
Abstract
BTK is a cytoplasmic protein-tyrosine kinase, whose corresponding gene was isolated in the early 1990s. BTK was initially identified by positional cloning of the gene causing X-linked agammaglobulinemia and independently in a search for new kinases. Given the phenotype of affected patients, namely lack of B-lymphocytes and plasma cells with the ensuing inability to mount humoral immune responses, BTK inhibitors were anticipated to have beneficial effects on antibody-mediated pathologies, such as autoimmunity. In contrast to, for example, the SRC-family of cytoplasmic kinases, there was no obvious way in which structural alterations would yield constitutively active forms of BTK, and such mutations were also not found in leukemias or lymphomas. In 2007, the first efficient inhibitor, ibrutinib, was reported and soon became approved both in the United States and in Europe for the treatment of three B-cell malignancies, mantle cell lymphoma, chronic lymphocytic leukemia and Waldenström’s macroglobulinemia. Over the past few years, additional inhibitors have been developed, with acalabrutinib being more selective, and recently demonstrating fewer clinical adverse effects. The antitumor mechanism is also not related to mutations in BTK. Instead tumor residency in lymphoid organs is inhibited, making these drugs highly versatile. BTK is one of the only 10 human kinases that carry a cysteine in the adenosine triphosphate-binding cleft. As this allows for covalent, irreversible inhibitor binding, it provides these compounds with a highly advantageous character. This quality may be crucial and bodes well for the future of BTK-modifying medicines, which have been estimated to reach annual multi-billion dollar sales in the future.
Similar content being viewed by others


Introduction
The development of the BTK inhibitors followed seminal studies that initially were made already in the 1930s. During this period, certain key discoveries paved the way for the subsequent achievements. I will return to the more historical aspects at the end of this review and begin with describing how studies of an immunodeficiency disease resulted in the identification of the target for the newly developed inhibitors, namely the BTK kinase. Owing to that the BTK gene carries more known mutations than any other kinase-encoding gene, I will briefly bring up some of the corresponding implications. Next, I will provide a short update of the BTK signaling pathway. Although numerous reports describe the signal transduction of BTK in mice and men, the evolution of this kinase dates back >600 million years, with certain evolutionary ramifications. The subsequent sections will describe the development of the new BTK inhibitors, and their role in the clinics, followed by a paragraph where the multifaceted role of BTK for tumor development is discussed. The most recent clinical advancements are also briefly reviewed.
X-linked agammaglobulinemia—the most classical genetic immunodeficiency
In 1952, Ogden C Bruton, in a landmark paper, reported on an immunodeficient boy, highly prone to infections.1 Although hereditary diseases accompanied by susceptibility to infections were published already prior to Bruton’s classical description, the uniqueness about this report was that it also identified an underlying disease mechanism.1 Thus, as reviewed in greater detail elsewhere,2, 3 Bruton’s patient was analyzed using serum electrophoresis, which at the time was a method only recently introduced into the clinic. Bruton anticipated that the reduced levels of gammaglobulin found in the patient might represent the cause of the disease and obtained proof for this concept by substituting the patient with gammaglobulin, which reduced the propensity for bacterial infections, a hallmark of this disorder.
At the time, this treatment represented the latest in the therapeutic arsenal, since manufacturing of gammaglobulin was developed by Edwin J Cohn during World War II in order to treat infected soldiers.4 Incidentally, Bruton used subcutaneous administration of the gammaglobulin, a route later abandoned for many years but which has been revived during the past decades.5, 6 Bruton’s report had a profound influence on the development of the field of primary immunodeficiencies with many new diseases being characterized over the next few years.
However, it was not until two decades after the initial report when it was demonstrated that lack of antibody-producing cells causes the humoral phenotype7, 8 in these patients. Among the agammaglobulinemias, the X-linked form, XLA, represents the most common disease9, 10 and it is with this disorder that Bruton’s name is associated. The disease is manifested at the stage when pro-B cells develop into pre-B cells, with all the subsequent stages essentially lacking,11 thereby causing an inability to mount humoral immune responses. Patients are thus devoid of mature B-lymphocytes as well as plasma cells, and lymphoid organs are reduced in size.
Disease gene cloning and the isolation of the BTK gene
In the 1980s, the hunt for new disease genes accelerated. This was aided by the continuous addition of new chromosomal markers enabling linkage analysis with increasing precision. At this time, we were more interested in T-cell leukemias, and incidentally, the identification of an inversion of the long arm of chromosome 14, inv(14)(q11 q32), as the most consistent chromosomal alteration in T-cell chronic lymphocytic leukemia (CLL),12, 13, 14, 15 currently referred to as T-cell prolymphocytic leukemia, set the stage for the subsequent characterization of the TCL1 locus at 14q32.1 by the groups of Carlo Croce and Giandomenico Russo.16 Serendipitously, the TCL1 open reading frame of 342 nt translates into a protein of 14 kD, whose activity induces B-CLL in IgH-Eμ-TCL1-transgenic mice17 and which has become a leading model for experimental CLL. Interestingly, this form of CLL is sensitive to BTK inhibitors and has also been used to identify components affected by ibrutinib, such as impaired C-X-C motif chemokine receptor 4 chemokine receptor surface expression18, 19, 20 as well as new experimental therapies in the form of anti-SLAMF6 combined with ibrutinib.21
In the 1980s, the nature of the gene causing XLA remained elusive, although we suggested that it may encode an enzyme.22 Also other predictions were made as reviewed in Sideras and Smith.3 The earliest studies demonstrated that XLA was not linked to the Xg blood group,23 whose locus maps to the end of the short arm of the X-chromosome. Following continued, refined mapping, as reviewed in Sideras and Smith,3 two groups, Guioli et al.24 and Kwan et al.,25 identified the DXS178 marker, which subsequently was used in the positional cloning strategy leading to the isolation of the BTK gene.26 The principle was to use a yeast artificial chromosome carrying the DXS178 marker to enrich cDNAs from B-cell progenitors and subsequently employ these as probes in Southern blotting of digested patient DNA in order to identify potential mutations from altered restriction fragment lengths. According to Dr Francis S Collins, director of the Human Genome project, BTK became the seventeenth disease gene to be cloned positionally (personal communication). In parallel, in the search for novel kinases, the group of Owen Witte identified a new target and, after mapping the corresponding gene to the X-chromosome, was able to link this discovery to XLA.27 The two reports complemented each other, but owing to the two different names that were used, a common name, BTK, was adopted and for the first time used in two back-to-back publications in the Science journal describing the genetic origin of the mouse xid mutation.28, 29
The identified gene encodes a cytoplasmic protein-tyrosine kinase of 659 amino acids with the following domain organization from the N-terminus: Pleckstrin homology (PH), Tec homology,30, 31 SRC homology 3 (SH3), SH2, and catalytic domain (SH1), as reviewed in Smith et al.32 and Mohamed et al.33 Amino acid 481 in the adenosine triphosphate-binding cleft of the kinase domain is a cysteine residue (Figure 1). As described below, this residue is crucial for the activity of the new BTK inhibitors, as it forms a covalent bond with the majority of these medicines.

Linear representation of the BTK kinase domain depicting all known missense mutations causing XLA.37 Substituted residues in orange correspond to those reported in 2005,39 whereas those marked in red are the ones appearing from 2005 up until now. Coils, helixes and β-strands are color-coded, as depicted. Residues buried (B), exposed (E) or being in an intermediate (I) position according to Valiaho et al.38 are indicated below the amino-acid sequence. Boxed residues correspond to substitutions identified in ibrutinib- (IMBRUVICA) or acalabrutinib-treated patients, with cysteine 481 to serine substitution being the predominant change.82, 90, 91, 92, 93, 94, 95
The spectrum of mutations in XLA
The first two XLA mutations were reported already in the cloning paper26 with patient A coming from a family in southern Sweden. Both of these were missense mutations, thereby immediately providing information about crucial residues. Two years after the cloning, we established the international database for mutations in the BTK gene, named BTKbase,34, 35 and this online resource is continuously being updated.36, 37, 38 Currently it contains a total number of 1374 public variants representing 717 unique public DNA variants. Figure 1 depicts all the known missense mutations in the kinase domain, which is the region with the highest variation density.39 The figure shows that the number of reported replacements since the Lindvall report39 has increased from 107 (affecting 73 residues) to 164 (affecting 92 residues).
We recently described new substitutions and characterized all possible single-nucleotide variation (SNAV) that caused amino-acid replacements in the kinase domain.38 Altogether 1495 single-nucleotide variations can appear and as many as 2/3 are predicted to be harmful, while in only 10% of the sites all substitutions likely would be tolerated. There is only a weak correlation among the 92 affected residues (Figure 1) to coils, β-strands and helixes, with 33–44% of these sites affected. Instead, the relation to accessibility is much stronger with 64% of buried and only 21% of intermediate and 14% of the exposed sites being affected. It is likely that catalytic domains in other kinases show a similar dependency, with buried residues being the most sensitive to replacements.
The BTK signaling pathway
Sequencing of the BTK gene immediately revealed that the corresponding protein was a previously unknown cytoplasmic tyrosine kinase, and soon thereafter signaling from the B-cell receptor (BCR) was implicated as part of the upstream pathway. Takata and Kurosaki40 were the first to demonstrate that phospholipase Cγ2 (PLCγ2) is a bona fide substrate for BTK. The current view is that SRC-family kinases, in particular LYN, activate the non-catalytic BCR through the phosphorylation motif-containing Igα and Igβ proteins. These subsequently serve as docking stations for the SYK kinase, which phosphorylates the adaptor BLNK/SLP-65 that interacts with BTK among other molecules. LYN-mediated phosphorylation of tyrosine 551 in BTK, a residue conserved among all cytoplasmic protein-tyrosine kinases and corresponding to tyrosine 416 (in man)/419 (in the mouse) in SRC, activates the kinase.41
Following the generation of cell membrane-bound phosphatidylinositol 3,4,5-trisphosphate (PIP3) by phosphatidylinositol-3-kinase, BTK translocates to the inner leaflet of the cell membrane. The most N-terminal domain of BTK, the pleckstrin homology domain specifically tethers to PIP342 and can also be used as a highly efficient probe for the detection of PIP3.43 Phosphorylation of the PLCγ2 substrate subsequently generates inositol-3,4,5-phosphate and also activates protein kinase C and calcium signaling.44 A crucial further downstream component is nuclear factor-κB, which is an important survival regulator for B-lymphocytes.45, 46, 47, 48
Inactivation of BTK signaling seems to be controlled by dual serine/threonine phosphorylation in the kinase domain, which attracts 14-3-3 proteins targeting phosphorylated BTK for degradation.49 Also other serine phosphorylations seem to have a negative effect,50, 51 suggesting that serine/threonine phosphorylations turn off signaling of this tyrosine kinase. Expression of the BTK gene is under the control of a promoter containing both general and hematopoietic transcription factor-binding sites, as published in this journal two decades ago.52 The above scenario is similar in mice and humans, even if there are some species differences. Thus, for reasons that are not known in detail, deficiency of BTK causes a much more severe phenotype in humans as compared with mice.28, 29
In Drosophila melanogaster, the corresponding kinase is known as Btk29A.53 Lack of Btk29A in flies causes a lethal phenotype with reduced size of the embryonic ring canals.54 Conversely, flies with an absent type 2-splice form are viable but with many organs affected. Thus the nervous system is influenced by the deficiency, with the lifespan being reduced by 85%. In transgenic flies carrying the human BTK cDNA under the control of a heat-shock promoter, the phenotype can be reverted upon induction,55 demonstrating functional conservation during evolution. The BTK kinase therefore seems to be highly adaptive, and during development, it has been functionally rewired.56 Unexpectedly, Btk29A acts a tumor suppressor during oocyte development, as lack of the type 2-splice form causes germ cell tumors through expansion of the cystoblast population.57 However, it is the expression of Btk29A in the somatic escort cell, which controls the cystoblast differentiation. Btk29A phosphorylates β-catenin (arm) controlling piwi expression, which is needed for the silencing of transposons in a Wnt-dependent signaling pathway. BTK expression seems to first appear in the unicellular choanoflagellate, suggesting that the ancestors of this kinase originated >600 million years ago.58
The development of inhibitors
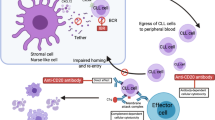
The first inhibitor for BTK was LFM-A13 [alpha-cyano-beta-hydroxy-beta-methyl-N-(2,5-dibromophenyl)propenamide], reported in 1999.59 Although this compound has been quite extensively studied, as evidenced by 62 citations appearing in PubMed, it was never used in a clinical setting. Instead, it was the compound initially referred to as ‘compound 4’,60 later known as PCI-32765, or ibrutinib (IMBRUVICA), and depicted in Figure 2, which set the stage. In 2010 Honigsberg et al.61 reported the beneficial effects in both the treatment of experimental leukemia and of autoimmunity. Thus it was found that PCI-32765 induced objective clinical responses in dogs with spontaneous B-cell non-Hodgkin lymphoma. Furthermore, in mice with collagen-induced arthritis, orally administered PCI-32765 reduced the level of circulating autoantibodies and completely suppressed disease and, furthermore, also inhibited autoantibody production and the development of kidney disease in a mouse lupus model. Importantly, occupancy of the catalytic site in BTK by PCI-32765 tightly correlated with blockade of BCR signaling and with in vivo efficacy. This was soon followed by reports on patient leukemia cells, which responded to the inhibitor in vitro as well as in the already mentioned IgH-Eμ-TCL1-transgenic mouse model for B-CLL.18, 62 Moreover, it was demonstrated that both adhesion and migration of patient CLL cells were affected.63, 64, 65

Chemical structure of ibrutinib (MW 440.5) and of acalabrutinib (MW 465.5), which both compete with the binding of adenosine triphosphate (ATP) to BTK. ATP (MW 507.2) is included for comparison.
The truly dramatic development began in 2013 following the trial reported in J Clin Oncol.66 A total of 56 patients with relapsed or refractory B-cell lymphoma or CLL received escalating oral doses of ibrutinib. Adverse events were mainly grades 1 and 2 in severity and self-limited. Consistent with the irreversible binding to cysteine 481, BTK occupancy was maintained for at least 24 h. The objective response rate was 60%, and complete response was noted in 16% of the patients. Rapid approval from the US Food and Drug Administration (FDA) followed: mantle cell lymphoma (MCL), November 2013; CLL, July 2014; and Waldenström’s macroglobulinemia (WM), January 2015; and the European Medicines Agency has also approved the drug for the treatment of these B-cell malignancies.67 The stunning development that ensued can be inferred from the number of citations according to PubMed. Up until the report in J Clin Oncol in 2013,66 there were 28 citations for ‘ibrutinib’; as of 26 July 2016, there are 629 citations, that is, a more than 20-fold increase over a 3-year period.
The role of BTK in tumor development
Based on the available literature, the role of BTK in tumor development is complex. I would first like to point out that activating BTK variants have not been reported in patients. The only BTK variations identified in cancer are those that occur after treatment with BTK inhibitors (depicted in Figure 1) and discussed in one of the following paragraphs. Experiments in mice, where inactivating mutations for the genes encoding BTK or BLNK/SLP-65 have been combined, clearly demonstrate that reduced BCR signaling can cause lymphomas.68 However, as this necessitates the introduction of several alterations in the DNA, this scenario is expected to be very rare in patients, and there are no convincing clinical reports to date. Moreover, certain amino-acid substitutions in BTK are oncogenic in transformation assays,33, 69 but their clinical relevance remains elusive. Reports on oncogenic forms of BTK outside the hematopoietic lineage also exist, but as BTK is normally not expressed in these cells, further studies are needed to confirm this possibility. Yet another option is to use BTK inhibitors to impair the activity of, for example, myeloid-derived suppressor cells, which are known to promote many tumors.70 This concept has been tested experimentally71 and could profoundly expand on the use of BTK inhibitors, if clinical correlates exist.
The current clinical landscape for BTK inhibitors
As mentioned, the first clinical report on a BTK inhibitor was published in January of 201366 and demonstrated a remarkable effect of ibrutinib on B-cell malignancies. Since then, the potent activity of this compound has been reported in several settings. I will begin with reviewing MCL, the first tumor for which ibrutinib treatment was FDA approved, followed by CLL, for which the largest number of treated patient exists, and end with a short paragraph on WM. Studies are also ongoing in other B-lymphocyte malignancies, such as diffuse large B-cell lymphoma,72 but will not be detailed here.
The effect on MCL
Only months after the very first clinical report on ibrutinib in 2013, this drug was shown to induce durable, single-agent efficacy in relapsed or refractory MCL.73 A phase III study comparing ibrutinib versus the mammalian target of rapamycin inhibitor, temsirolimus, in MCL, published in 2016, demonstrates that the BTK inhibitor yields significant improvement in progression-free survival and better tolerability in patients with relapsed or refractory MCL.74 However, long-term treatment outcome in MCL is not very good, despite unprecedented clinical activity, as primary and acquired resistance to ibrutinib is common.75 Thus there is a need for more efficient BTK inhibitors or for better combination therapies. In an open-label, phase II trial, it was shown that ibrutinib combined with rituximab, an anti-CD20 monoclonal, is active and well tolerated in patients with relapsed or refractory MCL.76 The triple combination of ibrutinib with the alkylating compound bendamustine and rituximab has also been investigated in studies, including but not focusing on MCL.72 The overall response rate in 12 MCL patients receiving ibrutinib together with bendamustine and rituximab was 94% with a complete remission in 76%, suggesting that combination therapies may be highly beneficial.77
The effect on CLL
Based on the excellent results in CLL from the first clinical report on ibrutinib,66 a large phase 1b-II trial followed, demonstrating similar activity in the cohort receiving 420 mg per day as in the one with the double dose.78 Several studies have shown a very potent effect of ibrutinib in elderly CLL patients.79 Ibrutinib treatment also yielded excellent results in patients with TP53 aberrations known to have dismal prognosis.80, 81 Like for MCL, a major interest is in combination therapies to be used when first-line treatments have failed. Among the ongoing combination phase III studies, the HELIOS trial has recently demonstrated that ibrutinib, when combined with bendamustine and rituximab as compared with bendamustine and rituximab alone shows 80% reduction in the risk of progression or death. Furthermore, the phase III RESONATE-2 trial shows a significant, 56% reduction for the risk of death with ibrutinib compared with chlorambucil, leading to the 2016 FDA approval of IMBRUVICA as first-line treatment for patients with CLL (http://www.janssen.com/fda-expands-imbruvica-label-include-new-data-two-key-phase-3-trials-adding-overall-survival-and).
The recent addition of the second-generation BTK inhibitor, acalabrutinib (Figure 2), has demonstrated that this compound may have certain advantages given its higher specificity for BTK.82 Thus the lack of inhibition of the related ITK kinase, expressed in both T and natural killer cells, could be of importance. Moreover, the absence of severe bleeds, thought to be secondary to effects on the TEC kinase in platelets, and the reported lack of atrial fibrillation, possibly caused by an effect on the receptor tyrosine kinases ErbB2/HER2/ErbB4/HER4,67 could also be beneficial. Importantly, the feared transformation of CLL to diffuse large B-cell lymphoma (Richter transformation), which is observed after ibrutinib treatment, albeit rarely, was so far not found in patients treated with acalabrutinib,82 although larger patient materials are needed in order to draw any firm conclusions. Therefore, the results from the ongoing phase III comparison between the two BTK inhibitors are eagerly awaited.
Apart from combining BTK inhibitors with well-established therapeutics, combinations could also be made with more recently developed compounds, such as the second-generation BCL2 inhibitor, venetoclax.83 Hence, it has been proposed that this combinatorial therapy may be advantageous, because these medicines rarely cause complete remission when used as single agents.84
The effect on WM
Ibrutinib treatment has demonstrated excellent effects in WM, with an overall response rate of 90.5% in 63 patients.85 Unexpectedly, it was recently proposed that the effect of ibrutinib on WM is not related to BTK but rather to an effect on the SRC family kinase HCK.86 Although the authors provide some support for this idea, it is not clear that a primary effect on BTK was ruled out, and for this reason it will be interesting to see how other BTK inhibitors with less activity on HCK fare in WM patients.
Other inhibitors for BTK
Also other BTK inhibitors have been used in the clinic, such as CC-292, formerly AVL-29287 and ONO/GS-4059. CC-292 is an irreversible binder, whereas ONO/GS-4059 binds reversibly. Limited information about their performance exists, but an overall response rate of 53% was recently observed in 84 in patients with CLL or SLL (small lymphocytic leukemia) receiving twice-daily dosing of CC-292.88 For ONO/GS-4059, 24/25 evaluable CLL patients (96%) responded, as did 11/12 patients with MCL (92%).89
Mutations causing resistance to BTK inhibitors
Resistance to BTK inhibitors can be caused by mutations. These variants frequently either affect BTK itself or downstream signaling components, preferentially BTK’s substrate PLCγ2. The most common resistance variant is the replacement of cysteine 481 with a serine residue, which has been observed both after ibrutinib and acalabrutinib treatment.82, 90, 91, 92, 93, 94 Figure 1 depicts all missense mutations observed in treated patients, including the replacement of C481 with serine. For PLCγ2, a few gain-of-function substitutions have been reported yielding a constitutively active enzyme, which no longer needs to be phosphorylated by BTK.82, 93 We have recently carried out mutation scanning for the BTK 481 codon and found that threonine substitution, which for genetic reasons should be rare, behaves similar to serine substitution causing resistance. We have also tried to explain the outcome of all other single-nucleotide exchanges for this codon.95 Recently, it was clearly demonstrated that resistance mutants exist prior to ibrutinib treatment and, furthermore, that genetic changes affecting non-BTK related components also might cause unresponsiveness.96
Brief historical outlook
The generation of the BTK inhibitors is a sign of how medical evolution takes place in discrete steps (Figure 3). A crucial step was the development of serum electrophoresis by Arne Tiselius in Uppsala in 1937,97 for which he was awarded the Nobel Prize in Chemistry in 1948 ‘for his research on electrophoresis and adsorption analysis, especially for his discoveries concerning the complex nature of the serum proteins’. Subsequently, Jan Waldenström, also residing in Uppsala, employed serum electrophoresis in his classical description of a tumor named after him.98

Timeline for developments and discoveries related to the earliest description of XLA, the BTK gene cloning and the development of the first BTK inhibitor used in the clinic. The corresponding references are provided in the paragraph entitled: ‘Brief historical outlook’.
Fifteen years after Tiselius’ original report, serum electrophoresis was applied at the Walter Reed General Hospital in Washington DC in the analysis of Bruton’s patient. Based on these results, Bruton, also in a single-authored paper, provided evidence that the missing factor in the patient was gammaglobulin,1 making XLA the first inherited immunodeficiency in which a disease mechanism was revealed. Over the next four decades, the development of molecular biology and genetics paved the way for the positional cloning effort, which identified the disease gene and detailed the first mutations.26 In 1993, on my way to present the BTK gene cloning at a Keystone conference in Taos, I, unexpectedly, had the great honor of meeting with Dr Bruton (Figure 4), as further detailed in Winkelstein and Smith.99 In 2007, the initial description of ibrutinib appeared in print60 with the first paper on its beneficial clinical effects published 6 years later. In 2015, 78 years after Tiselius’ landmark paper, this compound was reported to be highly efficient as treatment for patients with WM, thereby closing the circle (Figure 3). To this end, I could also mention that Professor Waldenström in the 1970s was one of my teachers in internal medicine at the Serafimer Hospital, another ingredient in the ‘full circle’.

Meeting with Dr Ogden C Bruton (sitting) in January 1993 when the author visited him after cloning of the BTK gene. (standing) Dr Jeffrey D Thomas (left), the author, CIES (center) and Mrs Kathryn D Bruton (right). Reproduced from Smith and Notarangelo2 with permission from the publisher.
Concluding remarks
The development of the BTK inhibitors demonstrates how basic research laid the foundation for clinical development in ways that cannot be predicted. Thus, while the possible use of such inhibitors for the treatment of neoplasms existed as one of many options, there was no direct evidence that this approach would be successful. Instead, as it turned out, BTK does not seem to be directly involved in tumor development but for certain leukemias and lymphomas is essential for the thriving of the malignant cells. Although BTK inhibitors have been said to revolutionize treatment, we are in an era of rapid development in the therapeutic arena. Numerous compounds are being investigated, both alone and in combinations. It is not possible to make any insightful predictions of compounds that will show the best profile in terms of efficacy and adverse effects, but certainly BTK inhibitors have demonstrated a good balance. The rare existence of a cysteine residue in the catalytic site, which is a signature of BTK and other members of this family of kinases, is definitely a major asset, as it enhances the ‘effective specificity’ by the irreversible binding to only those targets.
With the second generation of BTK inhibitors showing increased specificity, as well as permitting higher dosing, significant improvements have already been accomplished. This has also resulted in industrial acquisitions with AstraZeneca in February 2016 buying 55% of the share capital of Acerta Pharma for $2.5 billion US$ with a further unconditional payment of $1.5 billion. Although this review is focused on oncology, BTK inhibitors are certainly of great interest also for other disorders caused by B cells, including autoimmunity where resistance variants are not expected to occur. Thus the future for BTK inhibitors seems bright, but only the test of time will reveal whether these medicines will remain in the therapeutic arsenal for years to come.
References
Bruton OC . Agammaglobulinemia. Pediatrics 1952; 9: 722–728.
Smith CI, Notarangelo LD . Molecular basis for X-linked immunodeficiencies. Adv Genet 1997; 35: 57–115.
Sideras P, Smith CI . Molecular and cellular aspects of X-linked agammaglobulinemia. Adv Immunol 1995; 59: 135–223.
Cohn EJ . Chemical, physiological, immunological properties and clinical uses of blood derivatives. Praxis 1947; 36: 280.
Gardulf A, Hammarstrom L, Smith CI . Home treatment of hypogammaglobulinaemia with subcutaneous gammaglobulin by rapid infusion. Lancet 1991; 338: 162–166.
Jolles S, Orange JS, Gardulf A, Stein MR, Shapiro R, Borte M et al. Current treatment options with immunoglobulin G for the individualization of care in patients with primary immunodeficiency disease. Clin Exp Immunol 2015; 179: 146–160.
Naor D, Bentwich Z, Cividalli G . Inability of peripheral lymphoid cells of agammaglobulinaemic patients to bind radioiodinated albumins. Aust J Exp Biol Med Sci 1969; 47: 759–761.
Cooper MD, Lawton AR, Bockman DE . Agammaglobulinaemia with B lymphocytes. Specific defect of plasma-cell differentiation. Lancet 1971; 2: 791–794.
Conley ME, Dobbs AK, Farmer DM, Kilic S, Paris K, Grigoriadou S et al. Primary B cell immunodeficiencies: comparisons and contrasts. Annu Rev Immunol 2009; 27: 199–227.
Berglof A, Turunen JJ, Gissberg O, Bestas B, Blomberg KE, Smith CI . Agammaglobulinemia: causative mutations and their implications for novel therapies. Exp Rev Clin Immunol 2013; 9: 1205–1221.
Noordzij JG, de Bruin-Versteeg S, Comans-Bitter WM, Hartwig NG, Hendriks RW, de Groot R et al. Composition of precursor B-cell compartment in bone marrow from patients with X-linked agammaglobulinemia compared with healthy children. Pediatr Res 2002; 51: 159–168.
Zech L, Hammarstrom L, Smith CI . Chromosomal aberrations in a case of T-cell CLL with concomitant IgA myeloma. Int J Cancer 1983; 32: 431–435.
Zech L, Gahrton G, Hammarstrom L, Juliusson G, Mellstedt H, Robert KH et al. Inversion of chromosome 14 marks human T-cell chronic lymphocytic leukaemia. Nature 1984; 308: 858–860.
Rabbitts TH, Stinson A, Forster A, Foroni L, Luzzatto L, Catovsky D et al. Heterogeneity of T-cell beta-chain gene rearrangements in human leukaemias and lymphomas. EMBO J 1985; 4: 2217–2224.
Mengle-Gaw L, Willard HF, Smith CI, Hammarstrom L, Fischer P, Sherrington P et al. Human T-cell tumours containing chromosome 14 inversion or translocation with breakpoints proximal to immunoglobulin joining regions at 14q32. EMBO J 1987; 6: 2273–2280.
Virgilio L, Narducci MG, Isobe M, Billips LG, Cooper MD, Croce CM et al. Identification of the TCL1 gene involved in T-cell malignancies. Proc Natl Acad Sci USA 1994; 91: 12530–12534.
Bichi R, Shinton SA, Martin ES, Koval A, Calin GA, Cesari R et al. Human chronic lymphocytic leukemia modeled in mouse by targeted TCL1 expression. Proc Natl Acad Sci USA 2002; 99: 6955–6960.
Ponader S, Chen SS, Buggy JJ, Balakrishnan K, Gandhi V, Wierda WG et al. The Bruton tyrosine kinase inhibitor PCI-32765 thwarts chronic lymphocytic leukemia cell survival and tissue homing in vitro and in vivo. Blood 2012; 119: 1182–1189.
Woyach JA, Bojnik E, Ruppert AS, Stefanovski MR, Goettl VM, Smucker KA et al. Bruton's tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 2014; 123: 1207–1213.
Chen SS, Chang BY, Chang S, Tong T, Ham S, Sherry B et al. BTK inhibition results in impaired CXCR4 chemokine receptor surface expression, signaling and function in chronic lymphocytic leukemia. Leukemia 2016; 30: 833–843.
Yigit B, Halibozek PJ, Chen SS, O'Keeffe MS, Arnason J, Avigan D et al. A combination of an anti-SLAMF6 antibody and ibrutinib efficiently abrogates expansion of chronic lymphocytic leukemia cells. Oncotarget 2016; 7: 26346–26360.
Smith CI, Hammarstrom L . Cellular basis of immunodeficiency. Ann Clin Res 1987; 19: 220–229.
Sanger R, Race RR . The Xg blood groups and familial hypogammaglobulinaemia. Lancet 1963; 1: 859–860.
Guioli S, Arveiler B, Bardoni B, Notarangelo LD, Panina P, Duse M et al. Close linkage of probe p212 (DXS178) to X-linked agammaglobulinemia. Hum Genet 1989; 84: 19–21.
Kwan SP, Terwilliger J, Parmley R, Raghu G, Sandkuyl LA, Ott J et al. Identification of a closely linked DNA marker, DXS178, to further refine the X-linked agammaglobulinemia locus. Genomics 1990; 6: 238–242.
Vetrie D, Vorechovsky I, Sideras P, Holland J, Davies A, Flinter F et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 1993; 361: 226–233.
Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 1993; 72: 279–290.
Thomas JD, Sideras P, Smith CI, Vorechovsky I, Chapman V, Paul WE . Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science 1993; 261: 355–358.
Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L et al. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science 1993; 261: 358–361.
Smith CI, Islam KB, Vorechovsky I, Olerup O, Wallin E, Rabbani H et al. X-linked agammaglobulinemia and other immunoglobulin deficiencies. Immunol Rev 1994; 138: 159–183.
Vihinen M, Nilsson L, Smith CI . Tec homology (TH) adjacent to the PH domain. FEBS Lett 1994; 350: 263–265.
Smith CI, Islam TC, Mattsson PT, Mohamed AJ, Nore BF, Vihinen M . The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays 2001; 23: 436–446.
Mohamed AJ, Yu L, Backesjo CM, Vargas L, Faryal R, Aints A et al. Bruton's tyrosine kinase (Btk): function, regulation, and transformation with special emphasis on the PH domain. Immunol Rev 2009; 228: 58–73.
Smith CI, Vihinen M . Immunodeficiency mutation databases—a new research tool. Immunol Today 1996; 17: 495–496.
Vihinen M, Cooper MD, de Saint Basile G, Fischer A, Good RA, Hendriks RW et al. BTKbase: a database of XLA-causing mutations. International Study Group. Immunol Today 1995; 16: 460–465.
Vihinen M, Brandau O, Branden LJ, Kwan SP, Lappalainen I, Lester T et al. BTKbase, mutation database for X-linked agammaglobulinemia (XLA). Nucleic Acids Res 1998; 26: 242–247.
Valiaho J, Smith CI, Vihinen M . BTKbase: the mutation database for X-linked agammaglobulinemia. Hum Mutat 2006; 27: 1209–1217.
Valiaho J, Faisal I, Ortutay C, Smith CI, Vihinen M . Characterization of all possible single-nucleotide change caused amino acid substitutions in the kinase domain of Bruton tyrosine kinase. Hum Mutat 2015; 36: 638–647.
Lindvall JM, Blomberg KE, Valiaho J, Vargas L, Heinonen JE, Berglof A et al. Bruton's tyrosine kinase: cell biology, sequence conservation, mutation spectrum, siRNA modifications, and expression profiling. Immunol Rev 2005; 203: 200–215.
Takata M, Kurosaki T . A role for Bruton's tyrosine kinase in B cell antigen receptor-mediated activation of phospholipase C-gamma 2. J Exp Med 1996; 184: 31–40.
Rawlings DJ, Scharenberg AM, Park H, Wahl MI, Lin S, Kato RM et al. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science 1996; 271: 822–825.
Salim K, Bottomley MJ, Querfurth E, Zvelebil MJ, Gout I, Scaife R et al. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton's tyrosine kinase. EMBO J 1996; 15: 6241–6250.
Zoncu R, Perera RM, Balkin DM, Pirruccello M, Toomre D, De Camilli P . A phosphoinositide switch controls the maturation and signaling properties of APPL endosomes. Cell 2009; 136: 1110–1121.
Baba Y, Kurosaki T . Role of calcium signaling in B cell activation and biology. Curr Top Microbiol Immunol 2016; 393: 143–174.
Bajpai UD, Zhang K, Teutsch M, Sen R, Wortis HH . Bruton's tyrosine kinase links the B cell receptor to nuclear factor kappaB activation. J Exp Med 2000; 191: 1735–1744.
Petro JB, Rahman SM, Ballard DW, Khan WN . Bruton's tyrosine kinase is required for activation of IkappaB kinase and nuclear factor kappaB in response to B cell receptor engagement. J Exp Med 2000; 191: 1745–1754.
Derudder E, Herzog S, Labi V, Yasuda T, Kochert K, Janz M et al. Canonical NF-kappaB signaling is uniquely required for the long-term persistence of functional mature B cells. Proc Natl Acad Sci USA 2016; 113: 5065–5070.
Yu L, Mohamed AJ, Simonson OE, Vargas L, Blomberg KE, Bjorkstrand B et al. Proteasome-dependent autoregulation of Bruton tyrosine kinase (Btk) promoter via NF-kappaB. Blood 2008; 111: 4617–4626.
Mohammad DK, Nore BF, Hussain A, Gustafsson MO, Mohamed AJ, Smith CI . Dual phosphorylation of Btk by Akt/protein kinase b provides docking for 14-3-3zeta, regulates shuttling, and attenuates both tonic and induced signaling in B cells. Mol Cell Biol 2013; 33: 3214–3226.
Kang SW, Wahl MI, Chu J, Kitaura J, Kawakami Y, Kato RM et al. PKCbeta modulates antigen receptor signaling via regulation of Btk membrane localization. EMBO J 2001; 20: 5692–5702.
Yu L, Mohamed AJ, Vargas L, Berglof A, Finn G, Lu KP et al. Regulation of Bruton tyrosine kinase by the peptidylprolyl isomerase Pin1. J. Biol Chem 2006; 281: 18201–18207.
Muller S, Sideras P, Smith CI, Xanthopoulos KG . Cell specific expression of human Bruton's agammaglobulinemia tyrosine kinase gene (Btk) is regulated by Sp1- and Spi-1/PU.1-family members. Oncogene 1996; 13: 1955–1964.
Baba K, Takeshita A, Majima K, Ueda R, Kondo S, Juni N et al. The Drosophila Bruton's tyrosine kinase (Btk) homolog is required for adult survival and male genital formation. Mol Cell Biol 1999; 19: 4405–4413.
Guarnieri DJ, Dodson GS, Simon MA . SRC64 regulates the localization of a Tec-family kinase required for Drosophila ring canal growth. Mol Cell 1998; 1: 831–840.
Hamada N, Backesjo CM, Smith CI, Yamamoto D . Functional replacement of Drosophila Btk29A with human Btk in male genital development and survival. FEBS Lett 2005; 579: 4131–4137.
Nawaz HM, Kylsten P, Hamada N, Yamamoto D, Smith CI, Lindvall JM . Differential evolutionary wiring of the tyrosine kinase Btk. PLoS One 2012; 7: e35640.
Hamada-Kawaguchi N, Nore BF, Kuwada Y, Smith CI, Yamamoto D . Btk29A promotes Wnt4 signaling in the niche to terminate germ cell proliferation in Drosophila. Science 2014; 343: 294–297.
Ortutay C, Nore BF, Vihinen M, Smith CI . Phylogeny of Tec family kinases identification of a premetazoan origin of Btk, Bmx, Itk, Tec, Txk, and the Btk regulator SH3BP5. Adv Genet 2008; 64: 51–80.
Mahajan S, Ghosh S, Sudbeck EA, Zheng Y, Downs S, Hupke M et al. Rational design and synthesis of a novel anti-leukemic agent targeting Bruton's tyrosine kinase (BTK), LFM-A13 [alpha-cyano-beta-hydroxy-beta-methyl-N-(2, 5-dibromophenyl)propenamide]. J Biol Chem 1999; 274: 9587–9599.
Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC et al. Discovery of selective irreversible inhibitors for Bruton's tyrosine kinase. ChemMedChem 2007; 2: 58–61.
Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA 2010; 107: 13075–13080.
Herman SE, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011; 117: 6287–6296.
de Rooij MF, Kuil A, Geest CR, Eldering E, Chang BY, Buggy JJ et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 2012; 119: 2590–2594.
Davids MS, Burger JA . Cell trafficking in chronic lymphocytic leukemia. Open J Hematol 2012; 3.
de Rooij MF, Kuil A, Kater AP, Kersten MJ, Pals ST, Spaargaren M . Ibrutinib and idelalisib synergistically target BCR-controlled adhesion in MCL and CLL: a rationale for combination therapy. Blood 2015; 125: 2306–2309.
Advani RH, Buggy JJ, Sharman JP, Smith SM, Boyd TE, Grant B et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 2013; 31: 88–94.
Berglof A, Hamasy A, Meinke S, Palma M, Krstic A, Mansson R et al. Targets for ibrutinib beyond b cell malignancies. Scand J Immunol 2015; 82: 208–217.
Kersseboom R, Middendorp S, Dingjan GM, Dahlenborg K, Reth M, Jumaa H et al. Bruton's tyrosine kinase cooperates with the B cell linker protein SLP-65 as a tumor suppressor in Pre-B cells. J Exp Med 2003; 198: 91–98.
Li T, Tsukada S, Satterthwaite A, Havlik MH, Park H, Takatsu K et al. Activation of Bruton's tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH) domain. Immunity 1995; 2: 451–460.
Kumar V, Patel S, Tcyganov E, Gabrilovich DI . The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol 2016; 37: 208–220.
Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S et al. Myeloid-derived suppressor cells express Bruton's tyrosine kinase and can be depleted in tumor bearing hosts by ibrutinib treatment. Cancer Res 2016; 8: 2125–2136.
Younes A, Thieblemont C, Morschhauser F, Flinn I, Friedberg JW, Amorim S et al. Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 2014; 15: 1019–1026.
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 2013; 369: 507–516.
Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M et al. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomised, open-label, phase 3 study. Lancet 2016; 387: 770–778.
Martin P, Maddocks K, Leonard JP, Ruan J, Goy A, Wagner-Johnston N et al. Postibrutinib outcomes in patients with mantle cell lymphoma. Blood 2016; 127: 1559–1563.
Wang ML, Lee H, Chuang H, Wagner-Bartak N, Hagemeister F, Westin J et al. Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: a single-centre, open-label, phase 2 trial. Lancet Oncol 2016; 17: 48–56.
Maddocks K, Christian B, Jaglowski S, Flynn J, Jones JA, Porcu P et al. A phase 1/1b study of rituximab, bendamustine, and ibrutinib in patients with untreated and relapsed/refractory non-Hodgkin lymphoma. Blood 2015; 125: 242–248.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 2013; 369: 32–42.
O'Brien S, Furman RR, Coutre SE, Sharman JP, Burger JA, Blum KA et al. Ibrutinib as initial therapy for elderly patients with chronic lymphocytic leukaemia or small lymphocytic lymphoma: an open-label, multicentre, phase 1b/2 trial. Lancet Oncol 2014; 15: 48–58.
Farooqui MZ, Valdez J, Martyr S, Aue G, Saba N, Niemann CU et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single-arm trial. Lancet Oncol 2015; 16: 169–176.
Winqvist M, Asklid A, Andersson P-O, Karlsson K, Karlsson C, Lauri B et alA study from the Swedish CLL Group. Realworld results of ibrutinib in patients with relapsed or refractory chronic lymphocytic leukemia: data from 95 consecutive patients treated in a compassionate use program. Haematologica 2016, e-pub ahead of print 19 May 2016 doi:10.3324/haematol.2016.144576.
Byrd JC, Harrington B, O'Brien S, Jones JA, Schuh A, Devereux S et al. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. The N Engl J Med 2016; 374: 323–332.
Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med 2016; 374: 311–322.
Wilson WH . Progress in chronic lymphocytic leukemia with targeted therapy. N Engl J Med 2016; 374: 386–388.
Treon SP, Tripsas CK, Meid K, Warren D, Varma G, Green R et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med 2015; 372: 1430–1440.
Yang G, Buhrlage S, Tan L, Liu X, Chen J, Xu L et al. HCK is a survival determinant transactivated by mutated MYD88, and a direct target of ibrutinib. Blood 2016; 25: 3237–3252.
Evans EK, Tester R, Aslanian S, Karp R, Sheets M, Labenski MT et al. Inhibition of Btk with CC-292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther 2013; 346: 219–228.
Brown JR, Harb WA, Hill BT, Gabrilove J, Sharman JP, Schreeder MT et al. Phase 1 study of single-agent CC-292, a highly selective Bruton's tyrosine kinase inhibitor, in relapsed or refractory chronic lymphocytic leukemia. Haematologica 2016; 101: e295–e298.
Walter HS, Rule SA, Dyer MJ, Karlin L, Jones C, Cazin B et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood 2016; 127: 411–419.
Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS et al. Resistance mechanisms for the Bruton's tyrosine kinase inhibitor ibrutinib. N Engl J Med 2014; 370: 2286–2294.
Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P et al. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov 2014; 4: 1022–1035.
Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A et al. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med 2014; 370: 2352–2354.
Cheng S, Guo A, Lu P, Ma J, Coleman M, Wang YL . Functional characterization of BTK(C481S) mutation that confers ibrutinib resistance: exploration of alternative kinase inhibitors. Leukemia 2015; 29: 895–900.
Maddocks KJ, Ruppert AS, Lozanski G, Heerema NA, Zhao W, Abruzzo L et al. Etiology of ibrutinib therapy discontinuation and outcomes in patients with chronic lymphocytic leukemia. JAMA Oncol 2015; 1: 80–87.
Hamasy A, Wang Q, Blomberg KE, Mohammad DK, Yu L, Vihinen M et al. Substitution scanning identifies a novel, catalytically active ibrutinib resistant BTK cysteine 481 to threonine (C481T) variant. Leukemia 2016, e-pub ahead of print 10 June 2016 doi:10.1038/leu.2016.153.
Burger JA, Landau DA, Taylor-Weiner A, Bozic I, Zhang H, Sarosiek K et al. Clonal evolution in patients with chronic lymphocytic leukaemia developing resistance to BTK inhibition. Nat Commun 2016; 7: 11589.
Tiselius A . Electrophoresis of serum globulin. I. Biochem J 1937; 31: 313–317.
Waldenstrom J . Incipient myelomatosis or ‘essential’ hyper-globulinemia with fibrinogenopenia—a new syndrome? Acta Med Scand 1994; 117: 216–247.
Winkelstein JA, Smith CI. B-cell defects: from X-linked recessive to autosomal recessive agammaglobulinemia In: Etzioni AO, Ochs HD (eds). Primary Immunodeficiency Disorders. A Historic and Scientific Perspective. Academic Press: London, UK, 2014, pp 127–138.
Acknowledgements
This work was supported by the Swedish Cancer Society (CAN2013/389), the Swedish Medical Research Council (K2015-68X-11247-21-3) and the Swedish County Council (ALF-project 2012006). I thank Anna Berglöf, Qing Wang, Abdulrahman Hamasy and Rula Zain for their kind help and Anthony Wright, Birgitta Sander and Anders Österborg for valuable comments on the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The author declares no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Smith, C. From identification of the BTK kinase to effective management of leukemia. Oncogene 36, 2045–2053 (2017). https://doi.org/10.1038/onc.2016.343
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.343
This article is cited by
-
Battling BTK mutants with noncovalent inhibitors that overcome Cys481 and Thr474 mutations in Waldenström macroglobulinemia therapy: structural mechanistic insights on the role of fenebrutinib
Journal of Molecular Modeling (2022)
-
BTK gatekeeper residue variation combined with cysteine 481 substitution causes super-resistance to irreversible inhibitors acalabrutinib, ibrutinib and zanubrutinib
Leukemia (2021)
-
Development of BTK inhibitors for the treatment of B-cell malignancies
Archives of Pharmacal Research (2019)
-
Phase I/II Clinical Trial-Based Early Economic Evaluation of Acalabrutinib for Relapsed Chronic Lymphocytic Leukaemia
Applied Health Economics and Health Policy (2019)
-
Bruton’s tyrosine kinase potentiates ALK signaling and serves as a potential therapeutic target of neuroblastoma
Oncogene (2018)
