
Abstract
Differentiated epithelial structure communicates with individual constituent epithelial cells to suppress their proliferation activity. However, the pathways linking epithelial structure to cessation of the cell proliferation machinery or to unscheduled proliferation in the context of tumorigenesis are not well defined. Here we demonstrate the strong impact of compromised epithelial integrity on normal and oncogenic Myc-driven proliferation in three-dimensional mammary epithelial organoid culture. Systematic silencing of 34 human homologs of Drosophila genes, with previously established functions in control of epithelial integrity, demonstrates a role for human genes of apico-basal polarity, Wnt and Hippo pathways and actin dynamics in regulation of the size, integrity and cell proliferation in organoids. Perturbation of these pathways leads to diverse functional interactions with Myc: manifested as a RhoA-dependent synthetic lethality and Par6-dependent effects on the cell cycle. Furthermore, we show a role for Par6G as a negative regulator of the phosphatidylinositol 3′-kinase/phosphoinositide-dependent protein kinase 1/Akt pathway and epithelial cell proliferation and evidence for frequent inactivation of Par6G gene in epithelial cancers. The findings demonstrate that determinants of epithelial structure regulate the cell proliferation activity via conserved and cancer-relevant regulatory circuitries, which are important for epithelial cell cycle restriction and may provide new targets for therapeutic intervention.
Similar content being viewed by others

Introduction
Epithelial tissues are formed by organized, differentiated and mostly quiescent epithelial cells, which together produce the biological activity of epithelium. The highly cohesive epithelial tissues need to have a system that communicates from the level of epithelial structure to the level of individual epithelial cells, as to maintain their quiescent but simultaneously biologically active state. However, it is poorly understood, how the epithelial structure and individual cell components communicate with each other as to keep the tissue focused on biologically important tasks of quiescent epithelial tissue, such as secretion, and simultaneously prevent proliferative and apoptotic activities.1, 2 Better understanding of these mechanisms will shed new light into epithelial biology as well as mechanisms initiating and promoting tumorigenesis.
Epithelial structure arises from polarized epithelial cells that are bound to neighboring cells and attached to the extracellular matrix via specialized junctions. The polarity is formed when plasma membranes are organized into apical and basolateral domains, defined by asymmetric distribution of proteins managing intracellular polarity and pericellular cell–cell and cell–extracellular matrix contacts. Thus the inherent organization of epithelial cells provides a scaffold for correct positioning of the cohesive interactions, directional secretion and spatially controlled signaling pathways.3 Although the molecular pathways mediating crosstalk between the epithelial structure and the cell cycle of individual epithelial cells are in most part unclear, there are several known examples of proteins that can simultaneously control both epithelial integrity and the cell cycle machinery.1 For example, beta-catenin acts as an adhesion molecule in adherens junctions, whereas in Wnt signaling pool the same protein activates transcription in the nucleus to stimulate cell proliferation.4 Furthermore, tumor-suppressor NF2/Merlin may limit proliferation by engaging the adherens junctions protein E-cadherin to activated epithelial growth factor receptor in contact inhibited cells.5 The status of tight junctions (TJ) also controls signaling to proliferation machinery. For example, ZONAB is a Y-box transcription factor that shuttles between TJs, where it binds to ZO-1, and the nucleus where it acts as a cell proliferation-promoting transcription factor.6 ZO-2 has been reported to shuttle from the cytoplasmic surface of TJs to the nucleus where it interacts with transcription factors and YAP, the transcriptional coactivator of the Hippo pathway.7, 8
Genetic experiments in Drosophila provide compelling evidence that genes typically associated with regulation of the cell polarity and epithelial integrity also control cell proliferation. Inactivation of neoplastic tumor-suppressor genes (nTSG), which include the core cell polarity genes scribble, discs large 1 and lethal (2) giant larvae, causes not only loss of epithelial monolayer integrity but also extensive hyperproliferation and development of tumors. Furthermore, the tumorigenic effects resulting from loss of nTSG are dramatically enhanced when combined with oncogenes.9, 10, 11 However, while loss of epithelial integrity clearly promotes neoplastic proliferation, an intact epithelial integrity does not always curtail overproliferation. For example, inactivation of hyperplastic tumor-suppressor genes (hTSG) promotes increased cell proliferation and tissue overgrowth without disrupting the organized structure of epithelial monolayer.12 The hTSGs include genes such as pten, tsc1, tsc2 and merlin whose counterparts are frequently mutated in human cancers.
In humans, loss of epithelial integrity is a defining feature of malignant cancer. Therefore, it is an interesting question whether there are specific and prevalent genetic alterations that promote carcinogenesis by compromising epithelial integrity.2, 13, 14, 15, 16, 17 Several human genes of cell junction proteins, such as the CDH1 (E-cadherin) or CTNNB1 (beta-catenin), are frequently mutated or altered in cancer.17 Furthermore, DLG and SCRIB, encoded by human homologues of discs large 1 and scribble, are targeted to degradation by oncogenic papillomaviruses.18 In mice, targeted inactivation of SCRIB or LKB1/PAR4 and PARD3 promotes mammary tumorigenesis when combined with oncogenic Myc or Ras.19, 20, 21, 22 However, the overall contribution of compromised epithelial integrity to human tumor progression is still unclear.
Here we explored the impact of compromised epithelial integrity on proliferation control using loss-of-function genetics and a three-dimensional (3D) culture model of human mammary epithelial structures. The non-transformed mammary epithelial MCF10A cells proliferate in reconstituted basement membrane, forming polarized acinar structures with hollow lumen.23, 24 At the end of the morphogenesis, the acinar cells enter quiescence and become surprisingly proliferation resistant, restraining proliferative signals even from oncogenic Myc or Akt.19, 24 Therefore, MCF10A organoids provide a model to elucidate genetic mechanisms affecting epithelial cell cycle restriction and escape from this restriction. Our functional genetic approach demonstrates a role for conserved regulatory circuitries of epithelial integrity in cell cycle regulation of the organized mammalian epithelial structure. The results further indicate a role for Par6B and Par6G proteins in regulation of the Akt pathway and epithelial cell cycle restriction and they show that loss-of-function mutations frequently targets PARD6G in human cancer.
Results
Defining gene candidates at the intersection of epithelial integrity and proliferation control
To identify molecular pathways that may co-regulate epithelial integrity and cell proliferation, we mined genetic information related to well-established Drosophila epithelial phenotypes. The Interactive Fly database (http://www.sdbonline.org/fly/aimain/1aahome.htm; 47th Edition)25 was used as a source of annotated functional genetic data, and we enlisted genes present in at least one but commonly in several of the subcategories: tumor suppression, apico-basal cell polarity, tissue polarity, junctional complexes and their regulators, and asymmetric cell division. The search yielded 65 Drosophila genes, including nTSGs, hTSGs, members of the Hippo tumor-suppressor pathway, cell polarity and cell adhesion regulators, Ezrin–Radixin–Moesin (ERM) proteins and other signaling molecules.12, 26, 27, 28, 29 Their human homologs were identified from Ensembl (www.ensembl.org) and named as putative human epithelial integrity regulating (hEIR) genes. The structure of tight junctions (septate junctions in Drosophila) is different between flies and mammals, and for better coverage, the hEIR gene set was complemented with human TJ-specific genes. Notably, MCF10A cells do not have TJ structures but express functional TJ proteins.30 The final number of hEIR genes is 77 (Supplementary Table S1).
Computational analysis of tumor-suppressor function
The fly homologs of hEIR genes commonly exhibit tumor-suppressor functions, and therefore we sought to determine how commonly hEIR genes are targets of disabling genetic alterations in human cancer. Human TSGs are defined by mutations, deletions and/or loss of expression leading to complete loss or diminished protein activity in certain or many types of human cancers.31 However, no single criterion alone is sufficient to describe a TSG. To explore evidence for loss-of-function mutations in broader scope, we developed a computational method that integrates genomic and expression data available from public cancer gene databases. Briefly, the Tumor-Suppressor Predictor (TSP) program first gathers evidence for inactivating type of genetic alterations in cancer by taking into account gene expression profiles, somatic mutation frequencies and chromosomal deletions. Next, TSP uses these data to calculate a single gene-specific score value that reflects the total evidence of tumor-suppressor function for each gene (see Supplementary Material).
We calculated a TSP score for all human genes. The lowest score for reference TSGs was 0.0002, which was used as a threshold in the whole genome analysis (Figure 1a). The analysis identified 1252 human genes above the threshold (Supplementary Table S2) and 15 hEIR genes out of 77 (~20%) had a high TSP score value (>0.0002; Figures 1a and b). In addition, literature mining identified seven hEIR genes or their proteins, including GSK3B and TSC2, as targets of inactivation by oncogenic or transforming viruses (Supplementary Table S3). These results robustly indicate that at least one-fifth of the hEIR genes are frequently targeted by inactivating genetic changes in the examined cancers, which notion encouraged further functional analysis of hEIR genes in a reductionist MCF10A model of epithelial tumorigenesis.

Identification of epithelial structure regulating genes with tumor-suppressor function. (a) hEIR genes with high score for tumor-suppressor properties. All human genes, including hEIR genes, were ranked according to evidence for inactivating type of genetic alterations in cancer using the TSP algorithm. Left side of the column shows the rank and score for three classical TSGs and the right side shows hEIR genes with the highest rank and score. (b) Venn diagram indicating all human genes above the tumor-suppressor value threshold (1252), hEIR genes (77) and the overlapping genes (15). (c) A workflow illustrating the throughput of genes and shRNA constructs at different stages of the screen and the assay end points.
Short hairpin RNA (shRNA) screen to determine impact of hEIR gene loss on epithelial integrity and proliferation control
We employed an shRNA-mediated gene silencing approach to determine the impact of hEIR gene suppression on the formation of acinar architecture and establishment of the proliferation restriction in 3D MCF10A structures. A library of 219 lentiviral shRNA constructs, comprising 2–4 shRNA constructs per hEIR gene, was designed and validated in MCF10A cells (Supplementary Tables S4–S6). Off-targeting shRNAs were excluded as described in Supplementary Materials. In total, 52 validated shRNAs showed potent silencing activity against 34 hEIR genes (Supplementary Table S6, Supplementary Figure S1A). MCF10A cells were lentivirally transduced with the validated shRNA constructs in arrays—each transduced cell population, representing a single gene knockdown. Subsequently, the cells were seeded in Matrigel to form epithelial structures. Using this model system, we performed two independent screens (Figure 1c).
The first screen was designed to identify genes important for establishment of symmetric acinar morphology, size of the structures and cell cycle restriction (Figure 1c; first screen: 1.1). The set of 52 validated shRNAs, targeting 34 hEIR genes, were transduced in MCF10A-M cells (M stands for latent inactive MycER, see below). Microscopic examination revealed striking alterations in the size and symmetry of the structures on day 10 (Figure 2a), quantified using digital image analysis (Supplementary Figures S1B and C). In total, 16 hEIR gene knockdowns either increased or decreased the size of the structures (Figure 2b; ⩾20% change in the average size). Eleven hEIR gene knockdowns led to development of abnormally large acini (Figure 2b), and the most prominent overgrowth was observed in structures expressing diminished DVL3 (three shRNAs) or MPP5 (PALS1) (two shRNAs). Among the 11 hEIR gene knockdowns that increased acinar size, silencing of PARD6G, NUMB, DVL3, STK3 and MPP5 (PALS1) also induced misshapen morphology (Figure 2c; ⩾10% change in the average circularity). The size and circularity values corresponding to shRNA-induced acinar phenotypes are scatter plotted in Figure 2d, illustrating the relative differences. The DVL3 phenotype was further studied; the structures formed thick branches, which thrusted outwards from the structures. The protrusions commonly folded back and fused with other branches or the body of the structure (Supplementary Figures S2A and B; Supplementary Videos S1–S4). The uncontrolled branching phenotype was accompanied with disrupted apical polarity, whereas the basal polarity was still preserved (Figure 2e). Thus the defining differences between a controlled branching, for example, branching morphogenesis, and uncontrolled branching, caused by DVL3 loss, include loss of orientation in branching process and loss of apical polarity. Notably, all gene knockdowns that compromised the symmetric shape of structures in this study increased the acinar size, suggesting that establishment of epithelial integrity has an important role in limiting organoid growth in matrices.

Functional identification of genes regulating epithelial structure and cell cycle restriction. (a) Confocal images of hyperplastic and misshapen day-10 MCF10A epithelial structures. The acinar structures express shRNAs targeting MOB1B, GKS3B and DVL3 (shScra; scrambled control). The structures were immunostained for E-cadherin and nuclei to visualize acinar ultrastructure. Silencing of MOB1B and GKS3b leads to formation of large and symmetric acini whereas silencing of DVL3 to large and misshapen acini. Scale bar 20 μm. (b) Changes in acinar size (quantitated as area by Image J) of day-10 MCF10A epithelial structures expressing the indicated shRNAs. The area of control acini was set to 1, and the other area values (averages) are presented as relative to control. The averages represent at least 30 acinar structures per each shRNA. Twenty shRNA constructs resulted in ⩾0.2-fold change (cutoff) in epithelial structure size. Data are shown as fold change relative to control and s.e.m. Dashed lines indicates the 20% fold change relative to control (cutoff limit, see Supplementary Information). (c) Changes in symmetry (circularity) of day-10 MCF10A structures. The structures were quantitated as in panel (b). Six constructs resulted in >10% decrease in symmetry (cutoff limit, see Supplementary Information). Data are shown as fold change relative to control and s.e.m. (d) Scatter plot shows the relative changes in acinar size and symmetry values corresponding to 20 shRNAs indicated in panel (b). Green dot: control shRNA-transduced acini. Red dots: shRNA-transduced acini exhibiting >20% increase in size and >10% decrease in symmetry. (e) Cell polarity in hyperplastic and misshapen DVL3-deficient MCF10A epithelial structures. The structures were immunostained for E-cadherin and nuclei to visualize the ultrastructure and for golgi marker GM130 and α6-integrin to visualize the apico-basal polarity. Scale bar 20 μm. (f) Silencing of specific hEIR genes prevents epithelial cell cycle restriction. The MCF10A acini grown in Matrigel for 18 days were immunostained with proliferation marker Ki-67. Over 70% of control shRNA-transduced acini were Ki-67 negative, indicating quiescence. The acini containing ⩾1 Ki-67-positive cells were scored as Ki-67 positive. The acinar groups with enhanced Ki-67 positivity are grouped according to hTSG-like or nTSG-like phenotype. Mean and s.d. were calculated from three independent experiments and P-values by Student’s t-test. In 3D assays, ⩾50 acini were counted in each experiment. *P<0.5; **P<0.05.
We investigated whether the 11 hEIR genes, which limited acinar growth in matrix, have a role in suppressing the proliferative activity of cells in maturing organoids (Figure 1c; first screen: 1.2). By day 18, >70% of the MCF10A acini had normally entered quiescence in Matrigel culture (Figure 2f).24 Among the five gene knockdowns causing overgrowth and altered morphology, suppression of PARD6G, DVL3 and MPP5 (PALS1) (but not NUMB or STK3) extended proliferative activity of the acinar cells beyond day 18 (Figure 2f). Moreover, among the six gene knockdowns leading to an increase in acinar size but not affecting circularity, silencing of FZD1, MOB1B, TMOD3 and MOESIN extended proliferative activity (Figure 2f). Thus, the screen finds seven genes necessary for the establishment of epithelial cell cycle restriction in maturing MCF10A acinar structures. In Drosophila, inactivating gene mutations causing simultaneous loss of epithelial integrity and proliferation control define the group of nTSGs, whereas mutations only disrupting epithelial proliferation control define hTSGs. By analogy, PARD6G, DVL3 and MPP5 (PALS1) were functionally similar to Drosophila nTSGs and FZD1, MOB1B, TMOD3 and MOESIN showed hTSGs-like functions (Figure 2f).
Loss of hEIR gene function cooperates with oncogenic Myc
In the second screen, we explored whether the hEIR gene knockdowns cooperate with Myc to provide growth advantage for acinar structures and cells (Figure 1c; second screen: 2.1). The original set of 52 validated shRNAs was reintroduced into MCF10A-M cells. A conditionally active form of Myc, MycER, was activated with 4-hydroxytamoxifen on day 1 and the Myc activity was maintained until day 10. After 10 days of chronic Myc activation, the acini were 30% larger than controls (Supplementary Figure S3A) owing to Myc-dependent enhanced proliferative activity.24 Silencing of nine genes provided further growth advantage for MCF10A acini-expressing oncogenic Myc (Figures 3a and b). Among these genes, DVL3, MPP5 (PALS1), PARD6G, TMOD3, MOESIN, GSK3B and STK3 were identified as nTSG-like, hTSG-like or growth-limiting genes in the first screen. Two genes, PARD6B and EZRIN, were identified as genes regulating acinar size only in the second screen with Myc (Figure 3b). Silencing of EZRIN consistently showed suppressing effect on the epithelial cell cycle restriction (Supplementary Figure S3B; P=0.07), suggesting an hTSG-like function. The results show that loss of gene functions, which associate with the establishment of epithelial integrity and cell cycle restriction, generally facilitate Myc-promoted proliferation in mammary epithelial organoids.

Compromised epithelial integrity functionally interacts with Myc. (a) Confocal images of day-10 MCF10A structures expressing chronically activated Myc and shRNAs to DVL3, RHOA or MOESIN. Stainings for E-cadherin and nuclei visualize the acinar ultrastructure. Scale bar 20 μm. (b) Changes in the size of acinar structures were quantitated as in Figure 2b. The area value for control shRNA plus Myc expressing acini was set to 1, and the other area values (averages) are shown as relative to control. Data are shown as fold change relative to control and s.e.m. Dashed lines indicates the 20% fold change relative to control (cutoff). (c) Changes in the acinar symmetry (circularity) quantitated as in Figure 2c. Data are shown as fold change relative to control and s.e.m. Dashed lines indicates the 10% fold change relative to control (cutoff). (d) Scatter plot shows the relative changes in acinar size and symmetry values corresponding to 22 shRNAs indicated in Figure 3b. Green dot: Myc plus control shRNA. Red dots denote acini expressing Myc plus hEIR shRNA and exhibiting ⩾20% increase in size and ⩾10% decrease in symmetry values. Blue dots indicate hEIR shRNAs resulting in ⩾20% decrease in size and ⩾10% decrease in symmetry. (e) Synthetic lethal interaction between Myc and knockdown of RHOA. The acini were grown on Matrigel in the presence of chronic Myc activation and shRNA for RHOA or CDH1 for 10 days. Active caspase-3 antibody visualizes apoptosis and the basal α6-integrin acinar borders. Note the active caspase-3 positivity in RHOA-deficient but not in CDH1-deficient acini. Scale bar 20 μm. (f) Quantification of active caspase-3 (apoptosis) in RHOA-deficient acini with or without Myc activation. The acini were grown as in panel (e). Mean and s.d. were calculated from three independent experiments and P-values by Student’s t-test. In 3D assays, at least 50 acini were counted in each experiment. **P<0.05. NS, not significant.
Synthetic lethal interaction of RhoA inhibition and Myc
Surprisingly, while silencing of nine genes provided growth advantage for Myc-overexpressing acini, silencing of an almost equal number of genes (eight genes) had adverse effects on the growth of these acini (Figure 3b). Particularly, silencing of RHOA caused a marked drop in both acinar size and circularity (Figures 3c and d). A microscopic inspection revealed numerous disintegrated acini in RHOA-deficient acinar populations and, to a lesser extent, among CDC42-deficient acini, specifically when Myc was activated. Immunostaining with active caspase-3 antibody revealed a widespread Myc-dependent apoptosis in the RHOA-deficient structures (Figures 3e and f), thus exposing a strong synthetic lethal interaction between loss of RHOA and Myc activation.
Par6B and Par6G controls epithelial cell cycle restriction
The mature, quiescent MCF10A structures are exceedingly proliferation resistant, being able to prevent Myc-induced cell cycle re-entry. However, Myc-transformed cells can escape from quiescence in a laminin-free microenvironment or upon suppression of LKB1, which is required for formation of proper epithelial polarity, cell junctions and tissue structure in vitro and in vivo.21, 24 We used hEIR shRNAs that earlier showed cooperative activity with chronic Myc activation (Figure 1c; second screen: 2.1) to test whether any of these shRNAs would permit Myc-dependent disruption of the epithelial cell cycle restriction (Figure 1c; second screen: 2.2).
The structures expressing individual shRNA constructs were grown for 15 days to reach quiescence. Myc was acutely activated in the structures, and proliferation status was analyzed after 3 days of Myc activation (Figure 1c; second screen: 2.2). As expected, an acute activation of Myc alone was not sufficient to induce cell cycle re-entry. However, combination of Myc with knockdown of LLGL2, TMOD3, NUMB or PARD6B resulted in a prominent (>20%) increase in proliferative activity (Figure 4a). Notably, knockdown of NUMB or PARD6B (Figure 4b) did not have any cell cycle-promoting effects alone, indicating that the interaction with Myc was strictly synergistic. Only loss of PARD6B disrupted epithelial quiescence as efficiently as loss of LKB1 (PAR4) (Figures 4c and d). Notably, knockdown of the other PARD6 gene included in the screen, PARD6G, was alone able to sustain proliferation (Figure 4c). The results suggest that both Par6B and Par6G critically control the function of the epithelial cell cycle restriction machinery. Loss of Par6B activity cooperates with proliferation-promoting function of oncogenic Myc, whereas loss of Par6G can interfere with the cell cycle restriction alone.

Identification of gene deficiencies, which break epithelial cell cycle proliferation. (a) Analysis of proliferative activity in the presence of acute Myc activation and indicated shRNAs. Acinar structures were allowed to form in Matrigel for 15 days to reach quiescence and thereafter Myc was activated for 3 days. The acini containing ⩾1 Ki-67-positive cells were scored as positive. The Ki-67 positivity corresponding to the effect of indicated shRNA without Myc (Myc OFF) was normalized to 1 and the relative values corresponding to the effects of indicated shRNAs with Myc are presented. Dashed line indicates the 1.2-fold change (cutoff limit). (b) Proliferative activity in day-18 acini expressing the indicated shRNAs with or without acute Myc activation. Mean and s.d. are from three independent experiments and P-values by Student’s t-test. (c) Proliferative activity in day-18 acini expressing Myc (+ 4-hydroxytamoxifen (+4-OHT)) with shRNAs targeting either PARD6B or PARD6G. Note that silencing of PARD6G perturbs establishment of epithelial cell cycle restriction (Ki-67 in the absence and presence of Myc), whereas, in contrast, silencing of PARD6B allows cells to enter quiescence (Ki67 negative without Myc) but enables escape from the quiescence with Myc (Ki67 positivity with Myc). Scale bar 20 μm. (d) Comparison of the effects of PARD6B, PARD6G and LKB1 knockdowns on the proliferative activity of acini expressing acutely activated Myc (shPARD6B data are derived from the results presented in graph B). At least 50 acini were counted in each experiment. *P<0.5; **P<0.05; ***P<0.005. NS, not significant.
Par6B and Par6G controls cell cycle exit via protein kinase B/Akt pathway
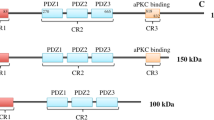
Par6 is a multimodular scaffold protein, which together with Par3 forms a complex with atypical protein kinase C (aPKC). This Par3-Par6-aPKC or 'PAR' complex is conserved among the metazoans, having a fundamental role in regulation of cell polarity.26 The PAR complex enables activation of aPKC and, particularly in mammary epithelial cells, T410 phosphorylated active PKCz has been linked to the polarizing functions of the PAR complex.32
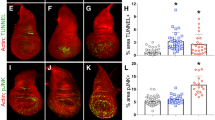
Knockdown of either PARD6B or PARD6G diminished the T410/412 phosphorylation of PKCz in MCF10A cells, indicating decreased PAR complex activity (Figure 5a). The effect was most clear when the basal phospho-aPKC activity was lowered by growth factor deprivation. Nevertheless, no major alterations were observed in apico-basal cell polarity, desmosomes or in the cytoskeleton structure of MCF10A acini (Figure 5b; Supplementary Figure S3C). We examined whether loss of PARD6B or PARD6G might promote activation of key proliferation-related signaling pathways and explored the status of protein kinase B1/Akt among several other pathways. On day 18, the MCF10A acini with or without Myc activation were negative for phospho-Akt (Figure 5c), whereas the PARD6G-deficient acinar structures retained the serine 473 phosphorylated status of Akt (Figure 5d; Supplementary Figure S3D). A weak phospho-Akt signal was observed in PARD6B-deficient structures, which signal was enhanced by activation of Myc (Figure 5d). Active Akt induces, via mammalian target of rapamycin complex 1 (mTORC1) complex, phosphorylation of ribosomal protein S6 (S6), which is a widely used marker of this pathway’s activation. PARD6G deficiency alone and PARD6B deficiency together with Myc induced a strong S6 phosphorylation (Figures 5e and f; Supplementary Figure S3E), indicating full activation of the Akt signaling pathway. Notably, however, the pattern of S6 phosphorylation was more sporadic than Akt phosphorylation, suggesting that the pathway is not completely linear.

Loss of Par6 activity deregulates Akt pathway. (a) Western blotting analysis shows the impact of PARD6B and PARD6G loss on phospho-PKCz (T410/412) in 2D cultures of MCF10A cells. The cells expressing the indicated shRNAs were grown in 2D culture for 24 h in the presence or absence of essential growth factors insulin and EGF (INS/EGF). Growth factor deprivation was used to reduce background phosphorylation. (b) Silencing of PARD6B or PARD6G does not affect apico-basal polarity in MCF10A acinar structures. The acinar structures expressing the indicated shRNAs were grown in Matrigel for 10 days and immunostained with GM130 golgi marker and α6-integrin to visualize apico-basal polarity. Scale bar 20 μm. (c) Confocal images demonstrating positive phospho-Akt (S473) staining in proliferative day-10 MCF10A acini and negative staining in quiescent day-18 acini. Scale bar 20 μm. (d) Quantification of phospho-Akt (S473) in day-18 PARD6B- and PARD6G-deficient acini with or without Myc activation. The acini containing ⩾1 phospho-Akt-positive cells were scored as phospho-Akt positive. Mean and s.d. were calculated from three independent experiments and P-values by Student’s t-test. (e) Confocal images demonstrating phospho-S6 staining pattern in day-10 and day-18 MCF10A acini. Scale bar 20 μm. (f) Quantification of phospho-S6 in day-18 PARD6B- and PARD6G-deficient acini with or without Myc activation. The acini containing ⩾1 phospho-S6-positive cells were scored as phospho-S6 positive. At least 50 acini were counted in each experiment unless otherwise indicated. Mean and s.d. were calculated from three independent experiments and P-values by Student’s t-test. (g) Western blotting showing the impact of PARD6B and PARD6G loss on Akt phosphorylation at T308 and S473 in growth factor-deprived cells. Cells were grown as in panel (a); in the right panel, cells were treated for 24 h with 4-hydroxytamoxifen (4-OHT) to activate Myc. (h) Loss of PARD6B or PARD6G expression impedes exit from the cell cycle. Control and MCF10A cells with silenced Par6 were deprived from essential growth factors insulin and EGF for 24 h, followed by analysis of Ki-67 positivity. Mean and s.e.m. were calculated from three independent experiments and P-values by Student’s t-test. (i) Western blotting shows the effect of PI3K inhibition (500 nM GDC-0941) and PDK1 inhibition (500 nM BX-795) on PARD6B and PARD6G loss-dependent phosphorylation of Akt at T308. Cells expressing the indicated shRNAs were deprived from growth factors for 24 h as in panel (a) and subsequently treated with inhibitors for 4 h. (j) PDK1 inhibition suppresses the cell cycle effects of PARD6B and PARD6G. MCF10A cells expressing the indicated shRNAs were deprived from growth factors for 24 h and subsequently treated with 500 nM BX-795 for another 24 h. *P<0.5; **P<0.05; ***P<0.005.
We further investigated the role of PARD6B and PARD6G in regulation of the Akt pathway using 2D cultures of MCF10A cells, because they are amenable for biochemical analysis. Activation of Akt is tightly controlled via phosphorylation of threonine 308 (T308) in the activation loop of the catalytic domain and serine 473 (S473) in the hydrophobic motif of the regulatory domain. Maximal Akt activity requires phosphorylation of both T308 and S473 residues.33, 34 In growth factor-deprived 2D cultures of MCF10A cells, T308 and S473 residues were not phosphorylated, indicating inactive Akt. However, both residues were strongly phosphorylated in the cells suffering loss of PARD6B or PARD6G (Figure 5g). Consistent with the notion of sustained Akt activity, Par6-deficient cells remained highly proliferative despite growth factor deprivation (Figure 5h). In growth factor-deprived 2D cultures, we could not detect similar effect of Myc activation on PARD6B-dependent Akt activity as in 3D culture (Figure 5g). Thus the cellular context appears important for PARD6B-dependent Akt regulation. Altogether, the results suggest that, in the absence of Par6 proteins, cells lose proper control on Akt activity and the cell cycle in proliferation-suppressive environment. Furthermore, in particular the PARD6B-dependent Akt control is strongly influenced by the context; loss of PARD6B unleashes strong Akt activity together with Myc in 3D culture while Myc requirement is bypassed in 2D cell culture.
In the cascade of events leading to Akt activation, phosphorylation at T308 induces a catalytically active conformation, which is stabilized by the phosphorylation of S473 by mTORC2.35 The major upstream events leading to phosphorylation of T308 occurs on the cytosolic side of the plasma membrane, where the activity of phosphatidylinositol 3′-kinase (PI3K) generates phosphatidylinositol (3-4-5)-trisphosphates as high affinity binding sites to pleckstrin homology domain of Akt. Once recruited and correctly positioned to membranes, Akt can be phosphorylated at T308 in the activation loop by phosphoinositide-dependent protein kinase 1 (PDK1).33 We studied whether Par6-deficiency promotes Akt activity via the canonical PI3K/PDK1 pathway by using highly selective small-molecule inhibitors to both PI3K and PDK1. Inhibition of PI3K abolished both growth factor and loss of Par6 (either PARD6B or PARD6G) mediated T308 phosphorylation of Akt. Interestingly, inhibition of PDK1 more specifically abolished the Par6 deficiency-induced T308 phosphorylation (Figure 5i). Furthermore, the proliferation restriction defect caused by loss of Par6G expression was partially rescued by inhibition of PDK1 (Figure 5j). In summary, the results suggest that Par6 proteins convey proliferation-suppressive signaling by negatively regulating the PI3K/PDK1/Akt pathway.
Par6G is frequently inactivated in epithelial cancers
Our initial searches of hEIR genes, which would be frequent targets of inactivating genetic changes, focused on four common cancers and identified PARD6G but not PARD6B (Figure 1a). To more comprehensively determine the status of PARD6B and PARD6G genes in human cancer, we expanded the analysis across 14 different cancer types (Supplementary Figure S3F). Interestingly, PARD6B is enriched in gain of function categories and, in concordance with published data, frequently upregulated in breast cancer (Figure 6a; Cunliffe et al.34). In addition, PARD6B is frequently involved in gene amplifications but is not commonly affected by deletions (Figure 6b). Strikingly, PARD6G shows the opposite being enriched in loss-of-function categories and affected by loss of heterozygosity in cancer cell lines and downregulation of mRNA in breast cancer (Figure 6c; Supplementary Figure S3F). Moreover, PARD6G is a commonly deleted gene in cancer (Figure 6d). Therefore, the evidence suggests that PARD6G but not PARD6B is frequently affected by loss-of-function-type genetic changes in epithelial cancers.

Genetic alterations affecting PARD6B and PARD6G in human cancer. (a) Mutation and expression status of PARD6B in six cancer types. The status of both PARD6B and PARD6G was analyzed across 14 different cancer types and all cancer types allowing comparison of PARD6B and PARD6G are shown (data exist for both genes at least in two databases; full analysis is shown in Supplementary Figure S3F). Highlighted with red are gain-of-function-type mutations (mRNA upregulation, DNA amplification and gain of copy number) present in ⩾10% of the analyzed samples. Highlighted with blue are loss-of-function changes (downregulation, mutation, deletion, loss of heterozygosity and copy number loss) present in ⩾10% of the analyzed samples. (b) cBioPortal analysis of the status of PARD6B in cancer genomics data sets. (c) Mutation and expression status of PARD6G in six cancer types. The analyses were performed as in panel (a). (d) cBioPortal (Memorial Sloan Kettering Cancer Center) analysis of the status of PARD6G in cancer genomics data sets.
Discussion
Here we identified biological pathways regulating the epithelial integrity and the cell cycle restriction by using an shRNA approach in a MCF10A model of glandular epithelial structure. In MCF10A model, the mammary acinus-like structures develop and upon maturation enter quiescence in laminin-rich matrix.36 We identified 11 genes that are needed to prevent overgrowth of the acinar structures. These genes encode proteins of Wnt (FZD1, DVL3, GSK3B), Notch (NUMB) and Hippo (LATS1, STK3 (MST2), MOB1B (MOBKL1A)) signaling pathways and also two protein modifiers of actin dynamics and actin cytoskeleton (Figure 7). Moesin encodes an ERM family member29 and TMOD3 (tropomodulin 3) encodes an actin filament capping protein implicated in cell shape regulation.37 Moreover, two classical apical polarity genes, PARD6G and MPP5 (PALS1) were identified as regulators of acinar size. These proteins are constituents of two evolutionary conserved apical polarity complexes: CRB comprised of Crumbs-PALS1-PATJ and PAR formed by Par3-Par6-aPKC.26 A defective epithelial cell cycle restriction was observed in acinar structures with silenced MPP5 (PALS1), DVL3, FZD1, MOB1B (MOBKL1A), TMOD3, MOESIN or PARD6DG. Four of these genes: FZD1, MOB1B (MOBKL1A), TMOD3, and MOESIN, were termed as hTSG-like genes, as loss of function increased acinar size without altering the rounded morphology of acinar structures (Figure 7). It should be noted, however, that FZD1 and MOB1B were borderline hits. The three remaining genes: PARD6G, DVL3, or MPP5 (PALS1), were termed as nTSG-like genes, as inactivation of these genes caused disorganized acinar morphology, considerable overgrowth of the acini and compromised cell cycle restriction. The shRNA screen impacts target genes with varying knockdown efficacy and for that reason many anticipated polarity proteins were missed in the screen. However, the positive hits, such as the nTSG-like genes identified in this study, warrant further studies on their role in regulation of the machinery that couples epithelial structure to the cell cycle.

Signaling pathways at the intersection of epithelial integrity and proliferation control. The epithelial structure and proliferation-regulating genes identified in this study encode proteins of Wnt, Notch and Hippo signaling pathways as well as regulators of apico-basal polarity (MPP5 (PALS1) and Par6 proteins) and actin dynamics (Moesin, Ezrin and TMOD3). Blue color denotes genes required for cessation of growth and proliferation in acinar structures. Green color denotes hTSG-like activity, in which case diminished gene activity causes overgrowth and extended proliferative activity in acinar structures without loss of symmetrical shape of the acini. Pink color marks the nTSG-like genes, which upon silencing cause overgrowth and extended proliferation in acini and disability to organize symmetric acinar architecture. Most cell cycle restriction/growth control-associated genes, when inhibited, strongly promoted acinar growth in the presence of Myc activity (right panel: transformed epithelial cell). However, depletion of RhoA or CDC42 selectively induces apoptosis and collapse of Myc-transformed acinar structures. Inactivation of PARD6B gene does not disturb the ability of maturing acini to establish quiescence but, together with Myc, permits reactivation of the cell cycle machinery in quiescent acinar structures. The present findings suggest an important role for both PARD6B and PARD6G genes (highlighted in figures) in regulation of the epithelial cell cycle restriction. In epithelial cancers, PARD6G is frequently targeted by loss-of-function mutations, whereas gain-of-function mutations are more characteristic for PARD6B.
Genetic defects, which deteriorate epithelial integrity and growth control, commonly enhance the tumorigenic potential of oncoproteins in epithelial tissues of both invertebrates and mammals.11, 20, 21, 22 Moreover, a recent study from Chatterjee et al.38 demonstrated that loss of cell polarity proteins potentiate the invasive properties of oncogenic ErbB2 challenged MCF10A cells. The present study exposes a growth-promoting cooperation between Myc and loss of nine hEIR gene functions. Surprisingly, silencing of nearly as many hEIR genes inhibited acinar growth when combined with Myc. Among these genes, E-cadherin has been implicated in cell proliferation control of MCF10A cells by an earlier study.39 Interestingly, inactivation of RHOA, and to lesser extent CDC42, induced widespread and strictly Myc-dependent apoptosis. RhoA and CDC42 belong to a family of Rho GTPases and both proteins have been previously implicated in anoikis,40, 41 occurring in anchorage-dependent cells detached from the extracellular matrix and thus ablated from critical integrin-dependent contacts with extracellular matrix.42 However, it remains to be clarified whether the unexpected synthetic lethal interactions with Myc found in the present study are attributable to adhesion-dependent survival or to cell-intrinsic mechanisms of apoptosis.43
Our findings suggest a critical role for PARD6B and PARD6G in suppression of epithelial cell proliferation. Both Par6 proteins are needed for suppression of Akt activity in proliferation-suppressive conditions and loss of this inhibition induces via canonical PI3K-PDK1 pathway an activation of Akt-mTOR pathway, one of the key proliferation-promoting pathways in mammals. Consistent with the notion of Par6 proteins as negative regulators of Akt1, earlier findings have shown that engineered Par6 activity perturbs insulin-stimulated Akt1 activity in myoblasts.44 Silencing of PARD6B exerted weaker impact on cell proliferation machinery than silencing of PARD6G, although the cell cycle-promoting effects by PARD6B loss were clear when combined with Myc activation. Evidently, assays with shRNA cannot fully answer the question of whether there are qualitative differences between these two proteins in cell cycle suppression, and furthermore, a possible role of Par6A activity needs to be more thoroughly examined by further studies. Nonetheless, the finding that loss of PARD6B or PARD6G expression in 2D culture has an equal impact on the Akt activity but unequal impact on the cell cycle is highly suggestive for a biological difference between the proteins. Loss of Par6 activity and consequent deregulation of Akt signaling is likely to influence tumorigenesis as deregulated PI3K-PDK1-Akt-mTOR signaling is one of the core cancer pathways as established in recent pan-can studies,45 and furthermore, a number of studies have demonstrated cooperation of the Akt pathway with Myc in tumorigenesis,46, 47 enhanced survival signaling,48, 49 metabolic reprogramming50 and via direct impact on cell cycle machinery.51, 52
Further evidence for different biological quality between PARD6B and PARD6G gene in tumorigenesis was obtained from analysis of gain- and loss-of-function-type alterations affecting these genes in cancer. Although loss-of-function changes strongly associated with PARD6G, almost opposite repertoire of genetic changes affected PARD6B in a panel of 14 studied tumor types. For example, in breast cancer, PARD6B gene is involved in amplifications and overexpression, whereas PARD6G is affected by gene deletions and diminished expression. Overexpression of PARD6B has been shown to promote proliferation via induction of mitogen-activated protein kinase/extracellular signal–regulated kinase pathway in earlier studies,53 and it is tempting to speculate that PARD6B primarily promotes tumorigenesis as an oncogene via its effects on the mitogen-activated protein kinase pathway, while PARD6G has a tumor-suppressor function attributable to repression of the Akt pathway. There is existing evidence for gene families having members with oncogenic and tumor-suppressor functions,54 and our present results suggest that the PARD6 family, which has been diverged during the evolution to three paralogs in mammals, may also promote tumorigenesis via gains and losses depending on the family member in question (Figure 7).
Materials and methods
Tumor-Suppressor Predictor
TSP algorithm searches evidence for loss of gene function in cancer by using information from The Cancer Genome Atlas, Catalogue of Somatic Mutations in Cancer and Tumorscape. The searches are focused on loss of gene expression, somatic mutation frequency and chromosomal deletions. TSP integrates information supporting loss of gene function to calculate a single gene-specific score value that summarizes the total evidence of tumor-suppressor function for each human gene. The TSP algorithms, source databases and methods of data derivation are described in the Supplementary Materials.
A validated lentiviral shRNA library for silencing epithelial integrity genes
shRNAs were designed to target the first and/or the longest ENSEMBL transcript of each hEIR gene and 1–3 shRNA target sequences were chosen per transcript. Altogether 136 shRNA oligos were cloned into pDSL_UGIH lentiviral shRNA vector (Alliance for Cellular Signaling, Berkeley, CA, USA) as described in the Supplementary Materials. The library was complemented with additional 40 shRNAs in pLKO lentiviral vector (Broad Institute TRC library, MISSION TRC-Hs 1.0 library; Sigma, St Louis, MO, USA) and with 43 shRNAs in pGIPZ mir-30-based vector (Open Biosystems, Fisher Scientific, Vantaa, Finland). All shRNA constructs were tested for gene-silencing activity in MCF10A MycER cells either by quantitative PCR or western blotting analysis prior 3D culture screens. A complete list of transcripts and genes in the hEIR library is presented in Supplementary Table S1. The validation procedures and shRNA constructs chosen for assays are described in Supplementary Materials and Supplementary Tables S3–S5. The shRNA constructs validated for assays represent about 23% of all constructs tested (Supplementary Table S4).
Lentiviral infections
Lentiviral particles were produced in Biomedicum Functional Genomics Unit's biosafety level 2 facilities. MCF10A-M cells were transduced with lentiviruses carrying individual shRNA constructs as described previously25 with minor modifications explained in the Supplementary Materials.
shRNA screen in 3D culture
MCF10A-M cells were cultured in reconstituted basement membrane (Matrigel, Becton Dickinson, Franklin Lakes, NJ, USA) as described previously.25 To analyze shRNA effects in 3D culture, MCF10A-M cells were infected with shRNA lentiviruses on 24-well plates and seeded on Matrigel in 8-chamber slides. The experimental set-up of the screen is described in Supplementary Materials. Experiments with each shRNA construct were performed in technical duplicates and repeated twice starting from new lentiviral infections.
Antibodies and reagents
The full list of antibodies and reagents is provided in Supplementary Materials.
Immunofluorescent staining and imaging
Immunofluorescent staining of 3D structures was carried out as described in Bordy25 and in the Supplementary Materials. Images of the immunostained structures were acquired by using Zeiss LSM Meta 510 and 780 confocal microscopes. For quantitative image analysis, images were acquired using Zeiss Axiovert 200 microscope equipped with Apotome system (Zeiss, Oberkochen, Germany). The images were arranged using Adobe Photoshop CS5 (San Jose, CA, USA).
Image J analysis and quantitation
For quantitative analysis, MCF10A-M structures were immunostained with E-cadherin antibody to visualize borders of the acini and the nuclei were counterstained with Hoechst. Digital images were analyzed using the Image J software (version 1.42q, National Institute of Health, Bethesda, MD, USA) and measured for shape descriptors (area, perimeter and circularity). Multiple images were counted from each experiment to obtain measurements from at least 30 acini per each transduced cell population. Two researchers independently analyzed duplicate slides in each experiment.
References
McCrea PD, Gu D, Balda MS . Junctional music that the nucleus hears: cell-cell contact signaling and the modulation of gene activity. Cold Spring Harb Perspect Biol 2009; 1: a002923.
Partanen JI, Nieminen AI, Klefstrom J . 3D view to tumor suppression: Lkb1, polarity and the arrest of oncogenic c-Myc. Cell Cycle 2009; 8: 716–724.
Bryant DM, Mostov KE . From cells to organs: building polarized tissue. Nat Rev Mol Cell Biol 2008; 9: 887–901.
MacDonald BT, Tamai K, He X . Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell 2009; 17: 9–26.
Curto M, McClatchey AI . Nf2/Merlin: a coordinator of receptor signalling and intercellular contact. Br J Cancer 2008; 98: 256–262.
Sourisseau T, Georgiadis A, Tsapara A, Ali RR, Pestell R, Matter K et al. Regulation of PCNA and cyclin D1 expression and epithelial morphogenesis by the ZO-1-regulated transcription factor ZONAB/DbpA. Mol Cell Biol 2006; 26: 2387–2398.
Gonzalez-Mariscal L, Bautista P, Lechuga S, Quiros M . ZO-2 a tight junction scaffold protein involved in the regulation of cell proliferation and apoptosis. Ann NY Acad Sci 2012; 1257: 133–141.
Oka T, Remue E, Meerschaert K, Vanloo B, Boucherie C, Gfeller D et al. Functional complexes between YAP2 and ZO-2 are PDZ domain-dependent, and regulate YAP2 nuclear localization and signalling. Biochem J 2010; 432: 461–472.
Bilder D, Li M, Perrimon N . Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 2000; 289: 113–116.
Brumby AM, Richardson HE . scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J 2003; 22: 5769–5779.
Pagliarini RA, Xu T . A genetic screen in Drosophila for metastatic behavior. Science 2003; 302: 1227–1231.
Hariharan IK, Bilder D . Regulation of imaginal disc growth by tumor-suppressor genes in Drosophila. Annu Rev Genet 2006; 40: 335–361.
Huang L, Muthuswamy SK . Polarity protein alterations in carcinoma: a focus on emerging roles for polarity regulators. Curr Opin Genet Dev 2010; 20: 41–50.
Lee M, Vasioukhin V . Cell polarity and cancer—cell and tissue polarity as a non-canonical tumor suppressor. J Cell Sci 2008; 121: 1141–1150.
Martin-Belmonte F, Perez-Moreno M . Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer 2012; 12: 23–38.
Royer C, Lu X . Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ 2011; 18: 1470–1477.
Tervonen TA, Partanen JI, Saarikoski ST, Myllynen M, Marques E, Paasonen K et al. Faulty epithelial polarity genes and cancer. Adv Cancer Res 2011; 111: 97–161.
Banks L, Pim D, Thomas M . Human tumour viruses and the deregulation of cell polarity in cancer. Nat Rev Cancer 2012; 12: 877–886.
Leung CT, Brugge JS . Outgrowth of single oncogene-expressing cells from suppressive epithelial environments. Nature 2012; 482: 410–413.
McCaffrey LM, Montalbano J, Mihai C, Macara IG . Loss of the Par3 polarity protein promotes breast tumorigenesis and metastasis. Cancer Cell 2012; 22: 601–614.
Partanen JI, Tervonen TA, Myllynen M, Lind E, Imai M, Katajisto P et al. Tumor suppressor function of Liver kinase B1 (Lkb1) is linked to regulation of epithelial integrity. Proc Natl Acad Sci USA 2012; 109: E388–E397.
Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB et al. Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell 2008; 135: 865–878.
Debnath J, Mills KR, Collins NL, Reginato MJ, Muthuswamy SK, Brugge JS . The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 2002; 111: 29–40.
Partanen JI, Nieminen AI, Makela TP, Klefstrom J . Suppression of oncogenic properties of c-Myc by LKB1-controlled epithelial organization. Proc Natl Acad Sci USA 2007; 104: 14694–14699.
Brody T . The interactive fly: gene networks, development and the internet. Trends Genet 1999; 15: 333–334.
Assemat E, Bazellieres E, Pallesi-Pocachard E, Le Bivic A, Massey-Harroche D . Polarity complex proteins. Biochim Biophys Acta 2008; 1778: 614–630.
Avruch J, Zhou D, Fitamant J, Bardeesy N, Mou F, Barrufet LR . Protein kinases of the Hippo pathway: regulation and substrates. Semin Cell Dev Biol 2012; 23: 770–784.
Brumby AM, Richardson HE . Using Drosophila melanogaster to map human cancer pathways. Nat Rev Cancer 2005; 5: 626–639.
Niggli V, Rossy J . Ezrin/radixin/moesin: versatile controllers of signaling molecules and of the cortical cytoskeleton. Int J Biochem Cell Biol 2008; 40: 344–349.
Fogg VC, Liu CJ, Margolis B . Multiple regions of Crumbs3 are required for tight junction formation in MCF10A cells. J Cell Sci 2005; 118: 2859–2869.
Hanahan D, Weinberg RA . Hallmarks of cancer: the next generation. Cell 2011; 144: 646–674.
Whyte J, Thornton L, McNally S, McCarthy S, Lanigan F, Gallagher WM et al. PKCzeta regulates cell polarisation and proliferation restriction during mammary acinus formation. J Cell Sci 2010; 123: 3316–3328.
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB et al. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Balpha. Curr Biol 1997; 7: 261–269.
Cunliffe HE, Jiang Y, Fornace KM, Yang F, Meltzer PS . PAR6B is required for tight junction formation and activated PKCzeta localization in breast cancer. Am J Cancer Res 2012; 2: 478–491.
Hemmings BA, Restuccia DF . PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol 2012; 4: a011189.
Debnath J, Muthuswamy SK, Brugge JS . Morphogenesis and oncogenesis of MCF-10 A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 2003; 30: 256–268.
Weber KL, Fischer RS, Fowler VM . Tmod3 regulates polarized epithelial cell morphology. J Cell Sci 2007; 120: 3625–3632.
Chatterjee S, Seifried L, Feigin ME, Gibbons DL, Scuoppo C, Lin W et al. Dysregulation of cell polarity proteins synergize with oncogenes or the microenvironment to induce invasive behavior in epithelial cells. PLoS One 2012; 7: e34343.
Chen A, Beetham H, Black MA, Priya R, Telford BJ, Guest J et al. E-cadherin loss alters cytoskeletal organization and adhesion in non-malignant breast cells but is insufficient to induce an epithelial-mesenchymal transition. BMC Cancer 2014; 14: 552.
Cheng TL, Symons M, Jou TS . Regulation of anoikis by Cdc42 and Rac1. Exp Cell Res 2004; 295: 497–511.
Ma Z, Myers DP, Wu RF, Nwariaku FE, Terada LS . p66Shc mediates anoikis through RhoA. J Cell Biol 2007; 179: 23–31.
Weaver VM, Lelievre S, Lakins JN, Chrenek MA, Jones JC, Giancotti F et al. beta4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2002; 2: 205–216.
Vleugel M, Hoogendoorn E, Snel B, Kops GJ . Evolution and function of the mitotic checkpoint. Dev Cell 2012; 23: 239–250.
Weyrich P, Neuscheler D, Melzer M, Hennige AM, Haring HU, Lammers R . The Par6alpha/aPKC complex regulates Akt1 activity by phosphorylating Thr34 in the PH-domain. Mol Cell Endocrinol 2007; 268: 30–36.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C et al. Mutational landscape and significance across 12 major cancer types. Nature 2013; 502: 333–339.
Clegg NJ, Couto SS, Wongvipat J, Hieronymus H, Carver BS, Taylor BS et al. MYC cooperates with AKT in prostate tumorigenesis and alters sensitivity to mTOR inhibitors. PLoS One 2011; 6: e17449.
Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 2004; 428: 332–337.
Kauffmann-Zeh A, Rodriguez-Viciana P, Ulrich E, Gilbert C, Coffer P, Downward J et al. Suppression of c-Myc-induced apoptosis by Ras signalling through PI(3)K and PKB. Nature 1997; 385: 544–548.
Link W, Rosado A, Fominaya J, Thomas JE, Carnero A . Membrane localization of all class I PI 3-kinase isoforms suppresses c-Myc-induced apoptosis in Rat1 fibroblasts via Akt. J Cell Biochem 2005; 95: 979–989.
Vander Heiden MG, Cantley LC, Thompson CB . Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009; 324: 1029–1033.
Bouchard C, Marquardt J, Bras A, Medema RH, Eilers M . Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J 2004; 23: 2830–2840.
Chan JC, Hannan KM, Riddell K, Ng PY, Peck A, Lee RS et al. AKT promotes rRNA synthesis and cooperates with c-MYC to stimulate ribosome biogenesis in cancer. Sci Signal 2011; 4: ra56.
Nolan ME, Aranda V, Lee S, Lakshmi B, Basu S, Allred DC et al. The polarity protein Par6 induces cell proliferation and is overexpressed in breast cancer. Cancer Res 2008; 68: 8201–8209.
Cameron ER, Neil JC . The Runx genes: lineage-specific oncogenes and tumor suppressors. Oncogene 2004; 23: 4308–4314.
Acknowledgements
We thank all the members of Klefström laboratory for discussions and critical comments. We thank Tiina Raatikainen and Tarja Välimäki for technical assistance and Biomedicum Imaging Unit and Biomedicum Functional Genomics Unit for core services and technical support. This study was funded by the Academy of Finland (to JK and SH), Center of Excellence in Cancer Genetics Research (to SH and ML), TEKES, Sigrid Jusélius Foundation, Finnish Cancer Organizations (to JK), Helsinki Graduate Program in Biotechnology and Molecular Biology (to EM) and Innovative Medicines Initiative Joint Undertaking under grant agreement no. 115188 (to JK).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Marques, E., Englund, J., Tervonen, T. et al. Par6G suppresses cell proliferation and is targeted by loss-of-function mutations in multiple cancers. Oncogene 35, 1386–1398 (2016). https://doi.org/10.1038/onc.2015.196
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2015.196
This article is cited by
-
Secreted breast tumor interstitial fluid microRNAs and their target genes are associated with triple-negative breast cancer, tumor grade, and immune infiltration
Breast Cancer Research (2020)
-
Phenotype-driven identification of epithelial signalling clusters
Scientific Reports (2018)
-
YAP Subcellular Localization and Hippo Pathway Transcriptome Analysis in Pediatric Hepatocellular Carcinoma
Scientific Reports (2016)
