effects of sevoflurane against hepatic ischemia-reperfusion injury by regulating microRNA-124-3p-mediated TRAF3/CREB axis
Cell Death Discovery (2022)
-
Oncogenic E3 ubiquitin ligase NEDD4 binds to KLF8 and regulates the microRNA-132/NRF2 axis in bladder cancer
Experimental & Molecular Medicine (2022)
-
RUNX3 methylation drives hypoxia-induced cell proliferation and antiapoptosis in early tumorigenesis
Cell Death & Differentiation (2021)
-
Expression patterns and prognostic value of RUNX genes in kidney cancer
Scientific Reports (2021)
