
Abstract
Progesterone (P4) has emerged as an important hormone-regulating mammary stem cell (MaSC) populations. In breast cancer, P4 and synthetic analogs increase the number of stem-like cells within luminal estrogen receptor (ER)- and progesterone receptor (PR)-positive breast cancers. These cells gain expression of de-differentiated cell markers CD44 and cytokeratin 5 (CK5), lose luminal markers ER and PR, and are more therapy resistant. We previously described that P4 downregulation of microRNA (miR)-29a contributes to the expansion of CD44high and CK5+ cells. Here we investigated P4 downregulation of miR-141, a member of the miR-200 family of tumor suppressors, in facilitating an increase in stem-like breast cancer cells. miR-141 was the sole member of the miR-200 family P4-downregulated at the mature miRNA level in luminal breast cancer cell lines. Stable inhibition of miR-141 alone increased the CD44high population, and potentiated P4-mediated increases in both CD44high and CK5+ cells. Loss of miR-141 enhanced both mammosphere formation and tumor initiation. miR-141 directly targeted both PR and signal transducer and activator of transcription 5A (Stat5a), transcription factors important for MaSC expansion. miR-141 depletion increased PR protein levels, even in cell lines where PR expression is estrogen dependent. Stat5a suppression via small interfering RNA or a small-molecule inhibitor reduced the P4-dependent increase in CK5+ and CD44high cells. These data support a mechanism by which P4-triggered loss of miR-141 facilitates breast cancer cell de-differentiation through deregulation of PR and Stat5a, two transcription factors important for controlling mammary cell fate.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
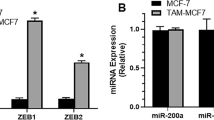

References
Ismail PM, Amato P, Soyal SM, DeMayo FJ, Conneely OM, O'Malley BW et alProgesterone involvement in breast development and tumorigenesis–as revealed by progesterone receptor ‘knockout’ and ‘knockin‘ mouse models. Steroids 2003; 68: 779–787.
Asselin-Labat ML, Vaillant F, Sheridan JM, Pal B, Wu D, Simpson ER et alControl of mammary stem cell function by steroid hormone signalling. Nature 2010; 465: 798–802.
Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL et alProgesterone induces adult mammary stem cell expansion. Nature 2010; 465: 803–807.
Gonzalez-Suarez E, Jacob AP, Jones J, Miller R, Roudier-Meyer MP, Erwert R et alRANK ligand mediates progestin-induced mammary epithelial proliferation and carcinogenesis. Nature 2010; 468: 103–107.
Schramek D, Leibbrandt A, Sigl V, Kenner L, Pospisilik JA, Lee HJ et alOsteoclast differentiation factor RANKL controls development of progestin-driven mammary cancer. Nature 2010; 468: 98–102.
Graham JD, Mote PA, Salagame U, van Dijk JH, Balleine RL, Huschtscha LI et alDNA replication licensing and progenitor numbers are increased by progesterone in normal human breast. Endocrinology 2009; 150: 3318–3326.
Tanos T, Sflomos G, Echeverria PC, Ayyanan A, Gutierrez M, Delaloye JF et alProgesterone/RANKL is a major regulatory axis in the human breast. Sci Transl Med 2013; 5: 182ra55.
Horwitz KB, Sartorius CA . Progestins in hormone replacement therapies reactivate cancer stem cells in women with preexisting breast cancers: a hypothesis. J Clin Endocrinol Metab 2008; 93: 3295–3298.
Horwitz KB, Dye WW, Harrell JC, Kabos P, Sartorius CA . Rare steroid receptor-negative basal-like tumorigenic cells in luminal subtype human breast cancer xenografts. Proc Natl Acad Sci USA 2008; 105: 5774–5779.
Sartorius CA, Harvell DM, Shen T, Horwitz KB . Progestins initiate a luminal to myoepithelial switch in estrogen-dependent human breast tumors without altering growth. Cancer Res 2005; 65: 9779–9788.
Axlund SD, Yoo BH, Rosen RB, Schaack J, Kabos P, Labarbera DV et alProgesterone-inducible cytokeratin 5-positive cells in luminal breast cancer exhibit progenitor properties. Horm Cancer 2013; 4: 36–49.
Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH et alAberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 2009; 15: 907–913.
Bocker W, Moll R, Poremba C, Holland R, Van Diest PJ, Dervan P et alCommon adult stem cells in the human breast give rise to glandular and myoepithelial cell lineages: a new cell biological concept. Lab Invest 2002; 82: 737–746.
Kabos P, Haughian JM, Wang X, Dye WW, Finlayson C, Elias A et alCytokeratin 5 positive cells represent a steroid receptor negative and therapy resistant subpopulation in luminal breast cancers. Breast Cancer Res Treat 2011; 128: 45–55.
Finnegan EF, Pasquinelli AE MicroRNA biogenesis: regulating the regulators. Crit Rev Biochem Mol Biol 2013; 48: 51–68.
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D et alMicroRNA expression profiles classify human cancers. Nature 2005; 435: 834–838.
Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G et alThe miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 2008; 10: 593–601.
Howe EN, Cochrane DR, Richer JK Targets of miR-200c mediate suppression of cell motility and anoikis resistance. Breast Cancer Res 2011; 13: R45.
Cochrane DR, Spoelstra NS, Howe EN, Nordeen SK, Richer JK MicroRNA-200c mitigates invasiveness and restores sensitivity to microtubule-targeting chemotherapeutic agents. Mol Cancer Ther 2009; 8: 1055–1066.
Hurteau GJ, Carlson JA, Spivack SD, Brock GJ Overexpression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res 2007; 67: 7972–7976.
Park SM, Gaur AB, Lengyel E, Peter ME The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev 2008; 22: 894–907.
Cochrane DR, Jacobsen BM, Connaghan KD, Howe EN, Bain DL, Richer JK Progestin regulated miRNAs that mediate progesterone receptor action in breast cancer. Mol Cell Endocrinol 2012; 355: 15–24.
Cochrane DR, Spoelstra NS, Richer JK The role of miRNAs in progesterone action. Mol Cell Endocrinol 2011; 357: 50–59.
Cittelly DM, Finlay-Schultz J, Howe EN, Spoelstra NS, Axlund SD, Hendricks P et alProgestin suppression of miR-29 potentiates dedifferentiation of breast cancer cells via KLF4. Oncogene 2013; 32: 2555–2564.
Reya T, Morrison SJ, Clarke MF, Weissman IL Stem cells, cancer, and cancer stem cells. Nature 2001; 414: 105–111.
Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A et alResidual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA 2009; 106: 13820–13825.
Chaffer CL, Brueckmann I, Scheel C, Kaestli AJ, Wiggins PA, Rodrigues LO et alNormal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proc Natl Acad Sci USA 2011; 108: 7950–7955.
Iliopoulos D, Hirsch HA, Wang G, Struhl K Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci USA 2011; 108: 1397–1402.
Edwards DP, Leonhardt SA, Gass-Handel E Novel mechanisms of progesterone antagonists and progesterone receptor. J Soc Gynecol Investig 2000; 7 (1 Suppl): S22–S24.
Richer JK, Lange CA, Manning NG, Owen G, Powell R, Horwitz KB Convergence of progesterone with growth factor and cytokine signaling in breast cancer. Progesterone receptors regulate signal transducers and activators of transcription expression and activity. J Biol Chem 1998; 273: 31317–31326.
Yamaji D, Na R, Feuermann Y, Pechhold S, Chen W, Robinson GW et alDevelopment of mammary luminal progenitor cells is controlled by the transcription factor STAT5A. Genes Dev 2009; 23: 2382–2387.
Neves R, Scheel C, Weinhold S, Honisch E, Iwaniuk KM, Trompeter HI et alRole of DNA methylation in miR-200c/141 cluster silencing in invasive breast cancer cells. BMC Res Notes 2010; 3: 219.
Guil S, Caceres JF The multifunctional RNA-binding protein hnRNP A1 is required for processing of miR-18a. Nat Struct Mol Biol 2007; 14: 591–596.
Liu C, Kelnar K, Vlassov AV, Brown D, Wang J, Tang DG Distinct microRNA expression profiles in prostate cancer stem/progenitor cells and tumor-suppressive functions of let-7. Cancer Res 2012; 72: 3393–3404.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100: 3983–3988.
Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D et alDownregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009; 138: 592–603.
Pillai MM, Gillen AE, Yamamoto TM, Kline E, Brown J, Flory K et alHITS-CLIP reveals key regulators of nuclear receptor signaling in breast cancer. Breast Cancer Res Treat 2014; 146: 85–97.
Kastner P, Krust A, Turcotte B, Stropp U, Tora L, Gronemeyer H et alTwo distinct estrogen-regulated promoters generate transcripts encoding the two functionally different human progesterone receptor forms A and B. EMBO J 1990; 9: 1603–1614.
Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev 1997; 11: 179–186.
Vafaizadeh V, Klemmt P, Brendel C, Weber K, Doebele C, Britt K et alMammary epithelial reconstitution with gene-modified stem cells assigns roles to Stat5 in luminal alveolar cell fate decisions, differentiation, involution, and mammary tumor formation. Stem Cells 2010; 28: 928–938.
Santos SJ, Haslam SZ, Conrad SE Estrogen and progesterone are critical regulators of Stat5a expression in the mouse mammary gland. Endocrinology 2008; 149: 329–338.
Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB et alThe STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 2011; 117: 3421–3429.
Reid BG, Jerjian T, Patel P, Zhou Q, Yoo BH, Kabos P et alLive multicellular tumor spheroid models for high-content imaging and screening in cancer drug discovery. Curr Chem Genomics Transl Med 2014; 8 (Suppl 1): 27–35.
Obr AE, Edwards DP The biology of progesterone receptor in the normal mammary gland and in breast cancer. Mol Cell Endocrinol 2012; 357: 4–17.
Thomson JM, Newman M, Parker JS, Morin-Kensicki EM, Wright T, Hammond SM Extensive post-transcriptional regulation of microRNAs and its implications for cancer. Genes Dev 2006; 20: 2202–2207.
Chatterjee S, Grosshans H Active turnover modulates mature microRNA activity in Caenorhabditis elegans. Nature 2009; 461: 546–549.
Grosshans H, Chatterjee S MicroRNAses and the regulated degradation of mature animal miRNAs. Adv Exp Med Biol 2010; 700: 140–155.
Schwab R, Speth C, Laubinger S, Voinnet O Enhanced microRNA accumulation through stemloop-adjacent introns. EMBO Rep 2013; 14: 615–621.
Gantier MP, McCoy CE, Rusinova I, Saulep D, Wang D, Xu D et alAnalysis of microRNA turnover in mammalian cells following Dicer1 ablation. Nucleic Acids Res 2011; 39: 5692–5703.
Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA et alAn HNF4alpha-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 2011; 147: 1233–1247.
Haga CL, Phinney DG MicroRNAs in the imprinted DLK1-DIO3 region repress the epithelial-to-mesenchymal transition by targeting the TWIST1 protein signaling network. J Biol Chem 2012; 287: 42695–42707.
Iliopoulos D, Lindahl-Allen M, Polytarchou C, Hirsch HA, Tsichlis PN, Struhl K Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol Cell 2010; 39: 761–772.
Li Z, Liu H, Jin X, Lo L, Liu J Expression profiles of microRNAs from lactating and non-lactating bovine mammary glands and identification of miRNA related to lactation. BMC Genomics 2012; 13: 731.
Vafaizadeh V, Klemmt PA, Groner B Stat5 assumes distinct functions in mammary gland development and mammary tumor formation. Front Biosci 2012; 17: 1232–1250.
Sato T, Tran TH, Peck AR, Girondo MA, Liu C, Goodman CR et alProlactin suppresses a progestin-induced CK5-positive cell population in luminal breast cancer through inhibition of progestin-driven BCL6 expression. Oncogene 2013; 33: 2215–2224.
Howell SJ, Anderson E, Hunter T, Farnie G, Clarke RB Prolactin receptor antagonism reduces the clonogenic capacity of breast cancer cells and potentiates doxorubicin and paclitaxel cytotoxicity. Breast Cancer Res 2008; 10: R68.
Rouet V, Bogorad RL, Kayser C, Kessal K, Genestie C, Bardier A et alLocal prolactin is a target to prevent expansion of basal/stem cells in prostate tumors. Proc Natl Acad Sci USA 2010; 107: 15199–15204.
Yamashita H, Nishio M, Ando Y, Zhang Z, Hamaguchi M, Mita K et alStat5 expression predicts response to endocrine therapy and improves survival in estrogen receptor-positive breast cancer. Endocr Relat Cancer 2006; 13: 885–893.
Clark GM, McGuire WL Prognostic factors in primary breast cancer. Breast Cancer Res Treat 1983; 3 (Suppl 1): S69–S72.
Wagner KU, Rui H Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. J Mammary Gland Biol Neoplasia 2008; 13: 93–103.
Pfaffl MW A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 2001; 29: e45.
Kabos P, Finlay-Schultz J, Li C, Kline E, Finlayson C, Wisell J et alPatient-derived luminal breast cancer xenografts retain hormone receptor heterogeneity and help define unique estrogen-dependent gene signatures. Breast Cancer Res Treat 2012; 135: 415–432.
Acknowledgements
We thank the University of Colorado Cancer Center Flow Cytometry and Tissue Culture Cores supported by P30CA046934 and the University of Colorado, Department of Pathology Sequencing Core for their technical assistance and services. This work was supported by Department of Defense BCRP grants W81XWH-11-1-0210 (CAS, JKR) and W81XWH-11-1-0101 (DMC) and NIH R01 CA140985 (CAS). PK was supported by NIH K08 CA164048.
Author Contributions
JF-S and PH performed most of the studies. DMC performed experiments in Figures 2a, b and 5c, d. PP performed experiments in Figure 3a. PK provided the PR exonic-binding site for miR-141 (Figure 4c) and technical advice. JF-S, DMC, BMJ, JKR and CAS contributed intellectual design and interpretation of results. JFS wrote the manuscript. BMJ, JKR and CAS provided editorial assistance. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
About this article

Cite this article
Finlay-Schultz, J., Cittelly, D., Hendricks, P. et al. Progesterone downregulation of miR-141 contributes to expansion of stem-like breast cancer cells through maintenance of progesterone receptor and Stat5a. Oncogene 34, 3676–3687 (2015). https://doi.org/10.1038/onc.2014.298
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2014.298
This article is cited by
-
The Role of Steroid Hormones in Breast and Effects on Cancer Stem Cells
Current Stem Cell Reports (2018)
-
Direct targeting sperm-associated antigen 9 by miR-141 influences hepatocellular carcinoma cell growth and metastasis via JNK pathway
Journal of Experimental & Clinical Cancer Research (2016)
-
Impact of Progesterone on Stem/Progenitor Cells in the Human Breast
Journal of Mammary Gland Biology and Neoplasia (2015)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}