
Abstract
The role of specificity protein 1 (Sp1) in controlling gene expression in lung tumor development and metastasis is not well understood. In this study, we showed that the Sp1 level was highly increased and required for lung tumor growth in transgenic mice bearing Kras-induced lung tumors under the control of doxycycline. Furthermore, the Sp1 level was highly upregulated in lung adenocarcinoma cells with low invasiveness and in patients with stage I lung cancer. We also demonstrated that Sp1 was downregulated in lung adenocarcinoma cells with high invasiveness and in patients with stage IV lung adenocarcinoma. Moreover, Sp1 inversely regulated migration, invasion and metastasis of lung adenocarcinoma cells in vivo. In addition, a decrease in the Sp1 level in highly invasive lung adenocarcinoma cells resulted from instability of the Sp1 protein. Furthermore, overexpression of Sp1 in highly invasive lung adenocarcinoma cells increased expression of E-cadherin, a suppressor of metastasis, and attenuated the translocation of β-catenin into the cellular nucleus that leads to tumor malignancy. Taken together, Sp1 level accumulated strongly in early stage and then declined in late stage, which is important for lung cancer cell proliferation and metastasis during tumorigenesis.
Similar content being viewed by others


Introduction
Lung cancer, predominantly lung adenocarcinoma, is the leading cause of cancer mortality. In particular, tumor metastasis is the most life-threatening event of lung cancer patients (Jemal et al., 2010). Despite surgical tumor resection of patients with stage I non-small cell lung cancer, 40–50% of these patients still die because of the occurrence of metastasis (Denlinger et al., 2010). Understanding the process that controls metastasis is beneficial for the development of efficient tumor therapies. In the first step of metastasis, malignant lung tumor cells dissociate from a primary tumor by losing cell–cell adhesion and exhibiting a mesenchymal phenotype with highly invasive and migratory abilities. The conversion of an epithelial phenotype to a mesenchymal phenotype is achieved by the downregulation of proteins related to tight junctions, such as E-cadherin, that are transcriptionally regulated by specificity protein 1 (Sp1) (Liu et al., 2005). Downregulation of E-cadherin is reportedly associated with metastasis and poor prognosis in lung cancer (Bremnes et al., 2002; Chao et al., 2009).
Sp1, which belongs to the specificity protein/Krüppel-like factor family of transcription factors that bind to the GC-rich promoter element through three Cys2His2-type zinc-fingers, is ubiquitously expressed in many tissues and regulates several cellular processes (Wierstra, 2008). Furthermore, Sp1 has been reported to contribute to tumorigenesis through regulating gene transcription related to growth and proliferation (Black et al., 2001). In particular, Sp1 was shown to mediate prostaglandin E2- and nicotine-induced growth of non-small cell lung cancer cells (Sun et al., 2009; Zheng et al., 2009). In addition, the level of Sp1 has been shown to increase during the process of transformation in a fibrosarcoma transformation model (Lou et al., 2005) and the downregulation of Sp1 by small interfering RNA was shown to reduce the extent of tumor growth in nude mice after they were implanted with N67 gastric cancer cells (Jiang et al., 2004). However, the role of Sp1 in metastasis remains unknown, although Sp1 was previously shown to enhance expression of antimetastatic proteins, such as KiSS-1 and RECK (Chang et al., 2004; Mitchell et al., 2007). Despite high upregulation of Sp1 in a variety of tumor strains clarified in our laboratory by tissue array (Wang et al., 2008), it remains unclear whether a high expression level of Sp1 is maintained during tumor malignancy. Furthermore, the role of Sp1 in lung cancer progression is also poorly studied. Thus, Sp1-controlled tumor development toward malignancy was investigated in the current study.
Results
Sp1 upregulation is involved in Kras-induced lung tumorigenesis
Although Sp1 accumulation was found in various tumor types in our previous study (Wang et al., 2008), the role of Sp1 accumulation in oncogene-induced tumorigenesis remains unknown. In the present study, we found that the Sp1 level was decreased in vitro upon treatment with FTI-276 (farnesyltransferase inhibitor-276), which is a Kras inhibitor (Supplementary Figure S1). To define the role of Sp1 during Kras-induced lung tumorigenesis in vivo, we established transgenic mice bearing lung tumors induced by oncogenic Kras4bG12D under the control of doxycycline (Supplementary Figure S2A) (Fisher et al., 2001). The genotype of the bitransgenic mice (Scgb1a1-rtTA/TetO-Kras4bG12D) was confirmed by PCR genotyping (Supplementary Figure S2B). To determine the effects of doxycycline on the pathological changes of lungs, lungs were excised for paraffin sections stained with hematoxylin and eosin 1–5 months after doxycycline (0.5 g/l) administration. The aberrant hyperplasia and lung tumor were induced in the doxycycline-treated group, as revealed by hematoxylin and eosin staining and counting pulmonary tumor nodules. As shown in Figures 1a and b, tumor formation was identified and pulmonary nodules were counted 3 months after doxycycline administration (Supplementary Figure S3). In addition, the expressions of Sp1, Kras, pErk and proliferating cellular nuclear antigen, the marker of hyperplasia, and Clara cell-specific protein (CCSP), the marker of lung epithelial cells, were all obviously increased with increasing time of doxycycline administration (Figure 1a). According to the H-score, which was used to semiquantitate the signal of immunohistochemical (IHC) staining, these proteins were significantly upregulated 1 month after doxycycline administration. One spot represents one mouse (Figure 1c and Supplementary Figure S4). Furthermore, western blotting revealed that doxycycline increased protein levels of Kras and Sp1 significantly (Figure 1d). These results indicate that Sp1 expression was increased in Kras-induced lung tumor development.

MMA prevents doxycycline-induced pathological changes in the lungs of bitransgenic mice. (a) Doxycycline-induced lung tumor formation accompanied by increases in Sp1 and Kras in bitransgenic mice. After treatment for 1–5 months, lungs were excised for paraffin sections. Sections were prepared for hematoxylin and eosin (HE) staining and Sp1, Kras, pErk, proliferating cellular nuclear antigen (PCNA) and CCSP immunohistochemical staining. Stained sections were visualized using light microscopy ( × 200). (b) On each excised lung, surface pulmonary tumor nodules were counted. One spot represents one mouse. (c) After IHC staining, the Sp1 signal on each slide was semiquantitated by H-score. (d) After doxycycline treatment, lungs were collected for protein extraction, and analyzed by western blotting with anti-Kras and Sp1 antibodies. Data of eight independent experiments are represented as mean±s.e.m. (*P<0.05). (e) Prevention of Kras-induced lung tumorigenesis under the control of doxycycline by MMA. (Upper panel) Time course of doxycycline and MMA treatment. (Lower panel) HE and IHC staining using anti-Sp1 and Kras antibodies of lungs from treated bitransgenic mice. (f) After doxycycline administration for 5 months with MMA treatment in the last 2 months, lungs were excised from mice and pulmonary tumor nodules were counted. (g) H-score for the Sp1 signal on each slide. (h) MMA treatment attenuated Sp1 upregulation in Kras-induced lung tumorigenesis. pSp1 represents phosphorylated Sp1 and data of eight independent experiments are represented as mean±s.e.m. (*P<0.05).
To determine whether inhibition of Sp1 transcriptional activity prevents Kras-induced lung tumorigenesis, we treated bitransgenic mice with mithramycin A (MMA) that can compete the Sp1-binding sites by intraperitoneal injection (0.5 mg/kg). Histological analysis revealed that administration of MMA for 2 months reduced the extent of lung tumor formation triggered by doxycycline (Figure 1e). Both pulmonary tumor nodules and Sp1 H-score were also significantly decreased in MMA-treated group (Figures 1f and g). In addition, MMA not only deprived the Sp1-binding activity, but also resulted in the downregulation of Sp1 detected by IHC staining and western blotting (Figures 1e and h), which indicates that Sp1 can regulate itself. Therefore, Sp1 upregulation might be involved in Kras-induced lung tumor growth.
Sp1 expression is decreased during lung tumor malignancy and low Sp1 expression correlates with poor prognosis of lung adenocarcinoma patients
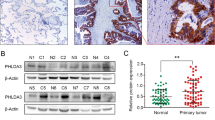
Previous studies have reported that Sp1 is involved in tumorigenesis through regulating gene expression related to cell growth and proliferation (Black et al., 2001; Lou et al., 2005). However, the role of Sp1 in the survival of cancer patients was unclear, especially in lung cancer patients. To determine the expression pattern of Sp1 in patients with lung adenocarcinoma, we collected clinically resected specimens of normal lung tissue and lung adenocarcinoma for IHC staining with an antibody against Sp1. In each clinical section, >30% of the cells expressing Sp1 were considered to have high expression by pathologists. Representative images of negative, low and high expression of Sp1 are shown in Figure 2a. Among 46 patients with stage I and II lung adenocarcinoma, 40 (87%) exhibited high Sp1 expression, whereas only 6 (13%) had low Sp1 expression. However, among 72 patients with stage IV lung adenocarcinoma, 29 (40.3%) exhibited high Sp1 expression, whereas 43 (59.7%) showed low Sp1 expression (Figure 2b and Table 1). Moreover, among all 118 patients, normal lung tissue of 105 (89%) exhibited negative and low expression. These results indicate that Sp1 expression was upregulated in the early phase and downregulated in the late phase of lung adenocarcinoma.

Sp1 expression is negatively correlated with survival of lung adenocarcinoma patients. (a) IHC staining of Sp1 in clinically resected normal lung tissue and lung adenocarcinoma. (b) Sp1 expression in different stages of lung adenocarcinoma patients and the corresponding normal lung tissue. (c, d) Kaplan–Meier analysis of overall survival in 118 patients with lung adenocarcinoma ranging from stages I to IV including 72 patients with stage IV. The Sp1 level was detected by IHC staining in resected lung adenocarcinoma. The P-value was determined by a two-sided log-rank test. (e) Sp1 expression in normal lung fibroblast (IMR) and CL-series cell lines with increasing invasiveness. Cells were harvested for whole-cell lysates and cellular proteins were immunoblotted with anti-Sp1 and tubulin antibodies. pSp1 represents phosphorylated Sp1 and data are representative of three independent experiments and are presented as mean±s.e.m. The P-value is indicated.
To determine whether Sp1 expression is associated with prognosis, we analyzed the correlation of Sp1 expression with the survival rate of patients. In Kaplan–Meier log-rank analysis, low Sp1 expression was significantly correlated with low survival in patients with lung adenocarcinomas from stages I, II and IV (Figure 2c; P<0.0001; hazard ratio 5.183). In addition, low Sp1 expression was also significantly associated with a low survival in patients in stage IV (Figure 2d; P=0.0021; hazard ratio 2.572). To further investigate the role of Sp1 in lung tumor malignancy, we examined Sp1 expression in a series of CL cell lines, which are lung adenocarcinoma cell lines with increasing invasiveness (Figure 2e) (Chu et al., 1997). Compared with expression in the IMR lung primary fibroblasts, the protein level of Sp1 was significantly increased in the CL series cell lines, indicating that Sp1 was overexpressed in lung adenocarcinoma. However, Sp1 expression in CL1-5, which had highest invasiveness in the series, exhibited significantly lower expression than that in CL1-0, which had lowest level of invasiveness, suggesting that Sp1 may negatively regulate tumor invasion.
Sp1 inhibits the invasive and migratory abilities of lung adenocarcinoma cells in vitro
To determine the effects of Sp1 on invasion and migration, we manipulated the Sp1 level in the CL cells. Because CL1-5 had lower Sp1 expression than CL1-0 but had the highest invasive ability of the CL panel (Figure 2e), we overexpressed Sp1 in CL1-5 cells by infection with adenovirus expressing green fluorescent protein (GFP)-Sp1 or GFP (for control). The expression level of GFP-Sp1 detected by western blotting was gradually increased with increasing infection dosage of GFP-Sp1 adenovirus for 48 h (Figure 3a). In addition, CCSP expression was increased with GFP-Sp1 adenovirus infection in CL1-5 cells, implying that Sp1 is involved in the switch to the epithelial phenotype. After infection with GFP or GFP-Sp1 adenovirus for 48 h, GFP-infected CL1-5 cells had a spindle-like morphology, whereas GFP-Sp1-infected cells exhibited an epithelial-like morphology and formed aggregates (Figure 3b). In addition, overexpression of GFP-Sp1 enhanced proliferation of CL1-5 cells (Supplementary Figure S5), implying that more Sp1 in cells will trigger proliferation-related gene expression. A Matrigel-combined in vitro invasion assay showed that the invasive ability of CL1-5 cells was decreased with increasing infection dosage of GFP-Sp1 (Figure 3c). To examine the effect of Sp1 on the migratory ability of cells, we used a wound-healing assay, a Transwell migration assay and time-lapse microscopy. We found that the migratory area of GFP-Sp1 adenovirus-infected CL1-5 cells was significantly smaller than that of GFP adenovirus-infected cells in the wound-healing assay (Figure 3d). In addition, the Transwell migration assay also indicated that the migratory ability of CL1-5 cells was attenuated with increasing infection dosage of GFP-Sp1 (Figure 3e). According to the video time-lapse microscopy analysis, after wounding a cellular monolayer, the migratory ability of CL1-5 cells was suppressed by GFP-Sp1 adenovirus infection (Figure 3f and Supplementary Movies). Based on these results, it is apparent that overexpression of Sp1 inhibited the invasive and migratory abilities of CL1-5 cells in vitro.

Sp1 negatively regulates invasive and migratory abilities of lung cancer cells. (a) CL1-5 cells were infected with adenovirus-GFP and increasing doses (1–20 m.o.i.) of adenovirus-GFP-Sp1. After incubation for 48 h, cells were harvested for whole-cell lysates and cellular proteins were immunoblotted with anti-Sp1, CCSP and actin antibodies. pSp1 represents phosphorylated Sp1. (Lower panel) The quantitated result for the ratio of exogenous GFP-Sp1 with endogenous Sp1, both of which were normalized by β-actin. (b) The morphologies of adenovirus-GFP- and GFP-Sp1-infected CL1-5 cells were observed using light microscope with × 100 magnification. (c) Effect of Sp1 on invasive ability. The in vitro invasive ability of CL1-5 cells infected with adenovirus-GFP or GFP-Sp1 was determined using Matrigel-combined Transwell chambers as described in Materials and methods. Data are representative of three independent experiments and are presented as mean±s.e.m. (*P<0.05). (d) Effect of Sp1 on the migratory ability of cells measured with a wound-healing assay. After infection with adenovirus-GFP or GFP-Sp1 for 48 h, confluent monolayers of CL1-5 were wounded with a pipette tip and incubated for an additional 24 h. The migratory area of cells was calculated for quantification. Data are representative of three independent experiments and are presented as mean±s.e.m. (*P<0.05). (e) Effect of Sp1 on migratory ability examined by a Transwell migration assay. After infection for 48 h, the migratory ability of adenovirus-GFP or GFP-Sp1-infected cells was determined using Transwell chambers as described in Materials and methods. Data are representative of three independent experiments and are presented as mean±s.e.m. (*P<0.05). (f) Effect of Sp1 on migratory ability monitored by video time-lapse microscopy. After infection with adenovirus-GFP or GFP-Sp1 for 48 h, the migratory area of CL1-5 cells was continually monitored for 24 h under a time-lapse microscopy at × 100 magnification.
Knockdown of Sp1 enhances the invasive and migratory abilities of lung adenocarcinoma cells in vitro
As both CL1-0 and CL1-5 came from the same patient's tumor (Chu et al., 1997), herein we found that there was an inverse proportion between Sp1 level and invasive activity in these two cells. Therefore, in addition to evaluating the effect of Sp1 overexpression on CL1-5 cells, we also determined the effect of Sp1 knockdown on invasion and migration in CL1-0 cells that express the highest level of Sp1 among the CL cell panel (Figure 2e). Sp1 knockdown was achieved by transfection of CL1-0 cells with Sp1 small hairpin RNA for 48 h (Figure 4a). A Matrigel-combined in vitro invasion assay revealed that Sp1 knockdown enhanced invasion of CL1-0 cells (Figure 4b). Moreover, Sp1 knockdown increased the migratory ability of the cells according to the wound-healing and Transwell migration assays (Figures 4c and d).

Sp1 knockdown enhances invasive and migratory abilities of CL1-0 cells. (a) CL1-0 cells were transfected with scrambled or Sp1 small hairpin RNA (shRNA). After incubation for 48 h, cells were harvested for whole-cell lysates and cellular proteins were immunoblotted with anti-Sp1 and actin antibodies. (b) Effect of Sp1 knockdown on invasive ability. The in vitro invasive ability of CL1-0 cells transfected with scrambled or Sp1 shRNA was determined using Matrigel-combined Transwell chambers as described in Materials and methods. Data are representative of three independent experiments and are presented as mean±s.e.m. (*P<0.05). (c) Effect of Sp1 knockdown on the migratory ability measured by wound-healing assay. After transfection with scrambled or Sp1 shRNA for 48 h, confluent monolayers of CL1-0 were wounded and incubated for an additional 24 h. Migratory area was calculated for quantification. Data are representative of three independent experiments and are presented as mean±s.e.m. (*P<0.05). (d) Effect of Sp1 knockdown on the migratory ability examined by Transwell migration assay. The migratory ability of scrambled or Sp1 shRNA-transfected cells was determined by Transwell chambers. Data are representative of three independent experiments and are presented as mean±s.e.m. (*P<0.05).
Sp1 inhibits metastasis of lung adenocarcinoma cells in vivo
In our in vitro experiments, Sp1 was shown to negatively regulate the invasive and migratory abilities of lung adenocarcinoma cells. To determine the effect of Sp1 on metastasis in vivo, we injected CL1-5 cells overexpressing Sp1 or CL1-0 with knocked down Sp1 into the lateral tail vein of severe combined immunodeficient mice. At 4 weeks after injection, all mice were killed, and the number of pulmonary metastatic tumor nodules in each was counted. Compared with the numerous pulmonary metastatic tumor nodules found in mice injected with CL1-5 cells expressing GFP, the mice injected with CL1-5 cells expressing GFP-Sp1 had fewer nodules (Figures 5a and e), indicating that overexpression of Sp1 inhibited metastasis of CL1-5 cells in vivo. IHC staining also showed that tumor formation in the lungs of mice injected with GFP-expressing CL1-5 cells was more obvious than that in mice injected with CL1-5 cells expressing GFP-Sp1 (Figure 5a). Moreover, the tumor part of lungs from mice injected with CL1-5 cells expressing GFP-Sp1 exhibited higher Sp1 expression (Figure 5a). The expression of GFP and GFP-Sp1 in CL1-5 cells injected into mice was detected by western blotting (Figure 5b). On the other hand, mice injected with CL1-0 cells with knocked down Sp1 had more pulmonary tumor nodules than mice injected with CL1-0 cells, indicating that Sp1 knockdown enhanced metastasis of CL1-0 cells in vivo (Figures 5c and f). In addition, the tumor part of lungs from mice injected with CL1-0 cells with knocked down Sp1, indeed, exhibited lower Sp1 expression. Sp1 knockdown of the CL1-0 cells injected into mice was confirmed by western blotting (Figure 5d). Taken together, cells with low Sp1 expression, such as CL1-0 with Sp1 knockdown and CL1-5 cells, exhibited strong metastatic ability in vivo in contrast to cells with high Sp1 expression, such as CL1-0 and CL1-5 cells with GFP-Sp1 overexpression (Figure 5g). Therefore, these results indicate that Sp1 negatively regulated metastasis of lung adenocarcinoma cells in vivo.

Sp1 suppresses lung adenocarcinoma cell metastasis in vivo. (a) After infection of GFP or GFP-Sp1 adenovirus for 48 h, CL1-5 cells ( × 106) were suspended in 100 μl of PBS and injected into the lateral tail vein of severe combined immunodeficient (SCID) mice. After 4 weeks, all mice were killed and the number of pulmonary tumor nodules was calculated 48 h after fixation of lungs with 10% formaldehyde. (Left panel) Representative images of lungs. Arrows indicate pulmonary metastatic tumor nodules. (Central panel) Hematoxylin and eosin (HE) staining of lungs. Arrows indicate pulmonary metastatic tumor. (Right panel) IHC staining using the anti-Sp1 antibody. (b) Confirmation of GFP and GFP-Sp1 expression of CL1-5 cells injected into mice by western blotting. (c) Effects of Sp1 knockdown on in vivo metastasis of CL1-0 cells. (Left panel) Representative images of lungs. (Central panel) HE staining of lungs. (Right panel) Sp1 IHC staining. (d) Confirmation of Sp1 knockdown in CL1-0 cells injected into mice by western blotting. (e, f) Quantitative result (five mice per group) of pulmonary metastatic tumor nodules 4 weeks after injection. Data are expressed as mean±s.e.m. (**P<0.01). (g) Sp1 expression in both CL1-0 and CL1-5 cells that were injected into the tail vein was compared. pSp1 represents phosphorylated Sp1.
Protein instability causes downregulation of Sp1 in highly invasive lung adenocarcinoma cells
Sp1 expression is important for cellular transformation and is increased in tumor cells. In our previous study, Sp1 was overexpressed in cervical cancer because of increased stability of Sp1 protein (Wang et al., 2008). Thus, we hypothesized that Sp1 stability plays an important role in maintaining a high Sp1 level in tumor cells. In addition to lower mRNA and protein levels of Sp1 in CL1-5 cells, phosphorylation of Sp1 at T739, which has been shown to enhance Sp1 protein stability (Chuang et al., 2008), was also decreased (Figure 6Aa and b), suggesting that Sp1 protein stability in CL1-5 cells was lower than that in CL1-0 cells. Further demonstrating that Sp1 protein stability was lower in the CL1-5 cells, high sumoylation and ubiquitination levels of Sp1 were detected in CL1-5 cells by western blotting (Figure 6Ab). As the association of Sp1 with ring finger factor-4 (RNF4), the E3 ligase of Sp1, has been reported (Wang et al., 2008), we then studied the interaction of these proteins, and the results indicated that Sp1 associated with RNF4 was enhanced in CL1-5 cells (Figure 6C). To further clarify whether both Sp1 sumoylation and ubiquitination are more obvious in CL1-5 cells, the anti-Sp1-mediated immunoprecipitation was performed. As shown in Figure 6D, the signal of SUMO-Sp1 was stronger in CL1-5 cells. In Figure 6E, after overexpression of myc-ubiquitin in the presence of MG132, which is a proteasome inhibitor, the ubiquitination signal was detected in the anti-Sp1 immunoprecipitates in CL1-5 cells, but not in CL1-0 cells. These results indicate that Sp1 protein stability was lower in the highly invasive lung adenocarcinoma cells because of higher sumoylation and ubiquitination. Because Sp1 protein was previously shown to autoregulate its own expression (Nicolas et al., 2001), we proposed a model explaining how Sp1 protein stability affects the Sp1 level in highly invasive lung adenocarcinoma cells (Figure 6F).

Downregulation of Sp1 expression in highly invasive lung adenocarcinoma cells is caused by instability of Sp1 protein. (Aa) Whole-cell extracts of cells were collected for western blotting with antibodies against Sp1, pSp1 (T739) and tubulin. Reverse transcriptase–PCR (RT–PCR) was used to determine the mRNA level of Sp1. (Ab) Sumoylation and ubiquitination in an equal Sp1 expression level of CL1-0 and CL1-5 cells. (Ac) The quantitated result for the ratio of pSp1 (T739) with Sp1 in CL1-0 and 1-5 cells. Data are representative of three independent experiments and are presented as mean±s.e.m. (**P<0.01). (B) After MG132 treatment (50 μM) for 6 and 12 h, cells were harvested as whole-cell extracts for western blotting. (C) The association of Sp1 with RNF4. The whole-cell extracts of cells were immunoprecipitated with an anti-RNF4 antibody or IgG, and immunoblotted with an anti-Sp1 antibody. (D) Sp1 sumoylation in CL1-5 cells. Whole-cell extracts of cells were immunoprecipitated with an anti-Sp1 antibody or IgG, and immunoblotted with the antibody against SUMO-1 or Sp1. SUMO-Sp1 is indicated. (E) Sp1 ubiquitination in CL1-5 cells. After overexpression of myc-ubiquitin in the presence of MG132 for 12 h, whole-cell extracts were immunoprecipitated with an anti-Sp1 antibody, and immunoblotted with the antibody against myc or Sp1. Ubi-Sp1 is indicated. pSp1 represents phosphorylated Sp1. (F) Proposed model of Sp1 downregulation caused by Sp1 protein instability in highly invasive lung adenocarcinoma cells.
Sp1 induces E-cadherin expression and blocks the translocation of β-catenin into nucleus in highly invasive lung adenocarcinoma cells
Downregulation of E-cadherin, a known suppressor of tumor cell invasion and migration, was previously shown to associate with tumor metastasis (Frixen et al., 1991; Bremnes et al., 2002). Sp1 has also been reported to regulate E-cadherin expression (Liu et al., 2005). Thus, we speculated that Sp1 inversely regulates tumor metastasis through inducing E-cadherin expression. As shown in Figure 7, E-cadherin expression was higher in CL1-0 cells that exhibit lower metastatic ability than in CL1-5. This phenomenon is the same with Sp1 expression. To confirm the previous study showing that Sp1-binding sites are important for E-cadherin promoter transcription, chromatin immunoprecipitation assay was used to determine whether Sp1 binds to E-cadherin promoter. As shown in Figure 7b, indeed, Sp1 obviously bound to E-cadherin promoter (−199/−450). Therefore, the effects of Sp1 on E-cadherin expression were studied. Sp1 small hairpin RNA-mediated Sp1 knockdown for 48 h significantly reduced E-cadherin expression in both CL1-0 and A549 cells (Figure 7c). Moreover, overexpression of GFP-Sp1 for 48 h significantly induced E-cadherin expression in CL1-5 cells (Figure 7d), indicating that Sp1 positively regulated E-cadherin expression.

Sp1 positively regulates E-cadherin expression and attenuates the translocation of β-catenin into the cell nucleus. (a) Whole-cell extracts of CL1-0 and CL1-5 cells were collected for western blotting with antibodies against E-cadherin, β-catenin and β-actin. (b) Binding of Sp1 to the promoter of E-cadherin was determined by chromatin immunoprecipitation (CHIP) assay. (c) Effect of Sp1 knockdown on the protein level of E-cadherin in lung adenocarcinoma cells with low invasiveness. After transfection of scrambled or Sp1 small hairpin RNA (shRNA) for 48 h, whole-cell extracts of CL1-0 and A549 cells were collected for western blotting with antibodies against E-cadherin and β-actin. Quantitated results are shown in the lower panel. Data are representative of three independent experiments and are presented as mean±s.e.m. (*P<0.05, **P<0.01). (d) Effect of Sp1 on the protein levels of E-cadherin and β-catenin in lung adenocarcinoma cells with high invasiveness. After infection with GFP or increasing dosage (1–20 m.o.i.) of GFP-Sp1 adenovirus for 48 h, whole-cell extracts of CL1-5 cells were collected for western blotting using antibodies against E-cadherin, β-catenin and β-actin. pSp1 represents phosphorylated Sp1. Quantitated results are shown in the lower panel. Data are representative of three independent experiments and are presented as mean±s.e.m. (*P<0.05, **P<0.01). (e) Effect of Sp1 on the translocation of β-catenin into nucleus. (Left panel) After infection with GFP or increasing doses of GFP-Sp1 adenovirus for 48 h, cytosolic and nuclear fractions were isolated for western blotting with antibodies against β-catenin, tubulin and histone H3. (Right panel) Data are representative of three independent experiments and are presented as mean±s.e.m. (*P<0.05, **P<0.01). (f) Immunofluorescent staining of nuclear GFP-Sp1 and β-catenin in CL1-5 cells. After infection of cells with adenovirus-expressing GFP-Sp1 for 48 h, cells on a coverslip were stained with an anti-β-catenin antibody and DAPI. Stained cells were photographed under a fluorescence microscope at × 1000 magnification. (g) Effect of Sp1 on the expression of T-cell factor-4 (Tcf4), a target gene of β-catenin. After infection of cells with adenovirus expressing GFP-Sp1 for 48 h, RNA was prepared for reverse transcriptase–PCR (RT–PCR). (h) The proposed model showing that Sp1 blocked β-catenin translocation into the cell nucleus through inducing E-cadherin, which was shown to anchor β-catenin near plasma membrane.
β-Catenin, which is the link between E-cadherin and cytoskeleton, also plays an important role in tumor malignancy. In previous studies, β-catenin has been shown to promote tumor malignancy through its translocation into the nucleus, and subsequently turning on gene transcription related to metastasis in colon cancer (Morin et al., 1997). However, increased expression of β-catenin is reportedly associated with good prognosis of patients with lung cancer (Hommura et al., 2002). Thus, it remains unclear whether β-catenin promotes tumor metastasis. In CL1-5 cells, β-catenin expression was higher than that in CL1-0 cells (Figure 7a). The β-catenin expression was apparently unaffected by Sp1 (Figure 7d). We also investigated the effect of Sp1 on the translocation of β-catenin by isolating cytosolic and nuclear fractions. In CL1-5 cells, overexpression of Sp1 significantly reduced nuclear β-catenin (Figure 7e); immunofluorescence analysis also revealed that the amount of nuclear β-catenin was decreased in GFP-Sp1-expressing cells (Figure 7f), indicating that β-catenin translocation into the nucleus was attenuated by Sp1. Consequently, β-catenin-mediated target gene expression, such as T-cell factor-4, was inhibited by GFP-Sp1 (Figure 7g) (Roose et al., 1999). Therefore, we propose a model in which Sp1 blocked β-catenin translocation into the nucleus through the induction of E-cadherin expression, which was shown to anchor β-catenin near the plasma membrane in lung adenocarcinoma cells (Figure 7h) (Rivard, 2009). These results suggest that Sp1 inversely regulated metastasis through inducing E-cadherin expression and attenuating β-catenin translocation into the nucleus.
According to our results, we conclude that Sp1 expression is highly increased during tumor formation and downregulated during metastasis (Figure 8a). In addition, we propose a model showing that Sp1 expression is maintained in the lowest level by protein instability triggered by sumoylation and ubiquitination. Furthermore, because of the increase of protein stability, Sp1 is upregulated in lung tumor cells to induce E-cadherin expression, which is a metastasis suppressor and anchors β-catenin near the plasma membrane. In highly invasive lung tumor cells, Sp1 downregulation, which is caused by protein instability, results in the loss of E-cadherin. Subsequently, β-catenin translocation into the nucleus for transcriptional regulation of genes related to invasion and migration is attenuated (Figure 8b).

Sp1 expression regulates lung tumor progression. (a) Sp1 expression is maintained at the lowest level in normal cells. Until cell transformation or tumor formation, Sp1 expression is highly upregulated. When tumor cells became more invasive or malignant, Sp1 expression is obviously decreased. (b) The lowest Sp1 level for basal gene transcription is maintained by protein instability triggered by sumoylation and ubiquitination. In lung tumor cells, the Sp1 level is upregulated for proliferation accompanied by the increased level of E-cadherin for adhesion. In highly invasive lung tumor cells, E-cadherin, which anchors β-catenin near the plasma membrane, is lost because of the downregulation of Sp1 level triggered by protein instability. Consequently, downregulation of Sp1 level increases the amount of nuclear β-catenin for gene transcription related to metastasis.
Discussion
Previous reports have shown that Sp1 transcriptionally induced expression of several metastatic suppressors, including tissue inhibitor of metalloproteinase-1 (Kwak et al., 2006), KiSS-1 (Mitchell et al., 2007), G-protein-coupled protein 54 (DeFino et al., 2010), bone morphogenetic protein-7 (Chen et al., 2010; Yi et al., 2010) and Src-suppressed C-kinase substrate (Bu et al., 2010), but the specific role of Sp1 in the process of metastasis remains unclear. In the present study, we first showed that the proportion of low Sp1 expression in patients with stage IV lung adenocarcinoma was higher than that in patients with stages I and II of lung adenocarcinoma. We also found that Sp1 negatively correlated with poor prognosis. Furthermore, the Sp1 level was downregulated in highly invasive lung adenocarcinoma cells, and inversely regulated cellular functions controlling metastasis. Moreover, we also confirmed that Sp1 upregulation was involved in lung tumor growth. These data suggest that upregulation of Sp1 occurred during the process of cellular transformation or tumor growth. When tumor cells became more invasive, the Sp1 level was downregulated for both inducing appropriate gene expression related to metastasis and reducing E-cadherin expression.
As we used the adenovirus to overexpress Sp1 in CL1-5 cells, in addition to the attenuation of cellular invasive activity, we also observed that the proliferation of cells was significantly increased. However, Sp1 knockdown with Sp1 small hairpin RNA (4 μg) enhanced the invasive ability but did not affect proliferation in CL1-0 cells. We speculate that Sp1 expression in CL1-0 cells with knocked down Sp1 is similar to that in CL1-5 cells, and the remaining Sp1 level is enough for proliferation in CL1-0 cells. To further address the effect of Sp1 downregulation on the invasiveness, CL1-0 cells were also treated with MMA for 24 h before invasion assay. Because of serious cell death and activation of proapoptotic proteins upon MMA treatment (data not shown), it is difficult to evaluate its effect on invasive activity of lung cancer cells. In addition, according to previous studies, MMA still contains other functions such as facilitating apoptosis through activating Fas pathway (Duverger et al., 2004; Leroy et al., 2006). Therefore, another effect(s) of MMA except affecting Sp1-binding activity might also be involved in the lung cancer tumorigenesis, such as downregulation of DNA methytransferase 1 and MUC5A, both of which are related to tumor malignancy (Lin et al., 2007; Lee et al., 2011).
In the bitransgenic mice bearing spontaneously developed lung tumor under the control of doxycycline, MMA treatment exhibited antitumor effect. Moreover, doxycycline-induced lung tumors in the bitransgenic mice still belong to the early phase of lung adenocarcinomas; thus, we did not detect any metastasis in the circulation or other organs with or without MMA treatment. Herein, we provide evidence that Sp1 upregulation is involved in lung tumor growth, but not for metastasis. MMA-inhibited lung tumor growth, in part, was contributed by MMA-mediated Sp1 downregulation through competing Sp1-binding sites on the promoter (Remsing et al., 2003; Maor et al., 2006). This finding further suggests that the therapeutic strategy targeting Sp1 is appropriate for patients with early-phase lung cancer, but not for patients with late-phase lung cancer.
In addition to upregulation of Sp1 mRNA (Kumar and Butler, 1999), protein stability also played an important role in increasing the Sp1 level in tumor cells (Wang et al., 2008). In this study, we provide evidence that the decrease of Sp1 in highly invasive lung adenocarcinoma cells was caused by Sp1 protein instability. Protein turnover of Sp1 was mediated through an ubiquitination pathway in a 26S-proteosome-dependent manner (Su et al., 1999). Our previous studies showed that Sp1 modified by sumoylation at lysine 16 or phosphorylation at threonine 278/739 had negative and positive effects on the protein stability, respectively (Nicolas et al., 2001; Chuang et al., 2008; Wang et al., 2008). In this study, we found that Sp1 was obviously sumoylated and ubiquitinated upon losing phosphorylation. Therefore, based on these data, it seems that protein instability of Sp1 by post-translational modification, rather than transcription, leads to cancer metastasis in the late stage. However, although it is known that RNF4 acts as an E3 ligase to modulate the Sp1 level in cancer metastasis, the detailed mechanism must be further elucidated.
In the promoter of E-cadherin, Sp1-binding sites were identified and were found to be important for transcription (Liu et al., 2005), suggesting that Sp1 may be involved in metastasis through regulating E-cadherin expression. Herein, we reported that Sp1 overexpression inhibited metastasis and increased E-cadherin expression, which suggests that Sp1 may inhibit metastasis through the E-cadherin-mediated maintenance of cell–cell adhesion.
In both A549 and CL1-5 cells, Sp1 knockdown led to the decrease of E-cadherin level, but the cell fate might be different. Our previous study showed that Sp1 overexpression causes cellular apoptosis of A549 (Chuang et al., 2009), but increases proliferation of CL1-5 cells. The discrepancy may be caused by A549 with wild-type p53 and CL1-5 with mutant p53 (p53R248W) (Tsai et al., 2006). In addition, previous reports showed that Sp1 cooperated with p53 to repress survivin, cyclin B1 and cdc25C, all of which are required for cell survival and proliferation (Innocente and Lee, 2005; St Clair and Manfredi, 2006; Esteve et al., 2007). These results suggest that Sp1 overexpression induces cellular proliferation in lung tumor cells with mutant p53, but induces p53-dependent apoptosis in that with wild-type p53.
The translocation of β-catenin into the nucleus was shown to promote metastasis in colon cancer, but high expression of β-catenin predicted good prognosis in lung cancer (Morin et al., 1997; Hommura et al., 2002). Thus, the role of β-catenin in tumor malignancy is controversial. In this study, we showed that β-catenin expression in highly invasive lung adenocarcinoma cells was significantly higher than in low invasive cells, which suggests that β-catenin is involved in lung tumor malignancy. Although Sp1 did not affect β-catenin expression, Sp1 attenuated translocation of β-catenin into the nucleus in highly invasive lung adenocarcinoma cells. This result might be caused by the Sp1-induced expression of E-cadherin shown to interact with β-catenin on the cell membrane (Schmalhofer et al., 2009).
In conclusion, Sp1 was required for lung tumor growth but it suppressed metastasis through inducing E-cadherin expression. The clinical implication of the present study is that inhibition of Sp1 is seemingly inappropriate for all patients with lung cancer ranging from stages I to IV.
Materials and methods
Cell culture and transfection
The human lung adenocarcinoma cell lines, CL-series cell lines, were kindly provided by Dr Pan-Chyr Yang (Chu et al., 1997). A549 cells were purchased from American Type Culture Collection (Manassas, VA, USA). CL cell lines and A549 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum, 100 μg/ml streptomycin sulfate and 100 U/ml penicillin G sodium at 37 °C and 5% CO2. Transfection with myc-ubiquitin was performed using Lipofectamine, 2000 (Invitrogen Life Technologies, Grand Island, NY, USA) according to the manufacturer’s instruction. For transfection, 9 μg of myc-ubiquitin plasmid DNA was incubated with 900 μl of Opti-MEM containing 9 μl of Lipofectamine for 30 min at room temperature. Subsequently, cells in the 10 cm culture dish were incubated with 3 ml of Opti-MEM containing myc-ubiquitin and Lipofectamine for 8 h at 37 °C in 5% CO2. After incubation with DMEM/10% fetal bovine serum for additional 18 h, cells were lysed for immunoprecipitation.
Generation of bitransgenic mice (Scgb1a1-rtTA/TetO-Kras4bG12D)
Two transgenic lines were acquired from Jackson Lab (Bar Harbor, MA, USA) and maintained at the National Laboratory Animal Center in Taiwan. The Scgb1a1-rtTA transgenic mice expressed the reverse tetracycline trans-activator (rtTA) protein under the control of Scgb1a1 (secretoglobin, family 1A, member 1) promoter. The TetO-Kras4bG12D transgenic mice expressed activated Kras (Kras4bG12D) under the regulation of a tetracycline-responsive promoter element (TRE; tetO). After mating, PCR genotyping was performed for detection of Scgb1a1-rtTA and TetO-Kras4bG12D in bitransgenic mice. Activated Kras expression in the bitransgenic mice was induced by administration of doxycycline (0.5 g/l) to the drinking water, starting at the age of 2 months.
Collection of clinical specimens from patients with lung adenocarcinoma
Clinical specimens were collected after clinical resection from 118 patients with lung adenocarcinoma ranging from stages I, II to IV who underwent surgery at the Department of Surgery of the National Cheng Kung University Hospital. The lung cancer stage was classified by pathologists. Collection of human specimens conformed to the human ethics and was approved by the Clinical Research Ethics Committee at National Cheng-Kung University Medical Center.
Histological analysis and immunohistochemistry
Lungs excised from bitransgenic mice or clinically resected specimens were fixed in 10% formaldehyde for 24 h, dehydrated and embedded in paraffin. Sections (5 μM) were cut and stained with hematoxylin and eosin. For immunohistochemistry, sections were dewaxed in xylene and rehydrated in a graded series of ethanols. Endogenous peroxidases were blocked by 0.3% hydrogen peroxide in phosphate-buffered saline (PBS) for 30 min. Histological sections, which was blocked by 3% bovine serum albumin in PBS, were incubated with appropriate diluted primary antibody for 2 h at room temperature. The immunoreactivity was visualized with a Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA) and photographed under Olympus BX-51 microscope (Olympus, Melville, NY, USA). To score IHC staining, H-score was used to evaluate expressions of Sp1, Kras, pErk, proliferating cellular nuclear antigen and CCSP in doxycycline-treated bitransgenic mice according to previous reports (McCarty et al., 1985; Hung et al., 2003). The H-score was calculated using the following formula: (Zero × percentage of negatively staining)+(percentage of weakly staining)+(twice percentage of moderately staining)+(thrice percentage of strongly staining). The range of summarization is from 0 to 300.
Generation of adenovirus-expressing GFP or GFP-Sp1
Adenoviruses carrying the complementary DNA encoding GFP or GFP-Sp1 were made according to the instructions of Adeno-X Expression System 2 kit (BD Biosciences, San Diego, CA, USA). The detail process is described in our previous study (Chuang et al., 2009).
In vitro Matrigel-combined invasion assay
Cellular invasive property of cells was analyzed by invasion assay using the 24-well plate Transwell system with an 8 μM pore size polycarbonate filter membrane (Corning Costar, Corning, NY, USA). The filter membrane was coated with 15 μg (45 μg/cm2) of Matrigel (BD Biosciences, San Diego, CA, USA). The cell suspensions were seeded to the upper compartment of the Transwell chamber at the cell density of 2 × 104 in 100 μl of medium. After 24 h, the filter membrane was fixed with methanol for 10 min. The opposite surface of the filter membrane facing the lower chamber was stained with 4,6-diamidino-2-phenylindole (DAPI; Invitrogen) for 3 min and the migrated cells were then counted under a fluorescent microscope. Assays was performed in duplicate and repeated three times.
Transwell migration assay
The cell migration assay was performed using Transwell system with an 8-μM pore size polycarbonate filter membrane. After overexpression of Sp1 in CL1-5 cells and knockdown of Sp1 in CL1-0 cells for 48 h, cells were trypsinized and suspended in serum-free DMEM. Upper wells were filled with cell suspensions (2 × 104) in serum-free DMEM and lower wells were filled with DMEM containing 10% fetal bovine serum. After incubation for 6 h at 37 °C, the lower side of filter membrane was fixed with 10% formaldehyde and stained with DAPI for 3 min. The migrated cells were counted under a fluorescent microscope. Assays was performed in duplicate and repeated three times.
Wound-healing assay
After overexpression of Sp1 in CL1-5 cells and knockdown of Sp1 in CL1-0 cells for 48 h, the linear wound of cellular monolayer was created by scratching confluent cell monolayer using a plastic pipette tip. Scratched cell monolayer was washed by PBS to remove debris. After incubation at 37 °C for 24 h, area of migration was photographed under light microscope for evaluation and monitored by time-lapse microscope (Olympus IX81-ZDC Zero Drift microscope; Olympus) photographing per 10 min for 24 h. Experiments were performed independently three times.
In vivo metastasis assay
The animal study was approved by the institutional animal care and use committee at the National Cheng-Kung University. After overexpression of Sp1 in CL1-5 cells and knockdown of Sp1 in CL1-0 cells for 48 h, cells were trypsinized and suspended in PBS for tail vein injection. A total of 106 cells in 100 μl of PBS were injected into the lateral tail vein of 8-week-old severe combined immunodeficient mice (five mice per group). Mice were killed after 4 weeks and excised lungs were fixed with 10% formaldehyde for 48 h. Finally, the number of pulmonary metastatic nodules on the surface of lung was counted and lungs were prepared for paraffin sections stained by hematoxylin and eosin.
Immunoprecipitation
Total cell extracts were prepared by incubating cells in RIPA lysis buffer (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 0.1% sodium dodecyl sulfate (SDS), 1% Nonidet P-40 and 0.25% deoxychlate) with freshly added inhibitors (1 mM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin and 1 μg/ml leupeptin) for 10 min at 4 °C. After centrifugation at 12 000 g for 10 min at 4 °C, supernatant was collected. For immunoprecipitation, supernatant was incubated with the anti-RNF4 (Sigma-Aldrich, St Louis, MO, USA) or anti-Sp1 (Millipore, Bedford, MA, USA) antibody at 4 °C for 1 h with rotation. After incubation with protein A agarose beads (Millipore) at 4 °C for 1 h with rotation, the beads were pelleted by centrifugation at 3000 r.p.m. at 4 °C for 5 min and washed with RIPA buffer three times. Proteins binding to the beads were eluted by 2 × Sample buffer (3.33% SDS, 116.67 mM Tris–HCl, pH 6.8, 10% glycerol, 0.004% bromophenol blue and 100 mM dithiothreitol) for electrophoresis.
Western blotting analysis
Proteins were subjected to one-dimensional SDS–polyacrylamide gel electrophoresis (4.5% stacking gel and 8–12.5% separating gel) and were transferred to a polyvinylidene difluoride membrane (Millipore) by using a transfer apparatus according to the manufacturer’s protocols (Bio-Rad, Richmond, CA, USA). Membranes were blocked with 3% nonfat dry milk in TBST buffer (10 mM Tris–HCl, pH 8.0, and 150 mM NaCl) containing 0.05% Tween-20, washed in the same buffer and probed with the anti-Sp1 (Millipore), anti-Kras (Proteintech Group, Chicago, IL, USA), anti-CCSP (Seven Hills Bioreagents, Cincinnati, OH, USA), anti-pSp1 (T739; Kelowna, Taiwan), anti-RNF4 (Sigma), anti-SUMO-1 (Epitomics, Burlingame, CA, USA), anti-β-catenin (Cell Signaling Technology, Boston, MA, USA), anti-E-cadherin (Cell Signaling Technology), anti-myc (Invitrogen), anti-histone H3 (Millipore), anti-tubulin (Sigma) or anti-β-actin (Millipore) antibody at 4 °C overnight. The secondary antibody (Millipore), goat anti-rabbit or anti-mouse immunoglobulin G linked with horse radish peroxidase, was added in the presence of the blocking agent and incubated with membranes for 1 h at room temperature. After three more washes, immune complexes were detected using chemiluminescence Alpha Innotech detection system (Alpha Innotech Corp., San Leandro, CA, USA). The intensity of the bands on western blots was analyzed using AlphaImager software (Alpha Innotech Corp.).
Chromatin immunoprecipitation assay
After crosslinking with 1% formaldehyde for 10 min at room temperature, cells (1 × 108) were washed with ice-cold PBS three times and whole-cell extracts were prepared with lysis buffer. The cellular DNA fragment approximate 500 bp was prepared by sonication. For immunoprecipitation, salmon sperm DNA-blocked cell lysate was incubated with 2 μg of anti-Sp1 antibody or IgG at 4 °C for 16 h with rotation. After incubation with 30 μl of protein A agarose beads at 4 °C for additional 2 h, beads were washed three times with high salt buffer (20 mM Tris–HCl, 500 mM NaCl, 2 mM EDTA and 0.5% NP-40) and another three times with low salt buffer (10 mM Tris–HCl, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40 and 0.01% SDS). Subsequently, proteins binding to beads were eluted by 500 μl of TE buffer containing 1% SDS, and crosslinks were reversed at 65 °C for 16 h. After protein digestion by 0.5 mg/ml proteinase K at 50 °C for 2 h, DNA was extracted by phenol/chloroform and precipitated by absolute alcohol. The purified DNA was analyzed by PCR using the primer for E-cadherin promoter (−199/−450): Forward, 5′-CAAAAAATCCCAAAAAACAA-3′; Reverse, 5′-ACCTGCCGGCCACAGCCAAT-3′.
Isolation of cytosolic and nuclear fractions
Cells were washed with ice-cold PBS and lysed in buffer A (10 mM Hepes, pH 7.9, 10 mM KCl and 1.5 mM MgCl2) supplemented with 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin and 1 μg/ml aprotinin. After incubation on the ice for 10 min, the cytosolic fraction was collected by centrifugation at 1200 r.p.m. for 5 min. The pellet was washed with ice-cold PBS three times. Next, the pellet was lysed by buffer C (150 mM NaCl, 1 mM EDTA, 20 mM Tris–Cl, pH 8.0 and 0.5% NP-40) supplemented with 1 mM phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin and 1 μg/ml aprotinin at 4 °C for 2 h. The nuclear fraction was collected by centrifugation at 14 000 r.p.m. at 4 °C for 10 min.
Immunofluorescent analysis
Cells were seeded onto coverslips and infected with adenovirus-expressing GFP-Sp1 for 24 h. After fixation with 4% paraformaldehyde (Sigma) in PBS for 10 min and permeabilization with 0.1% Triton X-100 for 10 min, cells on the coverslip were blocked with 1% bovine serum albumin for 1 h and stained with the antibody against β-catenin (1:200; Cell Signaling Technology) for 1 h at room temperature. Subsequently, cells on the coverslip were washed with PBS three times and stained with the cyanine (Cy)-5-conjugated donkey anti-rabbit antibody (1:200, Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 1 h at room temperature. Finally, cells were washed with PBS three times and mounted with 90% glycerol containing DAPI (Invitrogen) and photographed under Olympus BX-51 fluorescent microscope (Olympus) at × 1000 magnification.
Statistical analysis
Student's t-test was used to analyze the difference between two groups. Survival curves were calculated according to the Kaplan–Meier method and comparison of survival rate was performed using log-rank test. The P-value of <0.05 was considered statistically significant.
References
Black AR, Black JD, Azizkhan-Clifford J . (2001). Sp1 and kruppel-like factor family of transcription factors in cell growth regulation and cancer. J Cell Physiol 188: 143–160.
Bremnes RM, Veve R, Hirsch FR, Franklin WA . (2002). The E-cadherin cell-cell adhesion complex and lung cancer invasion, metastasis, and prognosis. Lung Cancer 36: 115–124.
Bu Y, Gao L, Gelman IH . (2010). Role for transcription factor TFII-I in the suppression of SSeCKS/Gravin/Akap12 transcription by Src. Int J Cancer 128: 1836–42.
Chang HC, Liu LT, Hung WC . (2004). Involvement of histone deacetylation in ras-induced down-regulation of the metastasis suppressor RECK. Cell Signal 16: 675–679.
Chao YC, Pan SH, Yang SC, Yu SL, Che TF, Lin CW et al. (2009). Claudin-1 is a metastasis suppressor and correlates with clinical outcome in lung adenocarcinoma. Am J Respir Crit Care Med 179: 123–133.
Chen J, Ye L, Xie F, Yang Y, Zhang L, Jiang WG . (2010). Expression of bone morphogenetic protein 7 in lung cancer and its biological impact on lung cancer cells. Anticancer Res 30: 1113–1120.
Chu YW, Yang PC, Yang SC, Shyu YC, Hendrix MJ, Wu R et al. (1997). Selection of invasive and metastatic subpopulations from a human lung adenocarcinoma cell line. Am J Respir Cell Mol Biol 17: 353–360.
Chuang JY, Wang YT, Yeh SH, Liu YW, Chang WC, Hung JJ . (2008). Phosphorylation by c-Jun NH2-terminal kinase 1 regulates the stability of transcription factor Sp1 during mitosis. Mol Biol Cell 19: 1139–1151.
Chuang JY, Wu CH, Lai MD, Chang WC, Hung JJ . (2009). Overexpression of Sp1 leads to p53-dependent apoptosis in cancer cells. Int J Cancer 125: 2066–2076.
DeFino MC, Wacker JL, Lyssand JS, Wang EH, Hague C . (2010). Differential regulation of GPR54 transcription by specificity protein-1 and partial estrogen response element in mouse pituitary cells. Biochem Biophys Res Commun 393: 603–608.
Denlinger CE, Ikonomidis JS, Reed CE, Spinale FG . (2010). Epithelial to mesenchymal transition: the doorway to metastasis in human lung cancers. J Thorac Cardiovasc Surg 140: 505–513.
Duverger V, Murphy AM, Sheehan D, England K, Cotter TG, Hayes I et al. (2004). The anticancer drug mithramycin A sensitises tumour cells to apoptosis induced by tumour necrosis factor (TNF). Br J Cancer 90: 2025–2031.
Esteve PO, Chin HG, Pradhan S . (2007). Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells. J Biol Chem 282: 2615–2625.
Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ et al. (2001). Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev 15: 3249–3262.
Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A et al. (1991). E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol 113: 173–185.
Hommura F, Furuuchi K, Yamazaki K, Ogura S, Kinoshita I, Shimizu M et al. (2002). Increased expression of beta-catenin predicts better prognosis in nonsmall cell lung carcinomas. Cancer 94: 752–758.
Hung TL, Chen FF, Liu JM, Lai WW, Hsiao AL, Huang WT et al. (2003). Clinical evaluation of HER-2/neu protein in malignant pleural effusion-associated lung adenocarcinoma and as a tumor marker in pleural effusion diagnosis. Clin Cancer Res 9: 2605–2612.
Innocente SA, Lee JM . (2005). p53 is a NF-Y- and p21-independent, Sp1-dependent repressor of cyclin B1 transcription. FEBS Lett 579: 1001–1007.
Jemal A, Siegel R, Xu J, Ward E . (2010). Cancer statistics, 2010. CA Cancer J Clin 60: 277–300.
Jiang Y, Wang L, Gong W, Wei D, Le X, Yao J et al. (2004). A high expression level of insulin-like growth factor I receptor is associated with increased expression of transcription factor Sp1 and regional lymph node metastasis of human gastric cancer. Clin Exp Metastasis 21: 755–764.
Kumar AP, Butler AP . (1999). Enhanced Sp1 DNA-binding activity in murine keratinocyte cell lines and epidermal tumors. Cancer Lett 137: 159–165.
Kwak HJ, Park MJ, Cho H, Park CM, Moon SI, Lee HC et al. (2006). Transforming growth factor-beta1 induces tissue inhibitor of metalloproteinase-1 expression via activation of extracellular signal-regulated kinase and Sp1 in human fibrosarcoma cells. Mol Cancer Res 4: 209–220.
Lee YC, Oslund KL, Thai P, Velichko S, Fujisawa T, Duong T et al. (2011). 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced MUC5AC expression: aryl hydrocarbon receptor-independent/EGFR/ERK/p38-dependent SP1-based transcription. Am J Respir Cell Mol Biol 45: 270–276.
Leroy I, Laurent G, Quillet-Mary A . (2006). Mithramycin A activates Fas death pathway in leukemic cell lines. Apoptosis 11: 113–119.
Lin RK, Hsu CH, Wang YC . (2007). Mithramycin A inhibits DNA methyltransferase and metastasis potential of lung cancer cells. Anticancer Drugs 18: 1157–1164.
Liu YN, Lee WW, Wang CY, Chao TH, Chen Y, Chen JH . (2005). Regulatory mechanisms controlling human E-cadherin gene expression. Oncogene 24: 8277–8290.
Lou Z, O'Reilly S, Liang H, Maher VM, Sleight SD, McCormick JJ . (2005). Down-regulation of overexpressed sp1 protein in human fibrosarcoma cell lines inhibits tumor formation. Cancer Res 65: 1007–1017.
Maor S, Mayer D, Yarden RI, Lee AV, Sarfstein R, Werner H et al. (2006). Estrogen receptor regulates insulin-like growth factor-I receptor gene expression in breast tumor cells: involvement of transcription factor Sp1. J Endocrinol 191: 605–612.
McCarty Jr KS, Miller LS, Cox EB, Konrath J, McCarty Sr KS . (1985). Estrogen receptor analyses. Correlation of biochemical and immunohistochemical methods using monoclonal antireceptor antibodies. Arch Pathol Lab Med 109: 716–721.
Mitchell DC, Stafford LJ, Li D, Bar-Eli M, Liu M . (2007). Transcriptional regulation of KiSS-1 gene expression in metastatic melanoma by specificity protein-1 and its coactivator DRIP-130. Oncogene 26: 1739–1747.
Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B et al. (1997). Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275: 1787–1790.
Nicolas M, Noe V, Jensen KB, Ciudad CJ . (2001). Cloning and characterization of the 5′-flanking region of the human transcription factor Sp1 gene. J Biol Chem 276: 22126–22132.
Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J . (2003). Inhibition of c-src transcription by mithramycin: structure-activity relationships of biosynthetically produced mithramycin analogues using the c-src promoter as target. Biochemistry 42: 8313–8324.
Rivard N . (2009). Phosphatidylinositol 3-kinase: a key regulator in adherens junction formation and function. Front Biosci 14: 510–522.
Roose J, Huls G, van Beest M, Moerer P, van der Horn K, Goldschmeding R et al. (1999). Synergy between tumor suppressor APC and the beta-catenin-Tcf4 target Tcf1. Science 285: 1923–1926.
Schmalhofer O, Brabletz S, Brabletz T . (2009). E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev 28: 151–166.
St Clair S, Manfredi JJ . (2006). The dual specificity phosphatase Cdc25C is a direct target for transcriptional repression by the tumor suppressor p53. Cell Cycle 5: 709–713.
Su K, Roos MD, Yang X, Han I, Paterson AJ, Kudlow JE . (1999). An N-terminal region of Sp1 targets its proteasome-dependent degradation in vitro. J Biol Chem 274: 15194–15202.
Sun X, Ritzenthaler JD, Zhong X, Zheng Y, Roman J, Han S . (2009). Nicotine stimulates PPARbeta/delta expression in human lung carcinoma cells through activation of PI3K/mTOR and suppression of AP-2alpha. Cancer Res 69: 6445–6453.
Tsai MF, Wang CC, Chang GC, Chen CY, Chen HY, Cheng CL et al. (2006). A new tumor suppressor DnaJ-like heat shock protein, HLJ1, and survival of patients with non-small-cell lung carcinoma. J Natl Cancer Inst 98: 825–838.
Wang YT, Chuang JY, Shen MR, Yang WB, Chang WC, Hung JJ . (2008). Sumoylation of specificity protein 1 augments its degradation by changing the localization and increasing the specificity protein 1 proteolytic process. J Mol Biol 380: 869–885.
Wierstra I . (2008). Sp1: emerging roles--beyond constitutive activation of TATA-less housekeeping genes. Biochem Biophys Res Commun 372: 1–13.
Yi T, Tan K, Cho SG, Wang Y, Luo J, Zhang W et al. (2010). Regulation of embryonic kidney branching morphogenesis and glomerular development by KISS1 receptor (Gpr54) through NFAT2- and Sp1-mediated Bmp7 expression. J Biol Chem 285: 17811–17820.
Zheng Y, Ritzenthaler JD, Sun X, Roman J, Han S . (2009). Prostaglandin E2 stimulates human lung carcinoma cell growth through induction of integrin-linked kinase: the involvement of EP4 and Sp1. Cancer Res 69: 896–904.
Acknowledgements
This work was supported by the National Cheng-Kung University project of the Program for Promoting Academic Excellence and Developing World Class Research Centers, together with Grants NSC 97-2320-B-006-016-MY3 and NSC 97-2311-B-006-002-MY3 obtained from the National Science Council, Taiwan. The clinical work in this study was supported by the Department of Health, Executive Yuan, Taiwan (Grants DOH99-TD-B-111-002 and DOH99-TD-B-111-003).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogene website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Hsu, TI., Wang, MC., Chen, SY. et al. Sp1 expression regulates lung tumor progression. Oncogene 31, 3973–3988 (2012). https://doi.org/10.1038/onc.2011.568
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2011.568
Keywords
This article is cited by
-
Sp1 promotes tumour progression by remodelling the mitochondrial network in cervical cancer
Journal of Translational Medicine (2023)
-
UBXN2A suppresses the Rictor-mTORC2 signaling pathway, an established tumorigenic pathway in human colorectal cancer
Oncogene (2023)
-
Tp53 haploinsufficiency is involved in hotspot mutations and cytoskeletal remodeling in gefitinib-induced drug-resistant EGFRL858R-lung cancer mice
Cell Death Discovery (2023)
-
Estradiol-mediated inhibition of Sp1 decreases miR-3194-5p expression to enhance CD44 expression during lung cancer progression
Journal of Biomedical Science (2022)
-
Sp1 induced gene TIMP1 is related to immune cell infiltration in glioblastoma
Scientific Reports (2022)
