
Abstract
Prostate cancer stem cells (CSCs) are defined by their extensive self-renewal, differentiation and tumor initiation properties. It is now clear that CSCs are involved in tumor growth and recurrence, and resistance to conventional treatments. The sonic hedgehog (Shh) pathway has a crucial role in stemness and tumorigenesis. Thus, the strategy that suppresses stemness and consequently tumorigenic potential of CSCs could be considered for the management of prostate cancer. The objectives of this study were to examine the molecular mechanisms, by which NVP-LDE-225/Erismodegib (smoothened inhibitor) regulates stem cell characteristics and tumor growth in prostate cancer. The effects of NVP-LDE-225 on CSC’s viability, sphere formation, apoptosis, epithelial–mesenchymal transition (EMT) and tumor growth in NOD/SCID IL2Rγ null mice were examined. NVP-LDE-225 inhibited cell viability and spheroid formation, and induced apoptosis by activation of caspase-3 and cleavage of poly-ADP ribose polymerase (PARP). NVP-LDE-225 induced expression of Bax and Bak, and inhibited the expression of Bcl-2, Bcl-XL, XIAP, cIAP1, cIAP2 and survivin. NVP-LDE-225 inhibited Gli transcriptional activity, Gli-DNA interaction and the expression of Gli1, Gli2, Patched1 and Patched-2 in prostate CSCs. Interestingly, NVP-LDE-225 induced PDCD4 and apoptosis and inhibited cell viability by suppressing miR-21. Furthermore, NVP-LDE-225 inhibited pluripotency-maintaining factors Nanog, Oct-4, c-Myc and Sox-2. The inhibition of Bmi-1 by NVP-LDE-225 was regulated by upregulation of miR-128. NVP-LDE-225 suppressed EMT by upregulating E-cadherin and inhibiting N-cadherin, Snail, Slug and Zeb1 by regulating the miR-200 family. Finally, NVP-LDE-225 inhibited CSC tumor growth, which was associated with the suppression of Gli1, Gli2, Patched-1, Patched-2, Cyclin D1, Bmi-1 and PCNA and cleavage of caspase-3 and PARP in tumor tissues derived from NOD/SCID IL2Rγ null mice. Overall, our findings suggest that inhibition of the Shh signaling pathway could therefore be a novel therapeutic option in treating prostate cancer.
Similar content being viewed by others


Introduction
The sonic hedgehog (Shh) signaling pathway has a major role in prostate cancer progression, and abnormal Shh signaling has been implicated in the tumorigenesis of prostate tumors.1 The normal function of the Shh ligand in the Shh pathway is to serve as a morphogen, inducing proper differentiation in embryogenesis.2, 3, 4 Genomic alterations of the Shh pathway have been shown to lead to the development of prostate cancer.5 Aberrant activation of the Shh pathway leads to an increase in cell survival and metastasis in cancer cells. Such aberrant activity includes inactivating mutations of Ptch1 or Sufu as well as activating mutations of Smo.6, 7, 8 The binding of the Shh ligand to its receptor, Patched, transmits the signal to activate Gli1 and Gli2.5, 9 Abnormal regulation of the Gli family of genes had been shown to lead to tumorigenesis. It has also been shown that expression of Gli2 in certain types of cancer cells leads to increased invasiveness and metastatic capabilities of those cells.10 Expression levels of Gli have been correlated with the expression levels of the Shh pathway as a whole, which suggests that the Gli family of transcription factors would serve as an indicator of Shh pathway activity.10
Cancer stem cells are believed to have important roles in tumor initiation, progression and drug resistance.11 At the initial stage of tumorigenesis, intrinsic and extrinsic factors cause intracellular genetic mutations and epigenetic alterations, resulting in generation of oncogenes that induce the production of prostate cancer stem cells (CSCs) and tumorigenesis.12 The CSCs can be produced from precancerous stem cells,13, 14, 15, 16, 17 cell de-differentiation18 and/or epithelial–mesenchymal transition (EMT).19, 20, 21 Malignant mesenchymal stem cells have been found in the niche of cancers,19, 20 and an epithelial–mesenchymal transition may be an early key step in the initiation of tumor microenvironment and tumorigenesis.21 The CSCs may expand by symmetric division, produce progenitor cells by asymmetric division and differentiate to multiple lineages of tumor cells, resulting in a rapid increase in tumor mass.
Acquisition of migratory properties is a prerequisite for cancer invasion into surrounding tissue. In cancer, acquisition of invasiveness requires a dramatic morphologic alteration, termed EMT, wherein cancer cells lose their epithelial characteristics of cell polarity and cell–cell adhesion, and switch to a mesenchymal phenotype.22, 23 Diverse signaling pathways regulate EMT including the Shh pathway.24 Induction of EMT functions in particular through downregulation of the epithelial adhesion protein E-cadherin (CDH1) and direct repression of Cdh1 has been shown to be under the control of transcriptional regulators ZEB1, ZEB2, TWIST1, SNAIL and SLUG, which also regulate a large number of other epithelial-related genes.25 Transcription factors of the ZEB protein family (ZEB1 and ZEB2) and several microRNA (miRNA) species (predominantly miR-200 family members) form a double-negative feedback loop, which controls EMT and mesenchymal–epithelial transition programs in both development and tumorigenesis. N-cadherin and fibronectin are mesenchymal markers. However, the molecular mechanism by which Shh pathway regulates EMT is not well understood.
MiRNAs are a class of small noncoding RNAs comprising ∼22 nucleotides in length. In general, miRNAs negatively regulate gene expression post-transcriptionally by binding to the 3′-untranslated region (UTR) of the targeted mRNA to inhibit gene translation. miRNAs have a critical role in developmental processes, stem cell maintenance and physiological processes, and are implicated in the pathogenesis of several human diseases, including prostate cancer.26 miRNAs also have a role in cancer by controlling the expression of certain oncogenes and tumor suppressor genes.27 miRNA profiling has revealed distinct expression signatures in various human cancers, including prostate. The functional significance of most of these alterations remains unclear.
The Polycomb-group transcriptional repressor Bmi-1 is a key regulator in several cellular processes, including stem cell self-renewal and cancer cell proliferation. Bmi-1 was first identified in 1991 as a frequent target of Moloney virus insertion in virally accelerated B-lymphoid tumors of E mu-myc transgenic mice.28 It is implicated in the modulation of self-renewal of stem cells, including hematopoietic,29 mammary30 and neural.31 vBmi-1 has been shown to maintain stem cell multipotency and self-renewal.32 Bmi-1 gene amplification and protein overexpression are also commonly found in various cancers. Bmi-1 is overexpressed in prostate cancer with adverse pathologic and clinical features.33, 34, 35 Tumors with Gleason scores of ⩾8 have a significant upregulation of Bmi-1, while the presence of Bmi-1 in lower-grade prostate cancer samples is highly predictive for prostate-specific antigen recurrence.36 Microarray meta-analyses have found that the presence of Bmi-1 in prostate cancer specimens often indicates metastatic disease and a high probability of unfavorable therapeutic outcome.37 Bmi-1 has been shown to be enriched in a population of prostate cancer cells with higher tumor-initiating capacities.33 Bmi-1 is a crucial regulator of self-renewal in adult prostate cells, and has important roles in prostate cancer initiation and progression.34 These studies suggest the functional involvement of Bmi-1 in prostate cancer progression and maintenance.
The purpose of this study was to examine the effects of NVP-LDE-225/Erismodegib (Smoothened inhibitor) on CSC characteristics and tumor growth. NVP-LDE-225 is in the early stage of clinical trials. Our data demonstrate that NVP-LDE-225 inhibits spheroid formation and self-renewal of CSC by suppressing the expression of pluripotency-maintaining factors (Nanog, Oct-4, Sox-2 and c-Myc). NVP-LDE-225 inhibits EMT by upregulating miR-200 and inhibiting transcription factors Snail, Slug and Zeb1. The inhibition of Bmi-1 by NVP-LDE-225 was regulated by induction of miR-128. NVP-LDE-225 also inhibits prostate CSC tumor growth by suppressing the Shh pathway, Bcl-2, cyclinD1, c-Myc and Bmi-1. Our data suggest that inhibition of the Shh signaling pathway is a potential therapeutic strategy for prostate cancer by targeting CSCs.
Results
NVP-LDE-225 induces apoptosis and inhibits cell viability in spheroids in prostate CSCs
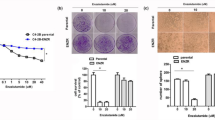
The Shh pathway is constitutively active in prostate cancer. We therefore first sought to inhibit this pathway by NVP-LDE-225, a smoothened inhibitor, and examine its effects on apoptosis and cell viability in spheroids. We measured the effects of NVP-LDE-225 on apoptosis in prostate CSCs by two assays, that is, annexin-propidium iodide (PI) and PI staining (Sub G0 cells). NVP-LDE-225 induced apoptosis in a dose-dependent manner as measured by both the assays (Figure 1a). The percentage of apoptotic cells was quantified, which demonstrated that NVP-LDE-225 induced apoptosis in a dose-dependent manner (Figure 1b). We next examined the effects of NVP-LDE-225 on cell viability in spheroids. NVP-LDE-225 inhibited cell viability in primary and secondary spheroids in a dose-dependent manner (Figure 1c).

Effects of NVP-LDE-225 on apoptosis, spheroid formation and cell viability in spheroids. (a) Effects of NVP-LDE-225 on apoptosis. Upper panel, CSCs were treated with NVP-LDE-225 (0, 1, 5 and 10 μM) for 48 h. At the end of the incubation period, apoptosis was measured by annexin-PI staining. Data are representative of three independent experiments. Lower panel, CSCs were treated with NVP-LDE-225 (0, 1, 5 and 10 μM) for 48 h. At the end of incubation period, apoptosis was measured by PI staining. Data are representative of three independent experiments. (b) Quantification of apoptosis. Data represent mean±s.d. Data are representative of three independent experiments. *, #, %=Significantly different from control, P<0.05. (c) Effects of NVP-LDE-225 on spheroids formation and cell viability. Upper panel, CSCs were seeded in suspension and treated with NVP-LDE-225 (0–10 μM) for 7 days. At the end of incubation period, spheroids were photographed. Data are representative of three independent experiments. Lower panel, cell viability in spheroids. CSCs were seeded in suspension and treated with NVP-LDE-225 (0–10 μM) for 7 days. At the end of incubation period, spheroids were collected and dissociated with Accutase (Innovative Cell Technologies, Inc.). For secondary spheroids, cells were reseeded and treated with NVP-LDE-225 for additional 7 days. Cell viability was measured by trypan blue assay. Data represent mean±s.d. *, %, # and ^=significantly different from control, P<0.05. (d) Expression of cleaved caspase-3 and PARP. CSCs were treated with NVP-LDE-225 for 48 h, and the expression of cleaved caspase-3 and PARP was determined by the western blot analysis.
We also examined the effects of NVP-LDE-225 on cleavage of caspase-3 and poly-ADP ribose polymerase (PARP), which are the hallmarks of apoptosis. As shown in Figure 1d, treatment of prostate CSCs resulted in an increase in the expression of cleaved caspase-3 and PARP. These data suggest that NVP-LDE-225 inhibits cell viability and spheroid formation, and induces apoptosis in a dose-dependent manner, and thus can be used for the treatment of prostate cancer by targeting CSCs.
Regulation of Bcl-2 and IAP family members by NVP-LDE-225
As Bcl-2 family members have a major role in cell survival and apoptosis, we sought to measure the effects of NVP-LDE-225 on the expression of Bcl-2, Bcl-XL, Bax and Bak by qRT–PCR and western blot analyses. NVP-LDE-225 inhibited the expression of Bcl-2 and Bcl-XL and induced the expression of Bax and Bak in a dose-dependent manner as measured by qRT–PCR (Figure 2a). These data were further confirmed by the western blot analysis. As shown in Figure 2b, NVP-LDE-225 inhibited the expression of Bcl-2 and Bcl-XL and induced the expression of Bax and Bak in a dose-dependent manner (Figure 2b).

NVP-LDE-225 regulates genes involved in apoptosis and cell survival. (a) CSCs were treated with NVP-LDE-225 (0–10 μM) for 36 h and the expression of Bak, Bax, Bcl-2 and Bcl-XL was measured by qRT–PCR. HK-GAPD was used as the endogenous normalization control. Data represent mean±s.d. *, %, #=Significantly different from control, P<0.05. (b) CSCs were treated with NVP-LDE-225 (0–10 μM) for 36 h and the expression of XIAP, survivin, cIAP1 and cIAP2 was measured by western blot analysis. (c) CSCs were treated with NVP-LDE-225 (0–10 μM) for 36 h and the expression of XIAP, cIAP1, cIAP2 and survivin was measured by the western blot analysis. Data represent mean±s.d. *, %, #=Significantly different from control, P<0.05. (d) CSCs were treated with NVP-LDE-225 (0–10 μM) for 36 h and the expression of XIAP, cIAP1, cIAP2 and survivin was measured by the western blot analysis.
As IAP family members have a major role in cell survival and apoptosis, we sought to measure the effects of NVP-LDE-225 on the expression of cIAP1, cIAP2, XIAP and survivin by qRT–PCR and western blot analysis. NVP-LDE-225 inhibited the expression of cIAP1, cIAP2, XIAP and survivin in a dose-dependent manner (Figures 2c and d). These data suggest that NVP-LDE-225 can inhibit cell survival and induce apoptosis through regulation of Bcl-2 family members and IAPs.
NVP-LDE-225 inhibits the components of the Shh pathway, Gli transcriptional activity and Gli nuclear translocation in prostate CSCs
As NVP-LDE-225 inhibited cell viability and induced apoptosis in prostate CSCs, we next examined the effect of NVP-LDE-225 on expression/translocation of Gli1 and Gli2 to the nuclei by immunofluorescence technique (Figure 3a). Prostate CSCs were treated with NVP-LDE-225, and the expression/translocation of Gli1 and Gli2 was observed under a fluorescence microscope. NVP-LDE-225 inhibited expression/translocation of Gli1 and Gli2 to the nuclei.

NVP-LDE-225 downregulates Shh-signaling pathway in CSCs. (a) NVP-LDE-225 inhibits expression of Gli1 and Gli2 in CSCs. The cells were seeded on fibronectin-coated coverslips and treated with NVP-LDE-225 (5 μM) for 48 h. Subsequently, cells were fixed with 4% paraformaldehyde, blocked in 5% normal goat serum and stained with Gli1 and Gli2 primary antibodies (1:100) for 16 h at 4 °C and washed with PBS. Afterwards, cells were incubated with fluorescently labeled secondary antibody (1:200) along with DAPI (1 mg/ml) for 1 h at room temperature and cells were mounted and visualized under a fluorescent microscope. (b) Gel shift asssay. Nuclear extracts were prepared and incubated with labeled Gli probe. Gelshift assay was performed as we described in Materials and methods. (c) Luciferase assay, CSCs were transduced with lentiviral particles expressing Gli-dependent luciferase reporter and treated with NVP-LDE-225 (0, 5 and 10 μM) for 48 h. Lysates were prepared, and luciferase activity was measured as described in Materials and methods. Normalized luciferase activity is presented as mean±s.d. *, % and #=Significantly different from control (P<0.05). (d) CSCs were treated with NVP-LDE-225 (10 μM) for 36 h. At the end of incubation period, RNA was extracted and the expression of Gli1, Gli2, Patched-1 and Patched-2 was measured by qRT–PCR. Data represent the mean±s.d (n=4). *, % and #=Significantly different from respective control (P<0.05). (e) Protein expression of Gli1, Gli2, Patched1 and Patched-2. CSCs were treated with NVP-LDE-225 for 48 h, and the expression of Gli1, Gli2, Patched1 and Patched-2 was determined by the western blot analysis.
The influence of NVP-LDE-225 on the Gli-DNA binding in CSCs was subsequently determined by electrophoretic mobility shift assay at 48-h treatment (Figure 3b). Treatment of CSCs with NVP-LDE-225 (0–10 μM) resulted in decreased Gli-DNA binding activity in a dose-dependent manner. Next we examined the effect of NVP-LDE-225 on Gli transcriptional activity. Prostate CSCs were transduced with a Gli-dependent luciferase reporter construct and treated with NVP-LDE-225 for 48 h (Figure 3c). NVP-LDE-225 inhibited Gli-dependent luciferase reporter activity in a dose-dependent manner. These data suggest that inhibition of Shh pathway by NVP-LDE-225 can inhibit Gli-DNA binding activity and Gli transcriptional activity.
As NVP-LDE-225 inhibited the expression/translocation of Gli1 and Gli2 to the nuclei, we next sought to examine its effects on various components of the Shh pathway in CSCs by qRT–PCR analysis (Figure 3d). NVP-LDE-225 inhibited the expressions of effectors (Gli1 and Gli2) and receptors (Patched-1 and Patched-2) of the Shh pathway in CSCs, as measured by qRT–PCR. The effects of NVP-LDE-225 on the expression of the Shh pathway were confirmed by western blot analysis. As shown in Figure 3e, NVP-LDE-225 inhibited the expression of Gli1, Gli2, Patched-1 and Patched-2 in prostate CSCs. These data suggest that NVP-LDE-225 can regulate prostate CSC characteristics by inhibiting various components of the Shh pathway.
NVP-LDE-225 inhibits the expression of genes involved in maintaining pluripotency
As NVP-LDE-225 inhibited the Shh pathway, we next examined the expression of genes that have roles in maintaining pluripotency. Prostate CSCs were exposed to NVP-LDE-225 (0–10 μM) for 36 h and the expression of Nanog, Oct-4, c-Myc and Sox-2 was measured by qRT–PCR. NVP-LDE-225 inhibited the expression of Nanog, Oct-4, c-Myc and Sox-2 in prostate CSCs in a dose-dependent manner (Figure 4a). Similarly, NVP-LDE-225 inhibited the expression of Nanog, Oct-4, c-Myc and Sox-2 in prostate CSCs in a dose-dependent manner as demonstrated by the western blot analysis (Figure 4b).

NVP-LDE-225 differentially regulates genes involved in self-renewal and pluripotency. (a) CSCs were treated with NVP-LDE-225 (0–10 μM) for 36 h and the expression of Nanog, Oct-4, c-Myc and Sox-2 was measured by qRT–PCR. HK-GAPD was used as the endogenous normalization control. Data represent mean±s.d. *, % and #=Significantly different from control, P<0.05. (b) Protein expression of Nanog, Oct-4, c-Myc and Sox-2. Prostate CSCs were treated with NVP-LDE-225 for 48 h, and the expression of Nanog, Oct-4, c-Myc and Sox-2 was determined by the western blot analysis. (c) Immunohistochemical examination of Nanog, Oct-4, c-Myc and Sox-2. Prostate CSCs were grown in suspension and treated with NVP-LDE-225 (5 μM) for 48 h. At the end of incubation period, spheroids were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked in 5% normal goat serum. After blocking, spheroids were stained with primary antibody (1:100) overnight at 4 °C, washed twice with PBS and incubated with fluorescently labeled secondary antibody (1:200) along with DAPI (1 mg/ml) for 1 h at room temperature. Finally, spheroids were mounted and visualized under a fluorescent microscope.
We confirmed the effects of NVP-LDE-225 on the expression of Nanog, Oct-4, c-Myc and Sox-2 in spheroids by immunocytochemistry (Figure 4c). NVP-LDE-225 inhibited the expression of Nanog, Oct-4, c-Myc and Sox-2 in prostate CSCs. These data suggest that inhibition of the Shh pathway can suppress the self-renewal capacity of CSCs by inhibiting the factors required for maintaining pluripotency.
NVP-LDE-225 inhibits Bmi-1 through upregulation of miR-128 in prostate CSCs
The polycomb-group gene Bmi-1 is overexpressed in prostate CSCs. The downregulation of Bmi-1 resulted in inhibition of clonogenic ability in vitro and tumor formation in vivo.34, 35, 36 Bmi-1 is required for spontaneous de novo development of the prostate tumor, and is considered as a key factor required for HH pathway-driven tumorigenesis.38 We therefore examined whether NVP-LDE-225 regulates the expression of Bmi-1 in prostate CSCs by immunohistochemistry and western blot analysis (Figures 5a and b). As shown in Figure 5a, NVP-LDE-225 inhibited the expression of Bmi-1 in spheroids. Similarly, NVP-LDE-225 inhibited the expression of Bmi-1 in spheroids in culture. These data indicate that NVP-LDE-225 may regulate stemness through Bmi-1, and thus suggest the requirement of Bmi-1 for cell survival.

NVP-LDE-225 downregulates Bmi-1 through upregulation of miR-128 in CSCs. (a) Immunohistochemical examination of Bmi-1. Prostate CSCs were grown in suspension and treated with NVP-LDE-225 (5 μM) for 48 h. At the end of the incubation period, spheroids were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked in 5% normal goat serum. After blocking, spheroids were stained with primary antibody (1:100) overnight at 4 °C, washed twice with PBS and incubated with fluorescently labeled secondary antibody (1:200) along with DAPI (1 mg/ml) for 1 h at room temperature. Finally, the spheroids were mounted and visualized under a fluorescent microscope. (b) Protein expression of Bmi-1. Prostate CSCs were treated with NVP-LDE-225 for 48 h, and the expression of Bmi-1 was determined by western blot analysis. (c) Top left, CSCs were treated with NVP-LDE-225 (0–10 μM) for 36 h and the expression of Bmi-1 was measured by qRT–PCR. Data represent mean±s.d. *, % and #=Significantly different from control, P<0.05. Top middle, CSCs were treated with NVP-LDE-225 (0–10 μM) for 36 h and the expression of miR-128 was measured by qRT-PCR. Data represent mean±s.d. *, % and #=Significantly different from control, P<0.05. Top right, 3′UTR-Bmi-1 luciferase activity. CSCs were transduced with 3′UTR-Bmi-1 luciferase construct and treated with NVP-LDE-225 for 36 h. Luciferase activity was measured as described in Materials and methods. Bottom left, CSCs were transduced with either control or anti-miR-128 viral particles. The expression of miR-128 was measured by qRT–PCR. Data represent mean±s.d. *=Significantly different from control, P<0.05. Bottom right, CSCs were transduced with either control (scrambled) or anti-miR-128 viral particles, and treated with NVP-LDE-225 (0–10 μM) for 36 h. At the end of the incubation period, the expression of Bmi-1 was measured by qRT(PCR. Data represent mean±s.d. *, % and #=Significantly different from control, P<0.05.
We next examined the mechanism by which NVP-LDE-225 inhibits Bmi-1 in prostate CSCs. As Bmi-1 is a direct target of miR-128,39, 40 we sought to examine whether miR-128 mediates the inhibitory effects of NVP-LDE-225 on Bmi-1 expression. NVP-LDE-225 inhibited the expression of Bmi-1 and induced the expression of miR-128 in CSCs (Figure 5c). In order to confirm whether miR-128 regulated the inhibitory effects of NVP-LDE-225 on Bmi-1, we silenced the expression of miR-128 by anti-miR-128. Prostate CSCs were transduced with anti-miR-128 and the expression of miR-128 was measured by qRT–PCR. Transduction of anti-miR-128 inhibited the expression of miR-128 in prostate CSCs. Overexpression of anti-miR-128 blocked the inhibitory effects of NVP-LDE-225 on Bmi-1 expression.
As NVP-LDE-225 induced the expression of miR-128 and inhibited the expression of Bmi-1, we sought to examine the 3′UTR-Bmi-1 activity by luciferase assay. miR-128 has been shown to bind 3′UTR of Bmi-1 and inhibit its expression. NVP-LDE-225 inhibited 3′UTR-Bmi-1-LUC activity in prostate CSCs. These data suggest that NVP-LDE-225 inhibits the expression of Bmi-1 by inducing the expression of miR-128.
NVP-LDE-225 inhibits motility, invasion and migration of CSCs
EMT has been increasingly recognized to occur during the progression of various carcinomas.22 It has been proposed that EMT is one of the key mechanisms through which metastasis occurs in different tumors, beginning with the disruption of intercellular contacts and the enhancement of cell motility, thus resulting in the release of cancer cells from the primary tumor. As CSCs appear to have a significant role in early metastasis,41 we sought to measure the effects of NVP-LDE-225 on the motility, migration and invasion of CSCs (Figures 6a and b). NVP-LDE-225 inhibited the motility, migration and invasion of prostate CSCs. These data suggest that NVP-LDE-225 can inhibit early metastasis of prostate CSCs.

Regulation of cell motility, migration, invasion and expression of EMT factors by NVP-LDE-225 in CSCs. (a) Motility Assay. Photomicrographs demonstrating the results of the in vitro motility of CSCs using scratch technique. CSCs were grown in monolayer, scratched and treated with or without NVP-LDE-225 for 24 or 48 h. Data are representative of three independent experiments. (b) Transwell migration assay (left panel): CSCs were plated in the top chamber of the transwell and treated with NVP-LDE-225 (0–10 μM) for 24 h. Cells migrated to the lower chambered were fixed with methanol, stained with crystal violet and counted. Data represent mean±s.d. *, % and #=Significantly different from respective controls, P<0.05. (c) Matrigel invasion assay, CSCs were plated onto the Matrigel-coated membrane in the top chamber of the transwell and treated with NVP-LDE-225 (0–10 μM) for 24 h. Cells invaded to the lower chambered were fixed with methanol, stained with crystal violet and counted. Data represent mean±s.d. *, # and & =Significantly different from respective controls, P<0.05. (d) CSCs were treated with NVP-LDE-225 (0–10 μM) for 48 h. The expression of E-cadherin, N-cadherin, Snail, Slug and ZEB1 was measured by western blot analysis. (e) CSCs were treated with NVP-LDE-225 (0–10 μM) for 36 h. At the end of the incubation period, the expression of E-cadherin and N-cadherin was measured by qRT–PCR. Data represent mean±s.d. *, % and #=Significantly different from respective controls, P<0.05. (f) CSCs were treated with NVP-LDE-225 (0–10 μM) for 36 h. At the end of the incubation period, the expression of Snail, ZEB1 and Slug was measured by qRT–PCR. Data represent mean±s.d. *, % and #=Significantly different from respective controls, P<0.05.
Tumor progression is frequently associated with the downregulation of E-cadherin22 and upregulation of vimentin and several transcription factors, including Snail, ZEB1 and Slug.42 We therefore measured the expression of E-cadherin, N-cadherin, Snail, Slug and ZEB1 by western blot analysis. NVP-LDE-225 induced the expression of E-cadherin and inhibited the expression of N-cadherin, Snail, Slug and ZEB1.
We next confirmed the regulation of cadherins by NVP-LDE-225 using qRT–PCR (Figure 6e). NVP-LDE-225 enhanced the expression of E-cadherin and inhibited the expression of N-cadherin, a phenomenon known as cadherin switch during EMT.
As NVP-LDE-225 inhibited EMT, we next examined the regulation of EMT inducing transcription factors Snail, Slug and Zeb1 (Figure 6f). NVP-LDE-225 inhibited the expression of Snail, Slug and Zeb1 as measured by qRT–PCR. These data suggest that NVP-LDE-225 can regulate early metastasis by modulating the expression of cadherins and EMT transcription factors.
EMT and mesenchymal–epithelial transition represent a mechanistic basis for epithelial cell plasticity implicated in cancer.22 Transcription factors of the ZEB protein family and several miRNA species (predominantly miR-200 family members) form a double-negative feedback loop, which controls EMT and mesenchymal–epithelial transition programs in both development and tumorigenesis. We therefore examined whether the miR-200 family mediates the effects of NVP-LDE-225 on EMT. NVP-LDE-225 induced the expression of miR-200a, miR-200b and miR-200c in CSCs (Figure 7a). Transduction of prostate CSCs with anti-miR-200 a/b/c blocked the inhibitory effects of NVP-LDE-225 on cell migration and invasion (Figures 7b and c). These data suggest that NVP-LDE-225 inhibits EMT by upregulating miR-200 family members.

NVP-LDE-225 inhibits migration and invasion by upregulating miR-200a, miR-200b and miR-200c. (a) CSCs were treated with NVP-LDE-225 (5 μM) for 36 h and the expression of miR-200a, miR-200b and miR-200c was measured by qRT–PCR. Data represent mean±s.d. *=Significantly different from control, P<0.05. (b) NVP-LDE-225 inhibits cell migration by upregulating miR-200a/b/c. CSCs were transduced with control (scrambled) or anti-miR-200a/b/c viral particles and treated with NVP-LDE-225 (0–10 μM) for 24 h. Cell migration was measured as described above. Data represent mean±s.d. *, % and #=Significantly different from control, P<0.05. (c) NVP-LDE-225 inhibits cell invasion by upregulating miR-200a/b/c. CSCs were transduced with control (scrambled) or anti-miR-200a/b/c viral particles and treated with NVP-LDE-225 (0–10 μM) for 24 h. Cell invasion was measured as described above. Data represent mean±s.d. *, % and #=Significantly different from control, P<0.05.
NVP-LDE-225 inhibits CSC tumor growth in NOD/SCID IL2Rγ mice
As NVP-LDE-225 inhibited cell viability, caused spheroid formation and induced apoptosis, we next examined its effects on CSC tumor growth in a humanized NOD/SCID IL2Rγ null mouse model. Prostate CSCs were injected subcutaneously into humanized NOD/SCID IL2Rγ null mice (harboring CD34+ peripheral blood stem/progenitor cells). After tumor formation, mice were treated with NVP-LDE-225 (20 mg/kg) intraperitoneally 3 days/week for 4 weeks. As shown in (Figure 8a), NVP-LDE-225 had no effect on body weight of mice. Interestingly, NVP-LDE-225 inhibited CSC tumor growth, as demonstrated by the significant reduction in tumor weight.

NVP-LDE-225 inhibits CSC tumor growth in NOD/SCID IL2Rγ null mice. (a) Effects of NVP-LDE-225 on CSC tumor growth in NOD/SCID IL2Rγ null mice. Tumor-bearing nude mice (seven mice/group) were injected with vehicle or NVP-LDE-225 (20 mg/kg) intraperitoneally three times per week for 4 weeks. Body weight was measured weekly. Tumor weights were measured at the end of the experiments. Data represent mean±s.d. *=Significantly different from respective control, P<0.05. A representative photograph of tumor size from control and drug-treated group is shown. (b) Gene expression in tumor tissues. Total RNA was extracted and the expression of Gli1, Gli2, Patched1, Patched2, Bcl-2, cyclin D1, c-Myc and Snail was measured by qRT–PCR. Data represent mean±s.d. *=Significantly different from control, P<0.05. (c) Tissue lysates were prepared from tumor tissues isolated from control and NVP-LDE-225-treated mice. Western blot analysis was performed to measure the expression of Gli1, Gli2, Patched1, Patched2, Cyclin D1, Bmi-1, PCNA, cleaved caspase-3 and PARP.
As NVP-LDE-225 inhibited CSC tumor growth in humanized NOD/SCID IL2Rγ null mice, we next examined the effects of NVP-LDE-225 on the expression of components of the Shh pathway (Gli1, Gli2, Patched-1 and Patched-2) and its downstream targets Bcl-2, Cyclin D1, c-Myc, Snail and Bmi-1 by qRT–PCR and western blot analysis. As shown in Figure 8b, NVP-LDE-225 inhibited the expression of Gli1, Gli2, Patched1, Patched-2, Bcl-2, Cyclin D1, c-Myc, Bmi-1 and Snail. We also confirmed the expression of these proteins by western blot analysis. As shown in Figure 8c, NVP-LDE-225 inhibited the expression of Gli1, Gli2, Patched1, Patched-2, Cyclin D1 and Bmi-1. NVP-LDE-225 also inhibited the expression of PCNA (proliferation marker) and induced the expression of cleaved caspase-3 and PARP (apoptosis marker).
We next confirmed the expression of these proteins by immunohistochemistry. As shown in Figure 9, NVP-LDE-225 inhibited the expression of Gli1, Gli2, Patched-1, Patched-2, PCNA, Bmi-1, c-Myc, Cyclin D1, Snail and Bcl-2. These in vivo data confirm our in vitro data, and suggest that NVP-LDE-225 can inhibit CSC tumor growth by regulating the Shh pathway and its downstream targets.

Immunohistochemical examination of tumor tissues derived from control and NVP-LDE-225-treated NOD/SCID IL2Rγ null mice. Tumor samples derived from control and treated mice (as described in Materials and methods) were subjected to immunohistochemistry with anti-Gli1, anti-Gli2, anti-Patched-1, anti-Patched-2, anti-PCNA, anti-Bmi-1, anti-cMyc, anti-Cyclin D1, anti-Snail or anti-Bcl-2 antibody.
Discussion
In the present study, we found that prostate CSCs consistently express various components of the Shh signaling pathway, including signaling molecules Gli1, Gli2, Patched-1 and Patched-2, suggesting that the Shh pathway is one of the ‘core’ signaling pathways or an autocrine mode of Shh signaling in these cells. NVP-LDE-225 (Erismodegib) is a selective antagonist of Smoothened (SMO). NVP-LDE-225 inhibited EMT, which was associated with inhibition in Snail, Slug, Zeb1 and N-Cadherin and upregulation in E-cadherin. NVP-LDE-225 also inhibited CSC’s tumor growth by regulating Bmi-1. Recently, NVP-LDE-225 has been used in topical creams for the treatment of basal cell carcinoma and has shown promise in its ability to effectively inhibit the Shh pathway.43 The inhibition of the Gli family of transcription factors by NVP-LDE-225 will decrease the transcription of genes associated with cell survival and proliferation in prostate cancer cells.
Growing evidence suggests that CSCs have aberrant or constitutively active self-renewal pathways that are controlled by genetic or epigenetic mechanisms and that lead to unrestrained proliferation. The Myc oncoproteins are highly amplified or constitutively expressed in prostate cancers.44, 45 Interestingly, overexpression of c-Myc has been correlated with a higher histological grade in prostate cancer.45, 46 NANOG and Oct-4 expressions positively correlated with increased prostate tumor Gleason score.47 Recent studies have demonstrated higher expression of c-Myc in CSCs relative to the bulk of tumor cells. Knockdown of c-Myc using small hairpin RNA (shRNAs) showed reduced cell proliferation, increased apoptosis and cell cycle arrest in the G0/G1 phase. Moreover, downregulation of c-Myc in the CSC population resulted in the inability to form spheroids or tumors in vivo.
Polycomb-group proteins regulate gene expression through modifications in chromatin structure.48 The polycomb-group gene Bmi-1 was found to be highly enriched in prostate CSCs and its downregulation resulted in inhibition of clonogenic ability in vitro and tumor formation in vivo.34, 35 Bmi-1 is required for spontaneous de novo development of a solid tumor arising in the prostate, and it is also essential for Hh pathway-driven tumorigenesis.38 Furthermore, Bmi-1 is a crucial regulator of self-renewal in adult prostate cells and has important roles in prostate cancer initiation and progression.34 In our study, NVP-LDE-225 inhibited the expression of Bmi-1, which may contribute to the self-renewal capacity of prostate CSCs. The inhibitory effects of NVP-LDE-225 on Bmi-1 were exerted through upregulation of miR-128. In another study using a panel of patient glioblastoma specimens, the upregulation of Bmi-1 expression and downregulation of miR-128 compared with normal tissue were demonstrated.39 Bmi-1 functions in epigenetic silencing of certain genes through epigenetic chromatin modification. In the same study, miR-128 expression caused a decrease in histone methylation (H3K27me(3)) and Akt phosphorylation and upregulation of p21/CIP1 levels, consistent with Bmi-1 downregulation.39
Elevated activation of Shh signaling has been shown to have important roles in proliferation, progression and metastasis of prostate cancer.49, 50 The Shh pathway regulates components of both cell-intrinsic and cell-extrinsic pathways of apoptosis. We have shown that NVP-LDE-225 inhibited pro-survival proteins, Bcl-2 and Bcl-XL, and pro-apoptotic proteins, Bak and Bax, in prostate CSCs. Bcl-2 family members exert their effects by regulating mitochondrial functions. Furthermore, NVP-LDE-225 inhibited the expression of XIAP, survivin, cIAP1 and cIAP2. In a recent report it has been demonstrated that GLI1, which has been shown to have a central role in Shh signaling in prostate cancer, can act as a corepressor to substantially block androgen receptor-mediated transactivation, at least in part, by directly interacting with the androgen receptor.51 These studies suggest that the Shh-GLI pathway might be one of determinants governing the transition of prostate cancer from an androgen-dependent to androgen-independent state by compensating, or even superseding androgen signaling.
EMT during embryogenesis, adult tissue homeostasis and carcinogenesis is characterized by class switch from E-cadherin to N-cadherin.23, 52 Accumulating evidence suggests that EMT has an important role during malignant tumor progression. During EMT, transformed epithelial cells can activate embryonic programs of epithelial plasticity and switch from a sessile, epithelial phenotype to a motile, mesenchymal phenotype. Induction of EMT can, therefore, lead to invasion of surrounding stroma, intravasation, dissemination and colonization of distant sites. It is now clear that sustained metastatic growth requires the dissemination of CSCs from the primary tumor followed by their re-establishment in a secondary site. Thus, EMT can confer metastatic ability on carcinomas. SNAI1 (Snail), SNAI2 (Slug), SNAI3, ZEB1, ZEB2 (SIP1), KLF8, TWIST1 and TWIST2 are EMT regulators repressing the CDH1 gene encoding E-cadherin. Hedgehog signals induce JAG2 upregulation for Notch-CSL-mediated SNAI1 upregulation, and also induce TGFβ1 secretion for ZEB1 and ZEB2 upregulation via TGFβ receptor and NF-κB. Hedgehog signaling activation indirectly leads to EMT through FGF, Notch, TGFβ signaling cascades and miRNA regulatory networks.24 Our results indicate a key and essential role of the Shh–Gli pathway in promoting prostate CSC tumor growth, stem cell self-renewal and metastatic behavior. NVP-LDE-225 inhibited EMT as demonstrated by inhibition in cell motility, invasion and migration. The inhibition of EMT was associated with suppression of EMT transcription factors (ZEB1, Snail and Slug) and cadherin switch (upregulation of E-cadherin and downregulation of N-cadherin) in CSCs, suggesting a potential role of NVP-LDE-225 in early metastasis. Targeting Gli-1/2 is thus predicted to decrease tumor bulk and eradicate CSCs and metastases.
In conclusion, we showed that the inhibition of Smo function by NVP-LDE-225 resulted in modulation of proliferation, EMT and apoptosis. Furthermore, NVP-LDE-225 inhibited CSC characteristics, which were associated with inhibition of Gli1 and Gli2, and regulation of Bcl-2 family members and IAPs. Inhibition of Bmi-1 through upregulation of miR128 appears to be one of the mechanisms by which NVP-LDE-225 regulates stemness and CSC tumor growth. In addition, the inhibition of EMT by NVP-LDE-225 was regulated by induction of the miR-200 family. Finally, NVP-LDE-225 inhibited CSC tumor growth, which was associated with the suppression of Gli1, Gli2, Patched-1, Patched-2, Cyclin D1, PCNA and cleaved caspase-3 and PARP in tumor tissues derived from NOD/SCID IL2Rγ null mice. Overall, our findings suggest that inhibition of the Shh signaling pathway is a potential therapeutic strategy for prostate cancer by targeting CSCs.
Materials and methods
Reagents
Antibodies against caspase-3, PARP, Gli1, Gli2, Patched-1, Patched-2, Bcl-2, Bcl-XL, Bax, Bak, XIAP, cIAP1, cIAP2, survivin and β-actin were obtained from Cell Signaling Technology (Danvers, MA, USA). Anti-CD44 antibody was purchased from BD Biosciences (San Jose, CA, USA). Anti-CD133 antibody was purchased from Miltenyi Biotec Inc. (Auburn, CA, USA). Enhanced chemiluminescence (ECL) Western blot detection reagents were from Amersham Life Sciences Inc. (Arlington Heights, IL, USA). NVP-LDE-225 was purchased from Chemie Tek (Indianapolis, IN, USA).
Isolation of cancer cells and cell culture
Human prostate tumor samples were minced and enzymatically dissociated with 1 mg/ml collagenase D (Roche, Indianapolis, IN, USA) and 1 μg/ml DNase I (Roche) for 1 h at 37 °C, and then sequentially filtered through 100 and 70-μm cell strainers (BD, Franklin Lakes, NJ, USA). After the lysis of red blood cells with Red Blood Cell Lysis Solution (Miltenyi Biotech Inc., Auburn, CA, USA), the filtered cells were grown in Stem Cell Growth Medium (Celprogen Inc., San Pedro, CA, USA) supplemented with 1% N2 (Life Technologies, Carlsbad, CA, USA), 2% B27 (Life Technologies), 20 ng/ml human basic fibroblast growth factor (Life Technologies), 100 ng/ml epidermal growth factor (Life Technologies) and 1% antibiotic-antimycotic (Life Technologies) on ultralow attachment culture dishes (Corning Inc., Corning, NY, USA) at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Dissociated single spheroid cells were filtered and double-stained with a phycoerythrin (PE)-conjugated monoclonal antibody against CD44 (G44-26; BD Bioscience) and an allophycocyanin (APC)-conjugated monoclonal antibody against CD133 (AC133; Miltenyi Biotec). Isotype-matched mouse immunoglobulins were used as controls. Stained cells were sorted using the FACS Aria II Cell Sorter (BD Bioscience). For serial passage, spheroid cells were dissociated into single cells with Accutase (Innovative Cell Technologies, Inc., San Diego, CA, USA) once a week and incubated under the culture conditions described earlier.
Lentiviral particle production and transduction
Packaged 293T cells were plated in 10-cm plates at a cell density of 5 × 106 a day before transfection in DMEM containing 10% heat-inactivated fetal bovine serum without antibiotics. Transfection of packaging cells and infection of prostate CSCs were carried out using standard protocols with some modifications. In brief, 293T cells were transfected with 4 μg of plasmid and 4 μg of lentiviral vector using lipid transfection (Lipofectamine-2000/Plus reagent, Life Technologies) according to the manufacturer’s protocol. Viral supernatants were collected and concentrated by adding PEG-it virus precipitation solution (SBI System Biosciences) to produce virus stocks with titers of 1 × 108–1 × 109 infectious units/ml. Viral supernatant was collected for 3 days by ultracentrifugation and concentrated 100-fold. Titers were determined on 293T cells. Prostate CSCs were transduced with lentivirus expressing scrambled (control) or shRNA against specific genes. Following transduction, the CSCs were washed three times with 1 × phosphate-buffered saline (PBS) and allowed to grow for three passages before screening for gene expression. Once decreased expression of the targeted gene was confirmed, the cells were used for experiments.
Cell viability and apoptosis assays
Accutase-dissociated single cells or fluorescence-activated cell sorting-sorted cells were seeded at a density of viable 1000 cells/well on 96-well ultra-low attachment plates (Corning Inc.) and treated with NVP-LDE-225 (0–10 μM) for 48 and 72 h. Cell viability was determined by the XTT assay. In brief, a freshly prepared XTT-PMS labeling mixture (50 μl) was added to the cell culture. The absorbance was measured at 450 nm with correction at 650 nm. The cell viability was expressed as OD (OD450–OD650).
The apoptosis was determined by fluorescence-activated cell sorting analysis of PI-stained cells. In brief, cells were dissociated, washed with PBS and resuspended in 200 μl PBS with 10 μl RNAase (10 mg/ml) and incubated at 37 °C for 30 min. After incubation, 50 μl PI solution (25 μg/ml) was added and cells were analyzed for apoptosis using FACS Calibur (BD Bioscience).
Motility assay
Scratch migration assay was used to study the horizontal movement of cells. A confluent monolayer of cells was established and then a scratch is made through the monolayer, using a standard 1–200 μl plastic pipette tip, which gives rise to an in vitro wound, washed twice with PBS and replaced in media with or without NVP-LDE-225. Cancer stem cells migrate into the scratch area as single cells from the confluent sides. The width of the scratch gap is viewed under the microscope in four separate areas each day until the gap is completely filled in the untreated control wells. Three replicate wells from a six-well plate were used for each experimental condition.
Transwell migration assay
For transwell migration assays, 1 × 105 prostate CSCs were plated in the top chamber onto the noncoated membrane (24-well insert; pore size, 8 μm; Corning Inc.) and allowed to migrate towards serum-containing medium in the lower chamber. Cells were fixed after 24 h of incubation with methanol and stained with Diff-Quick Fixative Solutions (Dade Behring, Newark, DE, USA). After 24 h, migration inserts were fixed and stained with Diff-Quick Fixative Solutions (Dade Behring).
Transwell invasion assay
For invasion assay, 1 × 105 cells were plated in the top chamber onto the Matrigel-coated Membrane (24-well insert; pore size, 8 μm; Corning Inc.). Each well was coated freshly with Matrigel (60 μg; BD Bioscience) before the invasion assay. Prostate CSCs were plated in medium without serum or growth factors and the medium supplemented with serum was used as a chemoattractant in the lower chamber. After 48 h, Matrigel-coated inserts were fixed and stained with Diff-Quick Fixative Solutions (Dade Behring). The number of cells invading through the membrane was counted under a light microscope (40 × , three random fields/well).
Tumor spheroid assay
For spheroid forming assay, cells were plated in six-well ultralow attachment plates (Corning Inc.) at a density of 1000 cells/ml in DMEM supplemented with 1% N2 (Life Technologies), 2% B27 (Life Technologies), 20 ng/ml human platelet growth factor (Sigma-Aldrich, St Louis, MO, USA), 100 ng/ml epidermal growth factor (Life Technologies) and 1% antibiotic-antimycotic (Life Technologies) at 37 °C in a humidified atmosphere of 95% air and 5% CO2. Spheroids (⩾20 μm in diameter) were collected after 7 days and dissociated with Accutase (Innovative Cell Technologies, Inc.). The CSCs obtained from dissociation were counted by Coulter counter using trypan blue dye.
Western blot analysis
Whole-cell lysates were extracted from cells using RIPA lysis buffer containing 1 × protease inhibitor cocktail. Cell lysates containing 50 μg of protein were loaded and separated on 10% Tris-HCl gel. Proteins from the gel were transferred on polyvinylidene difluoride membranes and subsequently blocked in blocking buffer (5% nonfat dry milk in 1 × Tris buffer saline) and incubated overnight with primary antibodies. Membranes were washed three times with Tris buffer saline-T for 10, 5 and 5 min each. After washing, membranes were incubated with secondary antibodies conjugated with horseradish peroxidase at 1:5000 dilution in Tris buffer saline for 1 h at room temperature. Membranes were again washed three times in Tris buffer saline-T and developed using ECL substrate. Protein bands were visualized on an X-ray film using an enhanced chemiluminescence system.
RNA isolation and mRNA expression analysis
Total RNAs were isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). Complementary DNAs were synthesized by oligo(dT)-priming methods. Real-time PCR was performed using the SYBR Green Supermix (Qiagen) according to the manufacturer’s instructions. Primers specific for each of the signaling molecules were designed using NCBI/Primer-BLAST and used to generate the PCR products. Expression levels of glyceraldehyde-3-phosphate dehydrogenase were used for normalization and quantification of gene expression levels. For the quantification of gene amplification, real-time PCR was performed using an ABI 7300 Sequence Detection System in the presence of SYBR-Green. The following gene-specific primers were used:
Smoothened (5′-TCGCTACCCTGCTGTTATTC-3′, 5′-GACGCAGGACAGAGTCTCAT-3′)
Patched1 (5′-TGACCTAGTCAGGCTGGAAG-3′, 5′-GAAGGAGATTATCCCCCTGA-3′)
Patched-2 (5′-AGGAGCTGCATTACACCAAG-3′, 5′-CCCAGGACTTCCCATAGAGT-3′)
Gli1 (5′-CTGGATCGGATAGGTGGTCT-3′, 5′-CAGAGGTTGGGAGGTAAGGA-3′)
Gli2 (5′-GCCCTTCCTGAAAAGAAGAC-3′, 5′-CATTGGAGAAACAGGATTGG-3′)
Myc (5′-CGACGAGACCTTCATCAAAA-3′, 5′-TGCTGTCGTTGAGAGGGTAG-3′
Nanog (5′-ACCTACCTACCCCAGCCTTT-3′, 5′-CATGCAGGACTGCAGAGATT-3′)
Sox-2 (5′-AACCCCAAGATGCACAACTC-3′, 5′-GCTTAGCCTCGTCGATGAAC-3′)
Oct-4 (5′-GGACCAGTGTCCTTTCCTCT-3′, 5′-CCAGGTTTTCTTTCCCTAGC-3′)
Snail (5′ACCCCACATCCTTCTCACTG-3′, 5′-TACAAAAACCCACGCAGACA-3′)
Slug (5′ACACACACACACCCACAGAG-3′, 5′-AAATGATTTGGCAGCAATGT-3′)
Zeb1 (5′-GCACAACCAAGTGCAGAAGA-3′, 5′-CATTTGCAGATTGAGGCTGA-3′)
E-cadherin (5′-TGCTCTTGCTGTTTCTTCGG-3′, 5′-TGCCCCATTCGTTCAAGTAG-3′)
N-cadherin (5′-TGGATGGACCTTATGTTGCT-3′, 5′-AACACCTGTCTTGGGATCAA-3′)
HK-GAPD (5′-GAGTCAACGGATTTGGTCGT-3′, 5′-TTGATTTTGGAGGGATCTCG-3′)
Gli reporter assay (p-GreenFire1 Lenti-Reporter)
Gli reporter activity was measured as we described elsewhere.53 In brief, cop-GFP and luciferase genes were cloned downstream of Gli-response element, containing four Gli binding motifs (pGreen Fire1-4xGli-mCMV-EF1-Neo; System Biosciences, Mountain View, CA, USA). Prostate CSCs were transduced with lentiviral particles and stable cells were selected. For transcription assay, CSCs (5–10 000 cells/well) were seeded in 12-well plates and treated with or without NVP-LDE-225 (0–10 μM) for up to 48 h. After incubation, CSCs were harvested and analyzed for luciferase reporter activity (Promega Corp., Madison, WI, USA).
Immunocytochemistry
Prostate CSCs were grown on fibronectin-coated coverslips (Beckton Dickinson, Bedford, MA, USA) in the presence or absence of NVP-LDE-225 (10 μM). Subsequently, cells were fixed with 4% paraformaldehyde for 15 min, permeabilized with 0.1% Triton X-100 in 1 × PBS, washed and blocked in 10% normal goat serum. After washing with PBS, cells were stained with Gli1 and Gli2 primary antibodies (1:100) for 16 h at 4 °C and washed with PBS. Afterwards, cells were incubated with fluorescently labeled secondary antibody (1:200) along with DAPI (1 mg/ml) for 1 h at room temperature. Finally, coverslips were washed and mounted using Vectashield (Vector Laboratories, Burlington, CA, USA). Isotype-specific negative controls were included with each staining. Stained cells were mounted and visualized under a fluorescent microscope. Imunohistochemistry of prostate tumor tissues was performed as we described elsewhere (35).
Antitumor activity of NVP-LDE-225
Human prostate CSCs (1 × 103 cells mixed with Matrigel, Becton Dickinson, Bedford, MA, USA, in 75 μl total volume, 50:50 ratio) were injected subcutaneously into the flanks of NOD/SCID IL2Rγnull mice (4–6 weeks old). After 2 weeks of CSC implantation, mice (seven mice/group) were treated with NVP-LDE-225 (0 and 20 mg/kg body weight) intraperitoneally three times per week for 4 weeks. At the end of the experiment, mice were euthanized, and tumors were isolated for biochemical analysis.
Statistical analysis
The mean and s.d. were calculated for each experimental group. Differences between groups were analyzed by one- or two-way analysis of variance, followed by Bonferoni’s multiple comparison tests using PRISM statistical analysis software (GrafPad Software, Inc., San Diego, CA, USA). Significant differences among groups were calculated at P<0.05.
References
Gupta S, Takebe N, Lorusso P . Targeting the Hedgehog pathway in cancer. Ther Adv Med Oncol 2010; 2: 237–250.
Alberta JA, Park SK, Mora J, Yuk D, Pawlitzky I, Iannarelli P et al. Sonic hedgehog is required during an early phase of oligodendrocyte development in mammalian brain. Mol Cell Neurosci 2001; 18: 434–441.
Matise MP, Wang H . Sonic hedgehog signaling in the developing CNS where it has been and where it is going. Curr Top Dev Biol 2011; 97: 75–117.
Pepicelli CV, Lewis PM, McMahon AP . Sonic hedgehog regulates branching morphogenesis in the mammalian lung. Curr Biol 1998; 8: 1083–1086.
Ruiz i Altaba A, Sanchez P, Dahmane N . Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer 2002; 2: 361–372.
Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet 2002; 31: 306–310.
Thomas WD, Chen J, Gao YR, Cheung B, Koach J, Sekyere E et al. Patched1 deletion increases N-Myc protein stability as a mechanism of medulloblastoma initiation and progression. Oncogene 2009; 28: 1605–1615.
Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res 2004; 64: 7794–7800.
Traiffort E, Angot E, Ruat M . Sonic Hedgehog signaling in the mammalian brain. J Neurochem 2010; 113: 576–590.
Haldipur P, Bharti U, Govindan S, Sarkar C, Iyengar S, Gressens P et al. Expression of sonic hedgehog during cell proliferation in the human cerebellum. Stem Cells Dev 2011; 21: 1059–1068.
Chen SY, Huang YC, Liu SP, Tsai FJ, Shyu WC, Lin SZ . An overview of concepts for cancer stem cells. Cell transplantation 2011; 20: 113–120.
Weinstein IB, Joe A . Oncogene addiction. Cancer Res. 2008; 68: 3077–3080 discussion 80.
D'Angelo RC, Wicha MS . Stem cells in normal development and cancer. Prog Mol Biol Transl Sci 2010; 95: 113–158.
Gao JX . Cancer stem cells: the lessons from pre-cancerous stem cells. J Cell Mol Med 2008; 12: 67–96.
Menakuru SR, Brown NJ, Staton CA, Reed MW . Angiogenesis in pre-malignant conditions. Br J Cancer 2008; 99: 1961–1966.
Dieter SM, Ball CR, Hoffmann CM, Nowrouzi A, Herbst F, Zavidij O et al. Distinct types of tumor-initiating cells form human colon cancer tumors and metastases. Cell Stem Cell 2011; 9: 357–365.
Davies EJ, Marsh V, Clarke AR . Origin and maintenance of the intestinal cancer stem cell. Mol Carcinog 2011; 50: 254–263.
Frankel TL, Burns W, Riley J, Morgan RA, Davis JL, Hanada K et al. Identification and characterization of a tumor infiltrating CD56(+)/CD16 (−) NK cell subset with specificity for pancreatic and prostate cancer cell lines. Cancer Immunol Immunother 2010; 59: 1757–1769.
Tellez CS, Juri DE, Do K, Bernauer AM, Thomas CL, Damiani LA et al. EMT and stem cell-like properties associated with miR-205 and miR-200 epigenetic silencing are early manifestations during carcinogen-induced transformation of human lung epithelial cells. Cancer Res 2011; 71: 3087–3097.
Hotz HG, Hotz B, Buhr HJ . Genes associated with epithelial-mesenchymal transition: possible therapeutic targets in ductal pancreatic adenocarcinoma? Anticancer Agents Med Chem 2011; 11: 448–454.
Said NA, Williams ED . Growth factors in induction of epithelial-mesenchymal transition and metastasis. Cells Tissues Organs 2011; 193: 85–97.
Thiery JP, Acloque H, Huang RY, Nieto MA . Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139: 871–890.
Gunasinghe NP, Wells A, Thompson EW, Hugo HJ . Mesenchymal-epithelial transition (MET) as a mechanism for metastatic colonisation in breast cancer. Cancer Metastasis Rev 2012; 31: 469–478.
Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res 2011; 71: 7061–7070.
Huber MA, Kraut N, Beug H . Molecular requirements for epithelial-mesenchymal transition during tumor progression. Curr Opin Cell Biol 2005; 17: 548–558.
Kloosterman WP, Plasterk RH . The diverse functions of microRNAs in animal development and disease. Dev Cell 2006; 11: 441–450.
Esquela-Kerscher A, Slack FJ . Oncomirs—microRNAs with a role in cancer. Nat Rev Cancer 2006; 6: 259–269.
van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A . Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 1991; 65: 737–752.
Park IK, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003; 423: 302–305.
Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res 2006; 66: 6063–6071.
Molofsky AV, He S, Bydon M, Morrison SJ, Pardal R . Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev 2005; 19: 1432–1437.
Fasano CA, Phoenix TN, Kokovay E, Lowry N, Elkabetz Y, Dimos JT et al. Bmi-1 cooperates with Foxg1 to maintain neural stem cell self-renewal in the forebrain. Genes Dev 2009; 23: 561–574.
Hurt EM, Kawasaki BT, Klarmann GJ, Thomas SB, Farrar WL . CD44+ CD24(−) prostate cells are early cancer progenitor/stem cells that provide a model for patients with poor prognosis. Br J Cancer 2008; 98: 756–765.
Lukacs RU, Memarzadeh S, Wu H, Witte ON . Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 2010; 7: 682–693.
Mimeault M, Batra SK . Frequent gene products and molecular pathways altered in prostate cancer- and metastasis-initiating cells and their progenies and novel promising multitargeted therapies. Mol Med 2011; 17: 949–964.
van Leenders GJ, Dukers D, Hessels D, van den Kieboom SW, Hulsbergen CA, Witjes JA et al. Polycomb-group oncogenes EZH2, BMI1, and RING1 are overexpressed in prostate cancer with adverse pathologic and clinical features. Eur Urol 2007; 52: 455–463.
Glinsky GV, Berezovska O, Glinskii AB . Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest 2005; 115: 1503–1521.
Michael LE, Westerman BA, Ermilov AN, Wang A, Ferris J, Liu J et al. Bmi1 is required for Hedgehog pathway-driven medulloblastoma expansion. Neoplasia 2008; 10: 1343–1349.
Godlewski J, Nowicki MO, Bronisz A, Williams S, Otsuki A, Nuovo G et al. Targeting of the Bmi-1 oncogene/stem cell renewal factor by microRNA-128 inhibits glioma proliferation and self-renewal. Cancer Res 2008; 68: 9125–9130.
Venkataraman S, Alimova I, Fan R, Harris P, Foreman N, Vibhakar R . MicroRNA 128a increases intracellular ROS level by targeting Bmi-1 and inhibits medulloblastoma cancer cell growth by promoting senescence. PLoS One 2010; 5: e10748.
Monteiro J, Fodde R . Cancer stemness and metastasis: therapeutic consequences and perspectives. Eur J Cancer 2010; 46: 1198–1203.
Iwatsuki M, Mimori K, Yokobori T, Ishi H, Beppu T, Nakamori S et al. Epithelial-mesenchymal transition in cancer development and its clinical significance. Cancer Sci 2010; 101: 293–299.
Skvara H, Kalthoff F, Meingassner JG, Wolff-Winiski B, Aschauer H, Kelleher JF et al. Topical treatment of Basal cell carcinomas in nevoid Basal cell carcinoma syndrome with a smoothened inhibitor. J Invest Dermatol 2011; 131: 1735–1744.
Benassi B, Flavin R, Marchionni L, Zanata S, Pan Y, Chowdhury D et al. MYC is activated by USP2a-mediated modulation of microRNAs in prostate cancer. Cancer Discov 2012; 2: 236–247.
Kim J, Roh M, Doubinskaia I, Algarroba GN, Eltoum IE, Abdulkadir SA . A mouse model of heterogeneous, c-MYC-initiated prostate cancer with loss of Pten and p53. Oncogene 2012; 31: 322–332.
Zafarana G, Ishkanian AS, Malloff CA, Locke JA, Sykes J, Thoms J et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer 2012; 118: 4053–4062.
Mathieu J, Zhang Z, Zhou W, Wang AJ, Heddleston JM, Pinna CM et al. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res 2011; 71: 4640–4652.
Lessard J, Sauvageau G . Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003; 423: 255–260.
Bragina O, Njunkova N, Sergejeva S, Jarvekulg L, Kogerman P . Sonic Hedgehog pathway activity in prostate cancer. Oncol Lett 2010; 1: 319–325.
Vezina CM, Bushman AW . Hedgehog signaling in prostate growth and benign prostate hyperplasia. Curr Urol Rep 2007; 8: 275–280.
Chen G, Goto Y, Sakamoto R, Tanaka K, Matsubara E, Nakamura M et al. GLI1, a crucial mediator of sonic hedgehog signaling in prostate cancer, functions as a negative modulator for androgen receptor. Biochem Biophys Res Commun 2011; 404: 809–815.
Lopez-Novoa JM, Nieto MA . Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med 2009; 1: 303–314.
Singh BN, Fu J, Srivastava RK, Shankar S . Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLoS One 2011; 6: e27306.
Acknowledgements
We thank our lab members for critical reading of the manuscript. This work was supported in part by the Kansas Bioscience Authority.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Nanta, R., Kumar, D., Meeker, D. et al. NVP-LDE-225 (Erismodegib) inhibits epithelial–mesenchymal transition and human prostate cancer stem cell growth in NOD/SCID IL2Rγ null mice by regulating Bmi-1 and microRNA-128. Oncogenesis 2, e42 (2013). https://doi.org/10.1038/oncsis.2013.5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/oncsis.2013.5
Keywords
This article is cited by
-
Hedgehog signaling in tissue homeostasis, cancers, and targeted therapies
Signal Transduction and Targeted Therapy (2023)
-
MicroRNA-128 acts as a suppressor in the progression of gastrointestinal stromal tumor by targeting B-lymphoma Mo-MLV insertion region 1
Clinical and Translational Oncology (2023)
-
Endothelial nitric oxide synthase (eNOS)-NO signaling axis functions to promote the growth of prostate cancer stem-like cells
Stem Cell Research & Therapy (2022)
-
Effects of an inhibitor of the SHH signaling pathway on endometrial cells of patients with endometriosis
BMC Molecular and Cell Biology (2022)
-
Understanding Abnormal SMO-SHH Signaling in Autism Spectrum Disorder: Potential Drug Target and Therapeutic Goals
Cellular and Molecular Neurobiology (2022)
