
Abstract
Background:
Obesity is a major health concern in the developed world, and increasing evidence suggests that exposures to common environmental substances may enhance the risk for the development of this disease.
Objectives:
The current study examines the effect of the ubiquitous plastic monomer bisphenol A (BPA) on the differentiation of primary human preadipocytes in vitro and the role of the estrogen and glucocorticoid receptors.
Methods:
In this study, the mechanism of BPA-induced adipogenesis in preadipocytes from donors with healthy body mass index in the absence of exogenous glucocorticoid was evaluated. The effects of estradiol, the estrogen-receptor (ER) antagonist ICI and the glucocorticoid receptor (GR) antagonist RU486 on BPA-induced adipogenesis were examined. The expression levels of key adipogenic factors were assessed.
Results:
Treatment of preadipocytes with 1–50 μM BPA induced a dose-dependent increase in differentiation and lipid accumulation as determined by lipid staining and triacylglyceride quantification. BPA also induced expression of the adipogenic markers aP2, adipsin, peroxisome proliferator-activated receptor γ and the CCAAT-enhancer-binding proteins α and β. Co-treatment of cells with ICI inhibited the BPA-induced increase in aP2 levels, while treatment with ICI or estradiol alone had no effect. Treatment of cells with the GR antagonist RU486 had no effect on BPA-induced differentiation as evaluated by aP2 levels.
Conclusions:
This study is one of the first to show that BPA induces human adipocyte differentiation in the absence of exogenous glucocorticoid through a non-classical ER pathway rather than through GR activation. These studies add to the growing evidence that endocrine-disrupting chemicals such as BPA have the potential to modulate adipogenesis and impact human biology.
Similar content being viewed by others


Introduction
Obesity is directly associated with a number of health complications, including diabetes, hypertension and heart disease (reviewed in Allender and Rayner1). Although unfavorable diet, lifestyle and genetic factors are associated with the increasing rates of obesity around the world, it is now suspected that exposure to environmental chemicals may be contributing to this epidemic by disrupting normal metabolism.2 Substances with ubiquitous human exposure, such as bisphenol A (BPA), may have an important role in fat cell formation and metabolism during development and adulthood by disrupting adipogenesis, lipid accumulation and contributing to obesity.3
BPA is an industrial chemical used in the manufacture of polycarbonate plastic, epoxy resins and thermal printing.4, 5 BPA is released from consumer products and has been detected in food, water and dust.6 Human exposure to BPA has been confirmed by its presence in blood, urine and adipose tissue.7, 8 Not only is BPA exposure associated with obesity9, 10 and diabetes11 but it has also been linked to modulation of adipocyte differentiation and function in rodent and human models.12, 13, 14 However, the molecular mechanism of action of BPA in human preadipocyte differentiation has yet to be determined.
Adipocyte differentiation in murine cells is regulated mainly by CCAAT-enhancer-binding protein (C/EBP) α, β and δ and peroxisome proliferator-activated receptor (PPAR) γ, which induce the expression of genes that lead to the development of the adipocyte phenotype which includes the formation of lipid droplets and adipokine release (reviewed in Tang and Lane15). In human preadipocytes, the transcriptional cascade that leads to a mature adipocyte phenotype is less understood than in murine models; however, evidence in the literature suggests that it involves similar transcription factors.16, 17 The mechanisms by which BPA affects adipocyte biology have yet to be determined; however, nuclear hormone receptors are believed to be involved.18 BPA has been linked to estrogen-receptor (ER) and glucocorticoid-receptor (GR) modulation, both of which can influence adipogenesis and lipid metabolism.19, 20, 21 Although estrogen has been shown to inhibit adipogenesis in vitro,22, 23, 24 ERα knockout mice are known to exhibit increased adipose tissue, supporting an anti-adipogenic role for ERα.25 BPA has also been shown to bind classical and non-classical ERs in several studies26, 27, 28, 29, 30 and therefore these receptors may have a role in BPA-induced adipogenesis.
Alternatively, BPA may be acting through the GR pathway. Glucocorticoids are involved in promoting adipogenesis in vitro, primarily through the activation of the C/EBPβ, leading to the expression of PPARγ and C/EBPα.31, 32 BPA was shown to promote adipogenesis through GR activation in murine 3T3-L1 preadipocytes21 and to increase the expression of 11β-hydroxysteroid dehydrogenase type 1 in primary adipocytes from overweight children resulting in the activation of GR and adipogenesis.14 Dexamethasone (DEX), a synthetic glucocorticoid, is universally used in adipocyte models of in vitro differentiation. The few studies showing effects of BPA on human preadipocyte differentiation have only examined the effect of BPA in the presence of glucocorticoid.14, 33 Although the addition of DEX is generally required to induce efficient differentiation, this raises some issues when attempting to explore a potential GR-related mechanism as DEX is a very potent GR agonist even at low concentrations.34
In the current study, the mechanism of BPA-induced adipogenesis in human preadipocytes was examined. The differentiation process requires the addition of a cocktail that initiates the adsipogenic transcriptional cascade. This cocktail includes high levels of insulin, which increases intracellular levels of cyclic adenosine monophosphate, 3-isobutyl-1-methylxanthine (IBMX), a PPARγ agonist (troglitazone) and high levels of glucocorticoids. Under these conditions, high percentages (∼80%) of the human preadipocytes accumulate lipid droplets. In order to determine the possible target of BPA action, components of the differentiation protocol were systematically eliminated. Here, we show for the first time that BPA induces lipid accumulation in human preadipocytes and increases expression of several key adipogenic markers in the absence of glucocorticoids. Moreover, BPA-induced differentiation was inhibited in the presence of the specific ER antagonist ICI-182,780, but not by a GR antagonist, despite the fact that estrogen had no agonistic effect in this model, suggesting that the mechanism of BPA action may be through a non-classical ER pathway.
Materials and methods
Adipocyte differentiation
Primary human preadipocytes (Zenbio, Inc., Research Triangle Park, NC, USA) from donors with body mass indexes ⩽24.99 kg m−2 were maintained in Preadipocyte Medium (ZenBio) at 37 °C and 5% CO2. For differentiation, confluent preadipocytes were treated with media containing 33 μM biotin, 17 μM pantothenate (both from Sigma-Aldrich, St Louis, MO, USA) and 100 nM insulin (Roche Applied Science, Laval, QC, Canada) for 14 days. In addition, 500 μM IBMX (Sigma-Aldrich) was also included in the differentiation media from day 0 to day 4. From day 2 until day 14, 5 μM troglitazone (Sigma-Aldrich) and the indicated concentrations of BPA were also included in the differentiation media, which was replenished every 2 days. As a positive control, cells were treated with 1 μM DEX (Sigma-Aldrich) starting on day 2 throughout differentiation with the same media as above instead of BPA. For the ER and GR antagonist studies, 1 nM estradiol, 1 μM ICI-182,780 or 1 μM RU486 (all Sigma-Aldrich) were also added as above with or without BPA.
Lipid staining and quantification
After 14 days of differentiation, cells were fixed with 4% formaldehyde and stained overnight with Oil Red O (Sigma-Aldrich) to visualize neutral lipid content as previously described.35 Cellular triacylglycerides (TGs) were quantified using a TG Assay Kit (Zenbio). TG levels were normalized to cellular protein content, which was quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific, Rockford, IL, USA).
Real-time PCR
Total RNA was extracted from differentiating cells treated as described at various time points using the RNeasy Kit (QIAGEN, Mississauga, ON, Canada). Peak expression time points for each gene were optimized and found to be 4 days post treatment for adipsin, PPARγ and CEBPβ and 6 days post treatment for aP2 and CEBPα. Genomic DNA was eliminated using the RNase-Free DNase Kit (QIAGEN). RNA quality was assessed using a BioAnalyzer (Agilent). Only samples with RNA integrity number values>8.0 were used. RNA (250–500 ng) were reverse transcribed into cDNA using iScript Advanced cDNA Synthesis Kit (Bio-Rad, Mississauga, ON, Canada). For each real-time PCR reaction, cDNA was amplified in a CFX96-PCR Detection System using the iQSYBR Green Supermix Kit (Bio-Rad). The primer pairs for each gene target were C/EBPα: Forward:5′-TGGACAAGAACAGCAACGAG-3′, Reverse 5′-CCATGGCCTTGACCAAGGAG-3′; C/EBPβ: Forward 5′-GAAGACCGTGGACAAGCACA-3′, Reverse 5′-ACAAGTTCCGCAGGGTGCTG-3′; PPARγ 1/2: Forward 5′-TCCGAGGGCCAAGGCTTCAT-3′, Reverse 5′-GCAAACCTGGGCGGTCTCCA-3′; aP2: Forward 5′-CATCAGTGTGAATGGGGATG-3′, Reverse 5′-GTGGAAGTGACGCCTTTCAT-3′; β-actin: Forward 5′-GACTTCGAGCAAGAGATGGC-3′, Reverse 5′-CCAGACAGCACTGTGTTGGC-3′; and adipsin: Forward 5′-CGAGCTGGCACCGGGAACTC-3′, Reverse 5′-TGCAGCTGTCCCGGCGATTG-3′. Standard curves were generated from the pooled cDNA obtained from cells treated with DEX from various time points. Primer efficiencies were ⩾90%, and specificity was confirmed by sequence blast and melting curve analysis. Reactions were normalized to β-actin expression, which was not affected by BPA treatment.
Western blotting analysis
Cells were washed in phosphate-buffered saline and lysed in buffer containing 50 mM Tris, 150 mM sodium chloride (NaCl), 1% IGEPAL, 5 mM EDTA (all from Sigma-Aldrich) and protease inhibitor cocktail (Roche Diagnostics, Laval, QC, Canada). Equal amounts of protein were resolved by SDS-PAGE (sodium dodecyl sulfate-polyacrylamide gel electrophoresis) and transferred to polyvinylidene membrane. Primary antibodies for aP2 (R&D, Minneapolis, MN, USA), perilipin (D1D8; Cell Signaling, Boston, MA, USA) and β-actin (13E5; Cell Signaling) and appropriate horseradish peroxidase-labelled secondary antibodies were used. Western blots were visualized using the ChemiDoc Imager and quantified using the Image Lab software (Bio-Rad).
Statistical analyses
All data were analyzed by Student’s t-test or analysis of variance with Holm–Sidak post-test analysis as indicated using SigmaPlot 11.0 (San Jose, CA, USA).
Results
BPA increases lipid accumulation in human preadipocytes
The differentiation of human preadipocytes requires well-defined inducers, including insulin, IBMX, troglitazone and DEX. Omission of any of these components during differentiation abolished the ability of the cells to assume a mature adipocyte phenotype (data not shown). Our initial experiments were designed to investigate whether BPA could replace any of these components, and it was found that BPA could only replace the DEX-mediated effects on differentiation (data not shown and Figure 1). We evaluated the effect of BPA on adipocyte differentiation, by assessing lipid accumulation using Oil Red O staining and TG quantification using a commercial assay. Cells treated with 25 or 50 μM BPA for 14 days showed increased Oil Red O lipid staining, indicating more differentiation compared with control cells treated with vehicle alone (ethanol; Figure 1a). Cells treated with 1 μM DEX as a positive control showed roughly 80–90% of the cells positive for lipid staining, indicating a high degree of adipocyte differentiation. Treatment of cells with 25 or 50 μM BPA also significantly stimulated TG accumulation (Figure 1b), inducing a 1.7–2.1-fold increase, respectively, in TG levels. Cells treated with DEX exhibited an almost 15.4-fold increase in TG levels relative to vehicle control (Figure 1b). No cell death was observed under any of the treatment conditions. The data show that BPA can induce adipocyte differentiation and lipid accumulation in the absence of exogenous glucocorticoids.

Bisphenol A (BPA)-treated human preadipocytes cells show increased lipid staining and cellular TG accumulation. Human preadipocytes were treated with ethanol (control), DEX or BPA at the indicated concentrations, and lipid accumulation was visualized using Oil Red O staining (a) or quantified using a commercial TG assay kit (b) at day 14 of differentiation. Pictures are representative of at least three independent experiments. Values are expressed as means±s.e.m. for three experiments performed in triplicate. *P<0.001 (n=3) relative to control calculated by one-way analysis of variance with Holm–Sidak post-test analysis.
BPA increases mRNA and protein expression of adipogenic markers
To further evaluate BPA-induced effects on differentiation, the mRNA expression of key adipogenic markers and transcription factors were evaluated using real-time PCR. Peak expression time points for each gene were optimized and found to be 4 days post-BPA treatment for adipsin, PPARγ and CEBPβ and 6 days post-BPA treatment for aP2 and CEBPα (data not shown). Treatment of cells with 25 or 50 μM BPA resulted in a statistically significant increase in the mRNA levels of the adipogenic marker aP2 by 3.1- and 3.9-fold, respectively, at 6 days post-BPA treatment (Figure 2). DEX treatment caused a dramatic 36.2-fold increase in aP2 expression relative to vehicle control. Adipsin mRNA levels were also induced 1.5-fold by 50 μM BPA at 4 days post-BPA treatment whereas DEX treatment resulted in a 6.3-fold increase in adipsin expression (Figure 2). The BPA-induced mRNA levels of key transcription factors involved in adipogenesis were also evaluated. Treatment of cells with 50 μM BPA or DEX caused a 2.0- and 7.0-fold increase, respectively, in PPARγ mRNA expression at 4 days post-BPA treatment (Figure 2). Expression of CEBPα was significantly increased by 1.8-fold at 6 days post-BPA treatment in response to 25 or 50 μM BPA, respectively. Similarly, DEX treatment resulted in a significant 15.9-fold increase in CEBPα expression (Figure 2). In contrast, CEBPβ mRNA expression was only slightly increased 1.5-fold at 4 days post-BPA treatment in response to 50 μM BPA, whereas DEX did not result in a statistically significant increase in CEBPβ levels (Figure 2). The data show that BPA induces human adipocyte differentiation as determined by the increased expression of adipogenic markers at the mRNA level and increases expression levels of transcription factors known to be involved in the transcriptional cascade leading to adipocyte differentiation.

Treatment of human preadipocytes with BPA induces mRNA expression of important adipogenic markers. Differentiation and treatment of preadipocytes with ethanol (control), 1 μM DEX or BPA was initiated as described. Total RNA was isolated at either 4 days (aP2 and CEBPα) or 6 days (adipsin, PPARγ and CEBPβ) post treatment and used for real-time PCR analysis of the relative mRNA expression of the adipogenic markers normalized to β-actin gene expression. Values are expressed as mean fold change relative to control±s.e.m. for five experiments. *P<0.05, **P<0.01 (n=5) relative to control calculated by Student’s t-test.
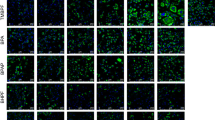
Next, we evaluated the effect of BPA treatment on the protein levels of the adipogenic markers aP2 and perilipin. The ability of increasing concentrations of BPA (from 0.01–50 μM) to potentiate the differentiation of human preadipocytes was examined at day 14 of differentiation. The data show that protein levels of the adipogenic marker aP2 were increased on day 14 by as low as 1 μM BPA; however, only 25 and 50 μM BPA treatments showed a statistically significant increase in aP2 protein levels relative to control (Figures 3a and b). Therefore subsequent experiments were performed in the presence of 25 μM BPA. We proceeded to investigate the protein expression levels of aP2 and perilipin during the differentiation process. The data indicate that BPA significantly induces aP2 expression by 1.8-fold compared with control as early as day 6 (Figure 3c) and by 3.2-fold at day 14. Perilipin protein levels were detectable only at day 14 and increased only by 1.5-fold following treatment with 25 μM BPA relative to control; however, the difference was not statistically significant (Figures 3e and f). As a positive control, DEX-treated preadipocytes showed high levels of both aP2 and perilipin protein levels by days 6 and 14 of differentiation (Figure 3g). These results clearly show BPA induces expression of key adipogenic markers at both the mRNA and protein levels.

Effects of BPA treatment on protein levels of the adipogenic markers aP2 and perilipin. Human preadipocytes were differentiated as described with increasing concentrations of BPA for 14 days, and protein levels of the adipogenic marker aP2 were assessed by western blotting (a) and densitometry (b) analyses. Human preadipocytes were also differentiated in the presence of 25 μM BPA or vehicle control for 6 or 14 days and protein levels of the adipogenic markers aP2 (c, d) and perilipin (e, f) were assessed by western blotting and densitometry analyses. (g) Cells were differentiated in the presence of 1 μM DEX, and protein levels of the adipogenic markers aP2 and perilipin were assessed at days 0, 6 and 14 of differentiation by western blotting analysis. Images are representative of at least three separate experiments. β-Actin was used as the gel loading control for all blots. All densitometry values are expressed as means±s.e.m. for at least three experiments. *P<0.05, **P<0.01 relative to control calculated by one-way analysis of variance with Holm–Sidak post-test analysis.
BPA-induced differentiation is inhibited by the ER antagonist ICI
Given that BPA has ER-binding activity at concentrations as low as 0.1 μM,36 the potential role of the ER in BPA-induced differentiation was investigated using the specific ER antagonist ICI. The protein levels of aP2 were examined at day 14 as a marker of differentiation in human preadipocytes following co-treatment with 25 μM BPA and 1 μM ICI. Co-treatment with ICI significantly inhibited BPA-induced aP2 protein levels by 75% (to background levels) at day 14 of differentiation (Figures 4a and b). Neither estradiol nor ICI treatment alone had a significant effect on differentiation or aP2 protein levels. This suggests that the mechanism of BPA-induced differentiation of human preadipocytes is likely mediated via a non-classical ER pathway.

The effect of the estrogen receptor antagonist ICI or GR antagonist RU486 on BPA-induced differentiation of human preadipocytes. (a) Preadipocytes were treated with ethanol (control), 1 nM estradiol or 25 μM BPA in the presence and absence of 1 μM of ICI, and the protein levels of the adipogenic marker aP2 were assessed by western blotting analysis at day 14 of differentiation. β-Actin was used as the gel loading control. (b) Densitometry analysis of aP2 protein levels from western blotting analysis in panel (a) at day 14 of differentiation. Values are expressed as means±s.e.m. for six separate experiments. *P<0.01 (n=6) relative to control calculated by one-way analysis of variance with Holm–Sidak post-test analysis. (c) Preadipocytes were treated with ethanol (control) or 25 μM BPA in the presence and absence of 1 μM RU486, and protein levels of the adipogenic marker aP2 were assessed by western blotting analysis at day 14 of differentiation. β-Actin was used as the gel loading control. (d) Densitometry analysis of aP2 protein levels from western blotting analysis in panel (c) at day 14 of differentiation. Values are expressed as means±s.e.m. for four separate experiments. *P<0.05 (n=4) relative to control calculated by one-way analysis of variance with Holm–Sidak post-test analysis.
BPA-induced differentiation is not affected by the GR antagonist RU486
The involvement of the GR in BPA-induced differentiation was also evaluated in the presence of a GR-specific antagonist. Human preadipocytes were co-treated with 25 μM BPA and 1 μM of the GR antagonist RU486, which has been reported to inhibit DEX-induced GR activation,37 throughout differentiation until day 14. In control experiments, 1 μM RU486 was able to inhibit differentiation of human preadipocytes treated with 10 nM DEX as determined by aP2 protein levels at day 14 (data not shown). The data in Figure 4 show that BPA treatment alone significantly increased aP2 protein levels relative to control, as shown earlier. However, BPA-induced aP2 protein levels were not significantly affected by co-treatment with RU486 relative to BPA alone (Figures 4c and d). Treatment of cells with RU486 on its own appeared to cause a small increase in aP2 protein levels as well compared with control, but the increase was not statistically significant. This is somewhat consistent with reports that RU486 alone has the potential to act as a GR agonist in adipocytes.38 These data suggest that BPA-induced differentiation is likely mediated through a non-classical ER-dependent mechanism rather than through the GR in human subcutaneous preadipocytes.
Discussion
The current study demonstrates that BPA induces differentiation of primary human preadipocytes in vitro in the absence glucocorticoid and examines several potential mechanisms of action of BPA-induced differentiation. This in vitro study is consistent with in vivo reports showing that BPA exposure is correlated with obesity in several human models.9, 39 Although transcriptional stimulation of the GR is believed to be crucial for the development of the adipocyte phenotype in these cells, addition of BPA causes increased fat accumulation and expression of mRNA and protein markers of adipocyte differentiation in the absence of a GR agonist. The fact that BPA-induced differentiation is not inhibited in the presence of an active concentration of the GR antagonist RU486 (Figures 4c and d) argues that the BPA effect is not mediated through activation of the GR in this model system. Interestingly, the adipogenic action of BPA is inhibited by a specific antagonist of the ER despite the lack of an effect of estrogen itself on differentiation in this model. This suggests that BPA induces adipocyte differentiation through a non-classical ER-mediated mechanism rather than through GR activation. These results indicate that BPA can contribute to the final maturation of cells committed to the adipocyte lineage and add to the growing body of evidence that BPA acts as an obesogen.
In this study, as opposed to many reports in the literature, we evaluated the effect of BPA on adipogenesis in human preadipocytes in the absence of exogenous glucocorticoids and examined its mechanism of action on differentiation and lipid accumulation. Several potential mechanisms have been suggested based on the ability of BPA to act as an estrogen through binding classical or non-classical ERs and its potential ability to activate GR.14, 40 Most of the studies investigating the effects of BPA on adipogenesis in vitro have done so using a differentiation cocktail containing DEX or cortisol, potent GR agonists and inducers of differentiation, making the interpretation of BPA-specific effects difficult. When DEX is present, it is very difficult to assess the mechanism of action and adipogenic effects of BPA, unless inhibitory effects are expected to be observed. Here, we report effects of BPA on human adipocyte differentiation in the absence of exogenous glucocorticoid.
The ability of BPA to promote adipogenesis is consistent with reports that examined mRNA expression of adipogenic markers, such as aP2, lipoprotein lipase and PPARγ.13, 41, 42 Here, we report at least a 2–3-fold increase in lipid accumulation and aP2 protein levels (Figures 1 and 3), consistent with previous studies in the 3T3-L1 murine cell model and human visceral preadipocytes.14, 21 The range of BPA concentrations used in cell culture models has varied. Some studies showed effects in the 0.1–1 μM BPA range,21, 33, 43 whereas others have gone as high as 80–100 μM BPA.14, 44 Our data show significant optimal effects on aP2 protein levels at 25–50 μM BPA but also as low as 100 nM (however, this concentration was not statistically significant) (Figure 3a). Very few studies have looked at BPA-induced adipogenesis in human preadipocytes. Most studies have examined adipocyte differentiation in murine cell lines, which differ from human adipocytes in many of the requirements for differentiation. Most murine cell lines require 7 days of differentiation, whereas the human preadipocytes require 14 days to fully differentiate. Moreover, the murine preadipocytes do not require the addition of a PPAR agonist (such as troglitazone) in addition to glucocorticoids to the differentiation cocktail, whereas human cells do not differentiate in the absence of troglitazone even when DEX is present (data not shown and Tomlinson et al.16). Due to these differences, we examined the effect of BPA in a human preadipocyte cell model, which is more relevant to human health.
The current study is the first to report that BPA induces differentiation of human preadipocytes entirely in the absence of glucocorticoid stimulation. Previous reports of the action of BPA in mediating adipocyte differentiation have shown effects only when BPA is added in combination with the potent GR agonist DEX or other synthetic glucocorticoids.21 One study also showed a BPA-induced increase in 11β-hydroxysteroid dehydrogenase type 1 in human omental fat cells14 leading to activation of GR. However, these reports all show an effect on adipogenesis in the presence of a GR agonist. As we examined a potential GR-related mechanism of BPA action, it was necessary to perform our studies in the absence of DEX in the differentiation cocktail. Our results clearly show that BPA induces human preadipocyte differentiation and can partially replace the effect of DEX on the differentiation of these cells. Moreover, BPA-induced adipogenesis was not inhibited by the GR antagonist RU486, further suggesting that the effect is not mediated via GR. It is possible that BPA may have a higher affinity for GR than RU486 as the antagonist did not inhibit the BPA effect but RU486 likely has a similar high affinity to GR as its natural ligand DEX.
The current study is also the first to report that BPA induces adipogenesis not only in the absence of exogenous glucocorticoid but that the mechanism is likely through a non-classical ER-mediated pathway rather than through the GR. Our data show that the ER agonist estradiol had no stimulatory effect on the differentiation of human preadipocytes, consistent with studies showing that estrogen does not have a positive effect on adipogenesis and is even considered to be anti-adipogenic.24 More importantly, we also show that ICI, a specific ER antagonist, was able to inhibit BPA-induced adipogenesis by 75% as measured by a decrease in aP2 protein levels (Figure 4a). This is intriguing as BPA is known to have estrogen-like properties and is able to bind to the ER; however, the fact that BPA has the opposite effect of estradiol on differentiation and is inhibited by an ER antagonist suggests that BPA is potentially acting through a non-classical ER pathway. Interestingly, it has been recently shown that diethylstilbestrol, a potent ERα activator, was adipogenic in mice and 3T3-L1 cells and that the effect could be inhibited by ICI.45 Human preadipocytes have been shown to express both ERα and ERβ.46 BPA has been shown to act through ERβ in β-islets and to signal through ERα to activate extracellular-regulated kinase 1/2 and increase insulin-induced genes.40 The ability of ICI to inhibit the effects of BPA on ERα and ERβ activation has been reported in HepG2 and HeLa cells.36 ICI has also been shown to inhibit BPA-induced proliferation in ovarian cancer cells.47 It remains to be determined in this system whether BPA binds the ER and activates non-classical ER-responsive genes that promote differentiation.
BPA may be regulating adipogenesis via other non-classical ER pathways. BPA has been shown to strongly bind the estrogen-related receptor γ (ERRγ) and may be acting through this family of receptors to activate downstream genes.27, 48 ERRγ expression was reported to be upregulated following treatment with BPA in an insect model,49 suggesting that BPA can affect ERR levels. Members of the ERR family have been shown to be involved in adipogenesis.50 ERRα-knockout mice have been reported to have reduced adipose tissue deposition.51 Moreover, activation of ERRα has been shown to upregulate PPARγ coactivator-1α (PGC-1β) expression, which promotes the formation of PPARγ/PGC-1β and sterol-regulatory-element-binding protein-1c/PGC-1β complexes, which are important adipogenic transcription factors.52 However, the influence of BPA and/or ICI on the activity of any member of the ERR family has yet to be thoroughly investigated. Another potential non-classical ER mechanism of BPA-mediated adipogenesis may involve G protein-coupled receptor 30 (GPR30), which is a membrane protein that binds estrogen and initiates intracellular signaling pathways. One study showed that BPA can signal through both GPR30 and ERα.53 However, the role of either GPR30 or the ERRs, which are expressed in adipose tissue,54 in BPA-induced adipogenesis remains to be characterized.
Conclusions
In this study, we show that BPA promotes differentiation and lipid accumulation in primary subcutaneous human preadipocytes. Moreover, this is one of the first reports showing that BPA can induce adipocyte differentiation in the absence of exogenous glucocorticoids. We also show that while estradiol has no positive effect on adipocyte differentiation, BPA-induced adipogenesis is inhibited by an ER antagonist, but not by a GR antagonist, suggesting that BPA is potentially acting through a non-classical ER pathway. Future studies should examine the roles of the classical ER, ERRs and GPR30 in the mechanism of action of BPA in adipogenesis.
References
Allender S, Rayner M . The burden of overweight and obesity-related ill health in the UK. Obes Rev 2007; 8: 467–473.
Decherf S, Demeneix BA . The obesogen hypothesis: a shift of focus from the periphery to the hypothalamus. J Toxicol Environ Health B Crit Rev 2011; 14: 423–448.
Vom Saal FS, Nagel SC, Coe BL, Angle BM, Taylor JA . The estrogenic endocrine disrupting chemical bisphenol A (BPA) and obesity. Mol Cell Endocrinol 2012; 354: 74–84.
Fleisch AF, Sheffield PE, Chinn C, Edelstein BL, Landrigan PJ . Bisphenol A and related compounds in dental materials. Pediatrics 2010; 126: 760–768.
Rubin BS . Bisphenol A: an endocrine disruptor with widespread exposure and multiple effects. J Steroid Biochem Mol Biol 2011; 127: 27–34.
Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV . Human exposure to bisphenol A (BPA). Reprod Toxicol 2007; 24: 139–177.
Dekant W, Volkel W . Human exposure to bisphenol A by biomonitoring: methods, results and assessment of environmental exposures. Toxicol Appl Pharmacol 2008; 228: 114–134.
Vandenberg LN, Chahoud I, Heindel JJ, Padmanabhan V, Paumgartten FJ, Schoenfelder G . Urinary, circulating, and tissue biomonitoring studies indicate widespread exposure to bisphenol A. Environ Health Perspect 2010; 118: 1055–1070.
Bhandari R, Xiao J, Shankar A . Urinary bisphenol A and obesity in US children. Am J Epidemiol 2013; 177: 1263–1270.
Harley KG, Schall RA, Chevrier J, Tyler K, Aguirre H, Bradman A et al. Prenatal and postnatal bisphenol A exposure and body mass index in childhood in the CHAMACOS cohort. Environ Health Perspect 2013; 121: 514–520.
Gong H, Zhang X, Cheng B, Sun Y, Li C, Li T et al. Bisphenol A accelerates toxic amyloid formation of human islet amyloid polypeptide: a possible link between bisphenol A exposure and type 2 diabetes. PLoS One 2013; 8: e54198.
Masuno H, Iwanami J, Kidani T, Sakayama K, Honda K . Bisphenol a accelerates terminal differentiation of 3T3-L1 cells into adipocytes through the phosphatidylinositol 3-kinase pathway. Toxicol Sci 2005; 84: 319–327.
Somm E, Schwitzgebel VM, Toulotte A, Cederroth CR, Combescure C, Nef S et al. Perinatal exposure to bisphenol a alters early adipogenesis in the rat. Environ Health Perspect 2009; 117: 1549–1555.
Wang J, Sun B, Hou M, Pan X, Li X . The environmental obesogen bisphenol A promotes adipogenesis by increasing the amount of 11beta-hydroxysteroid dehydrogenase type 1 in the adipose tissue of children. Int J Obes 2012; 37: 999–1005.
Tang QQ, Lane MD . Adipogenesis: from stem cell to adipocyte. Annu Rev Biochem 2012; 81: 715–736.
Tomlinson JJ, Boudreau A, Wu D, Atlas E, Hache RJ . Modulation of early human preadipocyte differentiation by glucocorticoids. Endocrinology 2006; 147: 5284–5293.
Lechner S, Mitterberger MC, Mattesich M, Zwerschke W . Role of C/EBPbeta-LAP and C/EBPbeta-LIP in early adipogenic differentiation of human white adipose-derived progenitors and at later stages in immature adipocytes. Differ Res Biol Divers 2013; 85: 20–31.
Wetherill YB, Akingbemi BT, Kanno J, McLachlan JA, Nadal A, Sonnenschein C et al. In vitro molecular mechanisms of bisphenol A action. Reprod Toxicol 2007; 24: 178–198.
Ben-Jonathan N, Steinmetz R . Xenoestrogens: the emerging story of bisphenol A. Trends Endocrinol Metab 1998; 9: 124–128.
Williams G . Aromatase up-regulation, insulin and raised intracellular oestrogens in men, induce adiposity, metabolic syndrome and prostate disease, via aberrant ER-alpha and GPER signalling. Mol Cell Endocrinol 2012; 351: 269–278.
Sargis RM, Johnson DN, Choudhury RA, Brady MJ . Environmental endocrine disruptors promote adipogenesis in the 3T3-L1 cell line through glucocorticoid receptor activation. Obesity 2010; 18: 1283–1288.
Dang ZC, van Bezooijen RL, Karperien M, Papapoulos SE, Lowik CW . Exposure of KS483 cells to estrogen enhances osteogenesis and inhibits adipogenesis. J Bone Miner Res 2002; 17: 394–405.
Homma H, Kurachi H, Nishio Y, Takeda T, Yamamoto T, Adachi K et al. Estrogen suppresses transcription of lipoprotein lipase gene. Existence of a unique estrogen response element on the lipoprotein lipase promoter. J Biol Chem 2000; 275: 11404–11411.
Okazaki R, Inoue D, Shibata M, Saika M, Kido S, Ooka H et al. Estrogen promotes early osteoblast differentiation and inhibits adipocyte differentiation in mouse bone marrow stromal cell lines that express estrogen receptor (ER) alpha or beta. Endocrinology 2002; 143: 2349–2356.
Couse JF, Korach KS . Reproductive phenotypes in the estrogen receptor-alpha knockout mouse. Ann Endocrinol 1999; 60: 143–148.
Alonso-Magdalena P, Ropero AB, Carrera MP, Cederroth CR, Baquie M, Gauthier BR et al. Pancreatic insulin content regulation by the estrogen receptor ER alpha. PLoS One 2008; 3: e2069.
Matsushima A, Kakuta Y, Teramoto T, Koshiba T, Liu X, Okada H et al. Structural evidence for endocrine disruptor bisphenol A binding to human nuclear receptor ERR gamma. J Biochem 2007; 142: 517–524.
Okada H, Tokunaga T, Liu X, Takayanagi S, Matsushima A, Shimohigashi Y . Direct evidence revealing structural elements essential for the high binding ability of bisphenol A to human estrogen-related receptor-gamma. Environ Health Perspect 2008; 116: 32–38.
Sheng ZG, Huang W, Liu YX, Zhu BZ . Bisphenol A at a low concentration boosts mouse spermatogonial cell proliferation by inducing the G protein-coupled receptor 30 expression. Toxicol Appl Pharmacol 2013; 267: 88–94.
Teng C, Goodwin B, Shockley K, Xia M, Huang R, Norris J et al. Bisphenol A affects androgen receptor function via multiple mechanisms. Chem Biol Interact 2013; 203: 556–564.
Abdou HS, Atlas E, Hache RJ . A positive regulatory domain in CCAAT/enhancer binding protein beta (C/EBPBeta) is required for the glucocorticoid-mediated displacement of histone deacetylase 1 (HDAC1) from the C/ebpalpha promoter and maximum adipogenesis. Endocrinology 2013; 154: 1454–1464.
Hamm JK, Park BH, Farmer SR . A role for C/EBPbeta in regulating peroxisome proliferator-activated receptor gamma activity during adipogenesis in 3T3-L1 preadipocytes. J Biol Chem 2001; 276: 18464–18471.
Hugo ER, Brandebourg TD, Woo JG, Loftus J, Alexander JW, Ben-Jonathan N . Bisphenol A at environmentally relevant doses inhibits adiponectin release from human adipose tissue explants and adipocytes. Environ Health Perspect 2008; 116: 1642–1647.
Lee MJ, Gong DW, Burkey BF, Fried SK . Pathways regulated by glucocorticoids in omental and subcutaneous human adipose tissues: a microarray study. Am J Physiol Endocrinol Metab 2011; 300: E571–E580.
Kuri-Harcuch W, Wise LS, Green H . Interruption of the adipose conversion of 3T3 cells by biotin deficiency: differentiation without triglyceride accumulation. Cell 1978; 14: 53–59.
Li Y, Burns KA, Arao Y, Luh CJ, Korach KS . Differential estrogenic actions of endocrine-disrupting chemicals bisphenol A, bisphenol AF, and zearalenone through estrogen receptor alpha and beta in vitro. Environ Health Perspect 2012; 120: 1029–1035.
Zhang P, O'Loughlin L, Brindley DN, Reue K . Regulation of lipin-1 gene expression by glucocorticoids during adipogenesis. J Lipid Res 2008; 49: 1519–1528.
Havel PJ, Busch BL, Curry DL, Johnson PR, Dallman MF, Stern JS . Predominately glucocorticoid agonist actions of RU-486 in young specific-pathogen-free Zucker rats. Am J Physiol 1996; 271 (3 Pt 2): R710–R717.
Pereira-Fernandes A, Demaegdt H, Vandermeiren K, Hectors TL, Jorens PG, Blust R et al. Evaluation of a screening system for obesogenic compounds: screening of endocrine disrupting compounds and evaluation of the PPAR dependency of the effect. PLoS One 2013; 8: e77481.
Soriano S, Alonso-Magdalena P, Garcia-Arevalo M, Novials A, Muhammed SJ, Salehi A et al. Rapid insulinotropic action of low doses of bisphenol-A on mouse and human islets of Langerhans: role of estrogen receptor beta. PLoS One 2012; 7: e31109.
Chamorro-Garcia R, Kirchner S, Li X, Janesick A, Casey SC, Chow C et al. Bisphenol A diglycidyl ether induces adipogenic differentiation of multipotent stromal stem cells through a peroxisome proliferator-activated receptor gamma-independent mechanism. Environ Health Perspect 2012; 120: 984–989.
Hsieh TJ, Lin T, Hsieh PC, Liao MC, Shin SJ . Suppression of glutamine:fructose-6-phosphate amidotransferase-1 inhibits adipogenesis in 3T3-L1 adipocytes. J Cell Physiol 2012; 227: 108–115.
Riu A, Grimaldi M, le Maire A, Bey G, Phillips K, Boulahtouf A et al. Peroxisome proliferator-activated receptor gamma is a target for halogenated analogs of bisphenol A. Environ Health Perspect 2011; 119: 1227–1232.
Linehan C, Gupta S, Samali A, O'Connor L . Bisphenol A-mediated suppression of LPL gene expression inhibits triglyceride accumulation during adipogenic differentiation of human adult stem cells. PLoS One 2012; 7: e36109.
Hao CJ, Cheng XJ, Xia HF, Ma X . The endocrine disruptor diethylstilbestrol induces adipocyte differentiation and promotes obesity in mice. Toxicol Appl Pharmacol 2012; 263: 102–110.
Joyner JM, Hutley LJ, Cameron DP . Estrogen receptors in human preadipocytes. Endocrine 2001; 15: 225–230.
Hwang KA, Kang NH, Yi BR, Lee HR, Park MA, Choi KC . Genistein a soy phytoestrogen, prevents the growth of BG-1 ovarian cancer cells induced by 17beta-estradiol or bisphenol A via the inhibition of cell cycle progression. Int J Oncol 2013; 42: 733–740.
Abad MC, Askari H, O'Neill J, Klinger AL, Milligan C, Lewandowski F et al. Structural determination of estrogen-related receptor gamma in the presence of phenol derivative compounds. J Steroid Biochem Mol Biol 2008; 108: 44–54.
Park K, Kwak IS . Molecular effects of endocrine-disrupting chemicals on the Chironomus riparius estrogen-related receptor gene. Chemosphere 2010; 79: 934–941.
Ijichi N, Ikeda K, Horie-Inoue K, Yagi K, Okazaki Y, Inoue S . Estrogen-related receptor alpha modulates the expression of adipogenesis-related genes during adipocyte differentiation. Biochem Biophys Res Commun 2007; 358: 813–818.
Luo J, Sladek R, Carrier J, Bader JA, Richard D, Giguere V . Reduced fat mass in mice lacking orphan nuclear receptor estrogen-related receptor alpha. Mol Cell Biol 2003; 23: 7947–7956.
Ju D, He J, Zhao L, Zheng X, Yang G . Estrogen related receptor alpha-induced adipogenesis is PGC-1beta-dependent. Mol Biol Rep 2012; 39: 3343–3354.
Sheng ZG, Zhu BZ . Low concentrations of bisphenol A induce mouse spermatogonial cell proliferation by G protein-coupled receptor 30 and estrogen receptor-alpha. Environ Health Perspect 2011; 119: 1775–1780.
Ben-Jonathan N, Hugo ER, Brandebourg TD . Effects of bisphenol A on adipokine release from human adipose tissue: Implications for the metabolic syndrome. Mol Cell Endocrinol 2009; 304: 49–54.
Acknowledgements
We thank Dr Michael Wade and Dr Errol Thomson for reviewing this manuscript. EA received grant fund from the Health Canada Chemical Management Plan. JGB is funded by a Natural Sciences and Engineering Research Council of Canada Visiting Scientist Fellowship.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Boucher, J., Boudreau, A. & Atlas, E. Bisphenol A induces differentiation of human preadipocytes in the absence of glucocorticoid and is inhibited by an estrogen-receptor antagonist. Nutr & Diabetes 4, e102 (2014). https://doi.org/10.1038/nutd.2013.43
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2013.43
Keywords
This article is cited by
-
BPA and BPS affect the expression of anti-Mullerian hormone (AMH) and its receptor during bovine oocyte maturation and early embryo development
Reproductive Biology and Endocrinology (2021)
-
Demystifying Bisphenol A-Induced Alterations in Hypothalamic-Pituitary-Ovarian Functions Leading to Polycystic Ovarian Syndrome
Proceedings of the Zoological Society (2021)
-
Bisphenol A enhances adipogenic signaling pathways in human mesenchymal stem cells
Genes and Environment (2020)
-
Porous β-cyclodextrin nanotubular assemblies enable high-efficiency removal of bisphenol micropollutants from aquatic systems
Nano Research (2020)
-
Possible Obesogenic Effects of Bisphenols Accumulation in the Human Brain
Scientific Reports (2018)
