
Abstract
Background:
Ethanol consumption during pregnancy can lead to a range of adverse developmental outcomes in children, termed fetal alcohol spectrum disorder (FASD). Central nervous system injury is a debilitating and widely studied manifestation of chronic prenatal ethanol exposure (CPEE). However, CPEE can also cause structural and functional deficits in metabolic pathways in offspring.
Objectives and Methods:
This study tested the hypothesis that CPEE increases whole-body adiposity and disrupts pancreatic structure in guinea pig offspring. Pregnant guinea pigs received ethanol (4 g kg−1 maternal body weight per day) or isocaloric-sucrose/pair-feeding (control) for 5 days per week throughout gestation.
Results:
Male and female CPEE offspring demonstrated growth restriction at birth, followed by a rapid period of catch-up growth before weaning (postnatal day (PD) 1–7). Whole-body magnetic resonance imaging (MRI) in young adult offspring (PD100–140) revealed increased visceral and subcutaneous adiposity produced by CPEE. At the time of killing (PD150–200), CPEE offspring also had increased pancreatic adipocyte area and decreased β-cell insulin-like immunopositive area, suggesting reduced insulin production and/or secretion from pancreatic islets.
Conclusion:
CPEE causes increased adiposity and pancreatic dysmorphology in offspring, which may signify increased risk for the development of metabolic syndrome and type 2 diabetes mellitus.
Similar content being viewed by others


Introduction
Maternal alcohol (ethanol) consumption during pregnancy can lead to a range of adverse developmental outcomes in children, which are collectively termed fetal alcohol spectrum disorder (FASD).1, 2 Central nervous system injury is a widely studied manifestation of FASD as it is often the most debilitating and permanent consequence of prenatal ethanol exposure.1 However, prenatal ethanol exposure may also damage other organ systems, including the heart,3, 4 lung5 and kidney.4, 6 In addition, recent research suggests that prenatal ethanol exposure causes structural and functional deficits in metabolic pathways in offspring.7, 8, 9, 10, 11, 12 Because of the range of comorbidities and challenges associated with a diagnosis of FASD, it is estimated that the total adjusted annual cost of FASD in Canada is $5.3 billion.13
It is well established that chronic heavy ethanol exposure in adult humans is a significant risk factor for symptoms of metabolic syndrome, including impaired glucose homeostasis, diabetes mellitus, hypertriglyceridemia, abdominal obesity and high blood pressure.14, 15 Conversely, relatively less is known about the consequences of chronic prenatal ethanol exposure (CPEE) on these same outcome measures during postnatal life of the offspring. In recent years, several studies have demonstrated that prenatal ethanol exposure leads to alterations of metabolic pathways, including impaired glucose metabolism, increased gluconeogenesis, insulin resistance and impaired insulin signaling.7, 8, 9, 10, 11, 12 Children with fetal alcohol syndrome have hyperinsulinemia and hyperglycemia in oral glucose tolerance tests compared with typically developing children.16 These results suggest that children with fetal alcohol syndrome are insulin resistant; however, the association between prenatal ethanol exposure and increased risk of metabolic syndrome in offspring has not been established.
In order to better understand and interpret the nature of ethanol metabolic teratogenicity, this study tested the hypothesis that CPEE increases whole-body adiposity and disrupts pancreatic structure in guinea pig offspring. Maternal ethanol consumption during gestation leads to hypertriglyceridemia in adult rat offspring, suggesting that CPEE affects lipid metabolism.17 In addition, previous research has demonstrated that high-dose ethanol exposure in adult rats disrupts pancreatic function by decreasing β-cell mass, phosphatidylinositol 3-kinase activity and GLUT-4 expression.18 However, the effects of CPEE on postnatal whole-body adiposity and pancreatic morphology have not been studied.
The guinea pig is a well-established animal model for the study of ethanol teratogenicity because its in utero development is more similar to the humans compared with other rodent species with respect to its trimester-equivalent gestation,19 placental morphology20 and extensive organ-system development, including the brain growth spurt.21 Furthermore, the pharmacokinetics of ethanol in the maternal–fetal unit are very similar between guinea pigs and humans.22
Materials and methods
Experimental animals
Female, nulliparous Dunkin-Hartley strain guinea pigs (Charles River Canada Inc., St Constant, QC, Canada), with body weight between 550 and 650 g were bred with male guinea pigs using an established procedure.23, 24 Gestational day 0 (GD0) was defined as the last day of full vaginal-membrane opening, and term was ∼GD68. Pregnant animals were housed individually in plastic cages at an ambient temperature of 23 °C with a 12-h light/dark cycle (lights on from 0700 to 1900 h). All animals were cared for according to the principles and guidelines of the Canadian Council on Animal Care, and the experimental protocol was approved by the Queen’s University Animal Care Committee.
Animal treatment regimens
Pregnant dams were randomly assigned to one of the two maternal treatment groups: ethanol or nutritional control (isocaloric-sucrose/pair-feeding (sucrose)). Each pregnant animal in the ethanol group received 4 g ethanol per kg maternal body weight per day as an aqueous ethanol solution (30% v/v, prepared in tap water), 5 days per week throughout treatment, with ad libitum access to standard guinea pig chow (Lab Diet 5025, Purina, St Louis, MO, USA). Each pregnant dam in the nutritional control group was paired to an individual ethanol-treated pregnant animal and received isocaloric sucrose (42% w/v, prepared in tap water) and food in the amount consumed daily by the ethanol-treated animal. Aqueous ethanol or sucrose solution was administered into the oral cavity of the pregnant animal using a syringe, with subsequent swallowing of the solution. Maternal treatments were given in two equally divided doses, 2 h apart, starting at 0900 h on each treatment day. Food intake and weight gain were recorded for each dam throughout pregnancy.
Maternal blood ethanol concentration
On GD57 or GD58, 120 μl of blood was collected from the marginal ear vein of each pregnant guinea pig and was treated with 38% (w/v) aqueous sodium citrate (anticoagulant). The blood sample was taken 1 h after the second divided dose of ethanol or sucrose. Maternal blood ethanol concentration was determined by an established gas–liquid chromatographic procedure using headspace gas analysis.25
Body weight gain and whole-body magnetic resonance imaging (MRI)
Starting at postnatal day 1 (PD1), offspring were weighed daily and monitored for general health. At PD21–22, offspring from each litter were weaned, separated by sex and housed in groups of 2–5 same-sex animals.
Between PD100 and PD140, adipose tissue volume was measured in randomly selected offspring by whole-body MRI based on previously described methods.26, 27, 28 Each guinea pig was anesthetized using an induction protocol of intraperitoneal injection of medetomidine (0.25 mg kg−1) followed by inhalation of 4% isoflurane in O2. The concentration of isoflurane was decreased to 2.5–3% for maintenance of anesthesia during the imaging session. Data were acquired with a 3T Siemens MAGNETOM Trio MRI system (Erlangen, Germany). Each guinea pig was placed in a small animal receiver coil with a gel heating pad for added mass to improve signal quality. A T1-weighted turbo spin-echo sequence (1.1 × 1.1 × 2.0 mm, TR: 6670 ms, TE: 13 ms, FoV: 280 mm, Slices: 60, turbo factor: 7, bandwidth: 130 Hz/Px, water suppressed) was acquired in the coronal orientation. A scan in the identical orientation, but without water suppression, was also acquired for reference. The volume (mm3) of total body adipose tissue was calculated for each guinea pig (custom software written in MATLAB, R2009b; The Mathworks Inc., Natick, MA, USA) and expressed as a proportion of total body weight. Total adipose tissue content was computed through the following steps: (1) images were thresholded to black and white; (2) connected segments were identified and the heating pad was removed; (3) morphologic opening and dilation were used to remove noise; (4) a morphologic dilation of the remaining image was then used to create a mask on the original volume; and (5) the remaining white voxels after masking of the original volume were counted and multiplied by the voxel volume. Following noise correction, MRI scans with maintained quantifiable background noise were removed from the analysis. For visceral and subcutaneous adiposity measures, the volumes were loaded into ITK-SNAP (University of Pennsylvania, Philadelphia, PA, USA), and masks were created manually for these regions.29 The voxels in each masked region were counted in MATLAB as described previously.
Pancreatic morphology and insulin-like immunoreactivity
At PD150–200, all offspring were anesthetized using inhalational halothane (Halocarbon Laboratories, River Edge, NJ, USA) and were killed by decapitation. The pancreas was excised from each offspring and weighed. The pancreas was stored in 10% (v/v) neutral buffered formalin at room temperature for 48 h, rinsed with water and then was stored in 70% (v/v) aqueous ethanol solution until analyzed.
Pancreatic tissue was paraffin embedded, and immunohistochemical detection of insulin was performed on 5-μm sections of pancreatic tissue from CPEE and nutritional control offspring. A total of 10 animals per group (5 male and 5 female offspring) were randomly selected from each of the maternal treatment groups (ethanol or sucrose control) for analysis. Tissue sections were deparaffinized in xylene, rehydrated and washed in phosphate-buffered saline. Endogenous peroxidase activity was quenched with methanol, followed by antigen retrieval in 10 mmol l−1 citrate buffer (pH 3.0), and blocking with 10% (v/v) normal goat serum and 1% (w/v) bovine serum albumin. Sections were incubated with the primary antibody, a polyclonal, guinea pig anti-swine insulin antibody (1:150 dilution; DakoCytomation, Carpinteria, CA, USA) overnight at 4 °C. Sections were then washed with phosphate-buffered saline, and immunostaining was identified using the Vectastain kit (Vector Laboratories, Burlingame, CA, USA), with diaminobenzidine as the chromogen. Tissue sections were counterstained with Harris’s hematoxylin, destained with acid alcohol, dehydrated and mounted with Permount (Fisher Scientific, Fair Lawn, NJ, USA). In all tissue sections, pancreatic morphology was analyzed in 20 random fields per section, from 2 sections per animal (separated by at least 40 μm) at × 10 magnification. Insulin-like immunopositive cells were identified using Image Pro Plus v.5.1 software (Media Cybernetics Inc., Silver Spring, MD, USA) for automated cell counting and the calculation of β-cell area. The percent β-cell area was calculated as: area of insulin-like immunopositive cells/total pancreas area × 100. Similarly, the percent adipocyte area and the percent exocrine tissue area were calculated. We also examined the insulin-like immunopositive area (that is, β-cell area) and area of adipocytes (that is, fat infiltration) within islets by averaging measurements from at least 164 random islets per animal.
Statistical analysis
Previous investigation of ethanol teratogenicity in the guinea pig has demonstrated that within-litter variability in various outcomes, including fetal body weight, is of similar magnitude to between-litter variability.30 In view of this, the data are presented as group mean±s.e.m. of the offspring in each of the maternal ethanol and sucrose control treatment groups. The data were analyzed using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA) or PASW Statistics 18 (SPSS Inc., Chicago, IL, USA). Initial statistical analyses were conducted to determine whether there was a significant effect of sex on outcome measures in offspring. In the absence of a sex effect, data for male and female offspring were grouped together. Maternal weight gain and food intake per kg body weight during pregnancy were recorded throughout gestation and analyzed every fifth day by two-way repeated-measures analysis of variance (ANOVA; independent variables: maternal treatment and gestational day), followed by post hoc analysis for a statistically significant F-statistic (P<0.05) using Student’s t-test with Bonferroni correction for multiple comparisons. Birth weight was analyzed using an unpaired Student’s t-test. Whole-body MRI and pancreatic morphology data of the offspring were analyzed for homogeneity of variance and then by two-way ANOVA (independent variables: maternal treatment and sex). Weight gain and body weight of the female and male offspring of the experimental groups were analyzed by two-way repeated-measures ANOVA (independent variables: maternal treatment and postnatal age). Two groups of data were considered to be statistically different when P<0.05.
Results
The mean maternal blood ethanol concentration was 281±15 mg dl−1 at 1 h after the second divided dose of ethanol on GD57. The effects of chronic maternal ethanol administration on pregnancy outcome are described in a previous publication by our group.23 Briefly, there was one maternal death, a single incident of spontaneous abortion and five stillbirths in the ethanol treatment group. There was one incident of perinatal death in each of the ethanol and isocaloric-sucrose/pair-fed nutritional control (sucrose) groups. Pregnant dams that were treated with ethanol had significantly longer gestation (P<0.05) compared with sucrose-treated pregnant animals, and there was no effect of maternal treatment on litter size (data not shown). Two-way ANOVA demonstrated a significant effect of gestational day (F(11, 242=5.32, P<0.0001), but not an effect of maternal treatment, on food intake during pregnancy (data not shown). There was a significant statistical interaction between maternal treatment and gestational day (F(11, 242=2.65, P<0.01), in which CPEE dams had decreased food intake on GD20. Two-way ANOVA also revealed significant effects of maternal treatment (F(1, 264=6.80, P<0.05) and gestational day (F(12, 264=30.72, P<0.0001) on maternal body weight gain, such that ethanol-exposed dams had decreased weight gain compared with sucrose dams (data not shown). There was no statistical interaction between maternal treatment and gestational day.
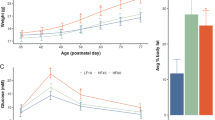
Body weight and weight gain were determined in offspring from birth until the time of killing (PD150–200). CPEE offspring had decreased birth weight compared with offspring of the sucrose control group (P<0.05; Figure 1a). Throughout the first week of the preweaning period (PD1–7), two-way repeated-measures ANOVA demonstrated that female CPEE offspring had increased weight gain compared with sucrose offspring (F(1, 245)=34.48, P<0.0001; Figure 1b). Two-way ANOVA also demonstrated a significant effect of day (F(6, 245)=68.01, P<0.0001), in which all female offspring gained weight less rapidly as they aged over the preweaning period. There was no statistical interaction between maternal treatment and day (F(6, 245)=0.72, P>0.05). For the remainder of the preweaning period (PD8–22), there was no difference between female CPEE and sucrose offspring in body weight gain. Similarly, for the male offspring, there were significant effects of maternal treatment (F(1, 245)=6.85, P<0.01) and day (F(6, 245)=58.00, P<0.0001) during the first week of preweaning (PD1–7), but no statistical interaction (F(6, 245)=0.33, P>0.05; Figure 1c). There was no difference between male CPEE and sucrose offspring in body weight gain for the remainder of the preweaning period (PD8–22).

Offspring birth weight (a). CPEE (ethanol) offspring were growth restricted at birth compared with isocaloric-sucrose/pair-fed (sucrose). The data are presented as group mean±s.e.m. of the offspring of each maternal treatment group with offspring from paired litters: ethanol (n=36 offspring; 22 male, 14 female) and sucrose (n=47 offspring; 23 male, 24 female) (*P<0.05). Weight gain during preweaning (PD1–22) (b, c). Female and male ethanol offspring demonstrated increased weight gain compared with sucrose offspring from PD1 to PD7 (females, P<0.0001; males, P<0.01; b, c, respectively). There was no difference in weight gain between ethanol and sucrose offspring over the rest of the preweaning period (PD8–22). For (b, c), the data are presented as group mean±s.e.m. of the offspring of each maternal treatment group with offspring from paired litters: ethanol (n=29 offspring; 16 male, 13 female) and sucrose (n=45 offspring; 21 male, 24 female).
Body weight was measured daily throughout adolescence (PD20–120). For the female offspring, two-way repeated-measures ANOVA demonstrated a significant effect of maternal treatment (F(1, 700)=4.30, P<0.05), such that female CPEE offspring demonstrated increased body weight compared with sucrose offspring (data not shown). Two-way ANOVA also demonstrated a significant effect of day (F(20, 700)=1558.70, P<0.0001), in which all female offspring had increased body weight over time. There was no statistical interaction between maternal treatment and day. In male offspring, there was an effect of day (F(20, 700)=2412.62, P<0.0001), reflecting the increase in body weight for all offspring from adolescence through adulthood (data not shown). However, there was no difference between the male CPEE and sucrose offspring in body weight from PD20 to PD120, nor was there a statistical interaction between the two factors.
Between PD100 and PD140, whole-body MRI analyses were conducted to determine adipose tissue volume in CPEE and sucrose control offspring (Figures 2 and 3). Following correction for background noise, 9 CPEE animals (4 males and 5 females) were removed from data analysis. Two-way ANOVA demonstrated a significant effect of maternal treatment (F(1, 47)=21.60, P<0.0001) on total adipose tissue volume per g body weight, such that CPEE offspring had increased adipose tissue volume compared with sucrose control offspring (Figure 3a). Two-way ANOVA also demonstrated a significant effect of sex (F(1, 47)=5.41, P<0.05), in which female offspring had increased total adipose tissue volume compared with male offspring. Adipose tissue volume was also determined in the visceral and subcutaneous regions of CPEE and sucrose control offspring. Two-way ANOVA demonstrated a significant effect of maternal treatment (F(1, 47)=13.91, P<0.001) on adipose tissue volume in the visceral region, such that CPEE offspring had increased adipose tissue volume compared with sucrose control offspring (Figure 3b). Two-way ANOVA also demonstrated a significant effect of sex (F(1, 47)=11.74, P<0.01), in which female offspring had increased visceral adipose tissue volume compared with male offspring. Similar to the total adipose tissue volume analysis, two-way ANOVA demonstrated a significant effect of maternal treatment (F(1, 47)=21.26, P<0.0001) on subcutaneous adipose tissue volume, such that CPEE offspring had increased adipose tissue volume compared with sucrose control offspring (Figure 3c). There was no effect of sex on subcutaneous adipose tissue volume.

MRI of CPEE and sucrose control offspring (PD100–140). Whole-body scans were performed using a high-resolution T1-weighted anatomical imaging protocol in a Siemens 3T whole-body MRI. A reference scan for each CPEE offspring (a) and sucrose control offspring (c) was performed before the water suppression scan. Water suppression scans were used for the determination of whole-body adipose tissue volume of CPEE offspring (b) compared with sucrose control offspring (d). CPEE offspring demonstrated increased total adipose tissue volume compared with sucrose control offspring.

Adipose tissue volume analysis by whole-body MRI (PD100–140). CPEE (ethanol) offspring had increased total (a), visceral (b), and subcutaneous (c) adipose tissue volume compared with nutritional control (sucrose) offspring (*P<0.001). Female offspring had increased visceral adipose tissue volume compared with male offspring (#P<0.05). The data are presented as group mean±s.e.m. of the offspring of each maternal treatment group with offspring from paired litters: ethanol (n=20 offspring; 12 males, 8 females) and sucrose (n=31 offspring; 16 males, 15 females).
Pancreatic morphology and insulin-like immunoreactivity were measured in adult tissue. Adipocyte infiltration in islets and surrounding tissue at × 10 magnification is shown in a CPEE animal (Figure 4a) compared with a sucrose control animal (Figure 4b). In the lobular pancreas, two-way ANOVA demonstrated a significant effect of maternal treatment on the percent adipocyte area, such that CPEE offspring had increased adipocyte area (F(1, 24)=4.27, P<0.05; Figure 4c). There was also a significant effect of sex (F(1, 24)=4.34, P<0.05) on the percent adipocyte area, where male offspring had increased adipocyte area. There was no statistical interaction between maternal treatment and sex. There was a main effect of maternal treatment (F(1, 24)=4.27, P<0.05) on the percent exocrine tissue in the whole pancreas, whereby CPEE offspring had decreased exocrine area. There was also a main effect of sex (F(1, 24)=4.34, P<0.05), where male offspring had decreased exocrine area, but there was no statistical interaction between maternal treatment and sex (Figure 4d). There was no difference in percent β-cell area in the whole pancreas of CPEE offspring compared with sucrose control offspring (data not shown). When only the pancreatic islet area was examined, however, two-way ANOVA revealed significant effects of maternal treatment on β-cell area per islet area (F(1, 35)=11.81, P<0.01) and adipocyte area per islet area (F(1, 35)=13.69, P<0.001), such that within islets, CPEE offspring had decreased β-cell area and increased adipocyte infiltration compared with sucrose control offspring (Figures 4e and f, respectively).

Pancreatic morphology (PD150–200). The × 10 magnification of adipocyte infiltration in islets and surrounding tissue in a CPEE (ethanol) animal (a). Pancreatic tissue from a nutritional control (sucrose) animal is shown at × 10 magnification (b). Ethanol offspring had increased adipocytes (c) and decreased exocrine tissue (d) in the lobular pancreas compared with sucrose offspring (*P<0.05). Male offspring had increased adipocytes and decreased exocrine tissue compared with female offspring (#P<0.05). The data are presented as group mean±s.e.m. of the offspring of each maternal treatment group with offspring from paired litters: ethanol (n=15 offspring; 7 males, 8 females) and sucrose (n=13 offspring; 7 males, 6 females). Pancreatic islet cell distribution (PD150–200) (boxed in red in a, b). Within each islet, CPEE (ethanol) offspring had fewer insulin-producing β-cells (arrows) (e) and more adipocytes (f) than nutritional control (sucrose) animals (P<0.05). The data are presented as group mean±s.e.m. of the offspring of each maternal treatment group with offspring from paired litters: ethanol (n=19 offspring; 10 males, 9 females) and sucrose (n=20 offspring; 10 males, 10 females).
Discussion
The results of the present study demonstrate that CPEE, via chronic maternal ethanol administration, causes metabolic teratogenicity in guinea pig offspring. Compared with nutritional control offspring, CPEE guinea pig offspring: (1) were growth restricted at birth and exhibited a greater rate of weight gain during the first week of postnatal life; (2) had a significantly greater proportion of total weight as visceral and subcutaneous adipose tissue in adulthood; (3) had increased adipocyte infiltration into the pancreas in adulthood; and (4) had decreased β-cell area and insulin-like immunoreactivity in the pancreas in adulthood. Increased adipocyte content in the pancreas as a consequence of CPEE, which may decrease β-cell mass, could potentially impair pancreatic function and increase the risk for development of metabolic syndrome.
Previous studies have suggested that CPEE can induce impairments in metabolism in rodent species.7, 8, 9, 10, 11, 12 Similarly, growth restriction at birth is one of the most frequently reported consequences of CPEE.31, 32, 33, 34 In the present study, male and female CPEE offspring demonstrated a rapid period of catch-up growth during preweaning (PD1–7). It has been well documented that fetal and early postnatal growth restriction and subsequent catch-up growth are associated with the development of metabolic syndrome.35, 36, 37, 38 The thrifty phenotype hypothesis postulates that poor nutrition in early life produces permanent changes in glucose/insulin metabolism.35, 36 The CPEE offspring in the present study were growth-restricted at birth with subsequent catch-up growth in the first week of postnatal life compared with the offspring of the isocaloric-sucrose/pair-fed control group. Chronic ethanol consumption is associated with alterations in intestinal microbial growth and increased intestinal permeability to endotoxins.39 CPEE in pregnant dams may have caused altered intestinal permeability, resulting in endotoxin exposure and nutritional deficiencies, which may have resulted in fetal growth restriction and organ-specific injury. Furthermore, the CPEE-induced effect of offspring growth restriction is not attributable solely to caloric restriction in pregnant females. It appears that CPEE, either alone or combined with decreased caloric intake, was responsible for the observed growth restriction and increased velocity of weight gain in early postnatal life. Our data are consistent with the findings of Chen and Nyomba,34 who showed that prenatal ethanol exposure in the rat produced growth restriction at birth followed by catch-up growth by 7 weeks of age. Furthermore, these offspring demonstrated impaired glucose tolerance and insulin resistance at 13 weeks of age.34 The data from our studies and others suggest that CPEE-induced growth restriction may be a risk factor for the development of metabolic syndrome and type 2 diabetes mellitus.
In the present study, MRI analyses revealed increased whole-body adiposity, along with increased visceral and subcutaneous adiposity, in adult CPEE guinea pig offspring. Visceral (central) obesity is a hallmark of metabolic syndrome and is a risk factor for type 2 diabetes and cardiovascular disease.40, 41, 42 Furthermore, Goodpaster et al.43 showed in humans that abdominal subcutaneous adiposity can predict insulin sensitivity independently of visceral adiposity.43 Ethanol consumption has been identified as a risk factor for the deposition of abdominal or visceral adipose tissue.44, 45, 46 Ethanol has been shown to suppress lipid oxidation, which results in the preferential deposition of nonoxidized lipids in the abdominal region.47, 48 Our study suggests that CPEE alters lipid metabolism in offspring and causes increased adipocyte deposition in both the visceral and subcutaneous regions. A study by Pennington et al.17 previously described the effects of prenatal ethanol exposure on lipid metabolism by examining rat offspring over 14 months for changes in triglyceride homeostasis. It was determined that prenatal ethanol exposure induces hypertriglyceridemia that markedly increases with the age of the offspring. Furthermore, the development of hypertriglyceridemia in offspring appears to be exacerbated by moderate restraint-induced maternal stress.17 Similarly, other studies in humans have determined that moderate to heavy ethanol consumption in adults significantly exacerbates preexisting hypertriglyceridemia.49, 50
In this study, adiposity was also assessed at the tissue level in the pancreas of adult guinea pig offspring. CPEE offspring demonstrated increased adipocyte area and decreased exocrine tissue area in the lobular pancreas. Furthermore, within the islets, CPEE offspring had increased adipocyte area and decreased insulin-like immunoreactivity area compared with nutritional control offspring. To date, there have been no studies to assess adiposity in the lobular pancreas or within islets following prenatal ethanol exposure. It is hypothesized that CPEE alters lipid metabolism, resulting in abnormal adipocyte accumulation in the pancreas. White adipose tissue is a critically important organ for whole-body glucose and lipid homeostasis.51 White adipose tissue is involved in clearance and storage of circulating lipids, thereby protecting other organs from ectopic lipid accumulation.51 It has been suggested that ectopic lipid accumulation in the pancreas and high content of free-fatty acids are involved in the eventual impairment of insulin secretion in subjects with type 2 diabetes mellitus.52, 53 A study by Tushuizen et al.54 showed that pancreatic lipid content is increased in overweight middle-aged Caucasian men with type 2 diabetes compared with nondiabetic men. In particular, pancreatic lipid content was reported to be inversely correlated with β-cell glucose sensitivity, which has been demonstrated to be a good predictor of progression to type 2 diabetes in nondiabetic subjects.54 Interestingly, in nondiabetic female Mexican-American teenagers, obesity was found to be associated with lipid accumulation in the pancreas without any impairment of insulin secretion.55 This suggests that pancreatic lipid accumulation may precede impairment in β-cell function, although it has not been determined whether pancreatic lipid accumulation is required for the subsequent development of type 2 diabetes in susceptible humans.54
The data from our study also suggest that β-cell function may be adversely affected by CPEE, resulting in decreased insulin production and/or secretion from islets. Relatively few studies have investigated the effect of prenatal ethanol exposure on pancreatic morphology and β-cell function. One study by Chen and Nyomba34 concluded that prenatal ethanol exposure impairs glucose tolerance in rat offspring by inducing both insulin resistance and β-cell dysfunction. Interestingly, pancreatic insulin content was decreased following prenatal ethanol exposure in 1-day-old rats, but not in 13-week-old rats.34 In addition, there was no effect of prenatal ethanol exposure on β-cell density or β-cell mass at either age.34
It is well established that heavy ethanol consumption in adults affects adversely pancreatic function and represents a potentially important, modifiable risk factor of type 2 diabetes.56, 57, 58 Lee et al.59 determined that excessive or chronic ethanol consumption leads to oxidative stress and mitochondrial dysfunction in pancreatic β-cells. Furthermore, a study by Nguyen et al.57 found that ethanol decreases insulin secretion from β-cell lines and isolated murine islets by interfering with muscarinic signaling and PKC activation. In addition, a study by Kim et al.58 in the rat determined that chronic ethanol consumption induces glucokinase downregulation, which results in β-cell apoptosis and disrupted β-cell function.56, 57, 58 It is unknown whether CPEE disrupts β-cell structure and function via a similar mechanism. Future studies should focus on the mechanism underlying the effect of CPEE on β-cell function and the relationship between altered β-cell function and the development of peripheral insulin resistance in offspring. In the future, peripheral insulin resistance should be measured in CPEE offspring using glucose tolerance testing, insulin tolerance testing, and the homeostatic model assessment (HOMA). Furthermore, future studies should investigate possible pharmacotherapy to improve or restore insulin sensitivity following CPEE.
In conclusion, this study has demonstrated for the first time that CPEE, via chronic maternal ethanol administration during gestation, increases whole-body adiposity and pancreatic adiposity in guinea pig offspring. Furthermore, CPEE reduces the β-cell insulin-like immunopositive area, suggesting that these animals may have impaired insulin production and/or secretion from pancreatic islets. The data suggest that CPEE impairs lipid metabolism and may therefore be a risk factor for the development of metabolic syndrome. Future studies should focus on the mechanism of CPEE-induced increased adiposity and pancreatic dysmorphology in adult offspring and on interventions that can prevent or reverse their occurrence.
References
Chudley AE, Conry J, Cook JL, Loock C, Rosales T, LeBlanc N et al. Fetal alcohol spectrum disorder: Canadian guidelines for diagnosis. CMAJ 2005; 172: S1–S21.
Sokol RJ, Delaney-Black V, Nordstrom B . Fetal alcohol spectrum disorder. JAMA 2003; 290: 2996–2999.
Goh JM, Bensley JG, Kenna K, Sozo F, Bocking AD, Brien J et al. Alcohol exposure during late gestation adversely affects myocardial development with implications for postnatal cardiac function. Am J Physiol Heart Circ Physiol 2011; 300: H645–H651.
Gray SP, Denton KM, Cullen-McEwen L, Bertram JF, Moritz KM . Prenatal exposure to alcohol reduces nephron number and raises blood pressure in progeny. J Am Soc Nephrol 2010; 21: 1891–1902.
Sozo F, Vela M, Stokes V, Kenna K, Meikle PJ, De Matteo R et al. Effects of prenatal ethanol exposure on the lungs of postnatal lambs. Am J Physiol Lung Cell Mol Physiol 2011; 300: L139–L147.
Probyn ME, Zanini S, Ward LC, Bertram JF, Moritz KM . A rodent model of low- to moderate-dose ethanol consumption during pregnancy: patterns of ethanol consumption and effects on fetal and offspring growth. Reprod Fertil Dev 2012; 24: 859–870.
Chen L, Nyomba BL . Glucose intolerance and resistin expression in rat offspring exposed to ethanol in utero: modulation by postnatal high-fat diet. Endocrinology 2003; 144: 500–508.
Chen L, Nyomba BL . Whole body insulin resistance in rat offspring of mothers consuming alcohol during pregnancy or lactation: comparing prenatal and postnatal exposure. J Appl Physiol 2004; 96: 167–172.
de la Monte SM, Wands JR . Role of central nervous system insulin resistance in fetal alcohol spectrum disorders. J Popul Ther Clin Pharmacol 2010; 17: e390–e404.
Elton CW, Pennington JS, Lynch SA, Carver FM, Pennington SN . Insulin resistance in adult rat offspring associated with maternal dietary fat and alcohol consumption. J Endocrinol 2002; 173: 63–71.
Yao XH, Chen L, Nyomba BL . Adult rats prenatally exposed to ethanol have increased gluconeogenesis and impaired insulin response of hepatic gluconeogenic genes. J Appl Physiol 2006; 100: 642–648.
Yao XH, Nyomba BL . Hepatic insulin resistance induced by prenatal alcohol exposure is associated with reduced PTEN and TRB3 acetylation in adult rat offspring. Am J Physiol Regul Integr Comp Physiol 2008; 294: R1797–R1806.
Stade B, Ali A, Bennett D, Campbell D, Johnston M, Lens C et al. The burden of prenatal exposure to alcohol: revised measurement of cost. Can J Clin Pharmacol 2009; 16: e91–102.
Lee K . Gender-specific relationships between alcohol drinking patterns and metabolic syndrome: the Korea National Health and Nutrition Examination Survey 2008. Public Health Nutr 2012; 15: 1917–1924.
Fan AZ, Russell M, Naimi T, Li Y, Liao Y, Jiles R et al. Patterns of alcohol consumption and the metabolic syndrome. J Clin Endocrinol Metab 2008; 93: 3833–3838.
Castells S, Mark E, Abaci F, Schwartz E . Growth retardation in fetal alcohol syndrome. Unresponsiveness to growth-promoting hormones. Dev Pharmacol Ther 1981; 3: 232–241.
Pennington JS, Shuvaeva TI, Pennington SN . Maternal dietary ethanol consumption is associated with hypertriglyceridemia in adult rat offspring. Alcohol Clin Exp Res 2002; 26: 848–855.
Zhao LN, Hao LP, Yang XF, Ying CJ, Yu D, Sun XF . The diabetogenic effects of excessive ethanol: reducing beta-cell mass, decreasing phosphatidylinositol 3-kinase activity and GLUT-4 expression in rats. Br J Nutr 2009; 101: 1467–1473.
Nishimura H, Shiota K . Summary of comparative embryology and teratology. In: Wilson C, Fraser FC, (eds) Handbook of Teratology. Plenum Press: New York, 1977. pp 119–154.
Martensson L . The pregnant rabbit, guinea pig, sheep and rhesus monkey as models in reproductive physiology. Eur J Obstet Gynecol Reprod Biol 1984; 18: 169–182.
Dobbing J, Sands J . Comparative aspects of the brain growth spurt. Early Hum Dev 1979; 3: 79–83.
Brien JF, Clarke DW . Disposition and fetal effects of ethanol during pregnancy. In: Kacew S, Lock S, (eds) Toxicologic and Pharmacologic Principles in Pediatrics. Hemisphere Publishing Corporation: New York, 1988. pp 199–222.
Dobson CC, Mongillo DL, Poklewska-Koziell M, Winterborn A, Brien JF, Reynolds JN . Sensitivity of modified Biel-maze task, compared with Y-maze task, to measure spatial learning and memory deficits of ethanol teratogenicity in the guinea pig. Behav Brain Res 2012; 233: 162–168.
Elvidge H . Production of dated pregnant guinea pigs without postpartum matings. J Inst Animal Tech 1972; 23: 111–117.
Steenaart NA, Clarke DW, Brien JF . Gas-liquid chromatographic analysis of ethanol and acetaldehyde in blood with minimal artifactual acetaldehyde formation. J Pharmacol Methods 1985; 14: 199–212.
Li X, Youngren JF, Hyun B, Sakkas GK, Mulligan K, Majumdar S et al. Technical evaluation of in vivo abdominal fat and IMCL quantification using MRI and MRSI at 3 T. Magn Reson Imaging 2008; 26: 188–197.
Hu HH, Hines CD, Smith DL, Reeder SB . Variations in T(2)* and fat content of murine brown and white adipose tissues by chemical-shift MRI. Magn Reson Imaging 2012; 30: 323–329.
Johnson DH, Flask CA, Ernsberger PR, Wong WC, Wilson DL . Reproducible MRI measurement of adipose tissue volumes in genetic and dietary rodent obesity models. J Magn Reson Imaging 2008; 28: 915–927.
Yushkevich PA, Piven J, Hazlett HC, Smith RG, Ho S, Gee JC et al. User-guided 3D active contour segmentation of anatomical structures: significantly improved efficiency and reliability. Neuroimage 2006; 31: 1116–1128.
Brien JF, Chan D, Green CR, Iqbal U, Gareri J, Kobus SM et al. Chronic prenatal ethanol exposure and increased concentration of fatty acid ethyl esters in meconium of term fetal Guinea pig. Ther Drug Monit 2006; 28: 345–350.
Iqbal U, Rikhy S, Dringenberg HC, Brien JF, Reynolds JN . Spatial learning deficits induced by chronic prenatal ethanol exposure can be overcome by non-spatial pre-training. Neurotoxicol Teratol 2006; 28: 333–341.
Shea KM, Hewitt AJ, Olmstead MC, Brien JF, Reynolds JN . Maternal ethanol consumption by pregnant guinea pigs causes neurobehavioral deficits and increases ethanol preference in offspring. Behav Pharmacol 2012; 23: 105–112.
Chen L, Zhang T, Nyomba BL . Insulin resistance of gluconeogenic pathways in neonatal rats after prenatal ethanol exposure. Am J Physiol Regul Integr Comp Physiol 2004; 286: R554–R559.
Chen L, Nyomba BL . Effects of prenatal alcohol exposure on glucose tolerance in the rat offspring. Metabolism 2003; 52: 454–462.
Hales CN, Barker DJ . Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 1992; 35: 595–601.
Barker DJ, Hales CN, Fall CH, Osmond C, Phipps K, Clark PM . Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 1993; 36: 62–67.
Bertram CE, Hanson MA . Animal models and programming of the metabolic syndrome. Br Med Bull 2001; 60: 103–121.
Cettour-Rose P, Samec S, Russell AP, Summermatter S, Mainieri D, Carrillo-Theander C et al. Redistribution of glucose from skeletal muscle to adipose tissue during catch-up fat: a link between catch-up growth and later metabolic syndrome. Diabetes 2005; 54: 751–756.
Purohit V, Bode JC, Bode C, Brenner DA, Choudhry MA, Hamilton F et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: summary of a symposium. Alcohol 2008; 42: 349–361.
Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR et al. A transgenic model of visceral obesity and the metabolic syndrome. Science 2001; 294: 2166–2170.
Despres JP, Lemieux I . Abdominal obesity and metabolic syndrome. Nature 2006; 444: 881–887.
Wajchenberg BL . Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr Rev 2000; 21: 697–738.
Goodpaster BH, Thaete FL, Simoneau JA, Kelley DE . Subcutaneous abdominal fat and thigh muscle composition predict insulin sensitivity independently of visceral fat. Diabetes 1997; 46: 1579–1585.
Kim KH, Oh SW, Kwon H, Park JH, Choi H, Cho B . Alcohol consumption and its relation to visceral and subcutaneous adipose tissues in healthy male Koreans. Ann Nutr Metab 2012; 60: 52–61.
Yokoyama H, Hiroshi H, Ohgo H, Hibi T, Saito I . Effects of excessive ethanol consumption on the diagnosis of the metabolic syndrome using its clinical diagnostic criteria. Intern Med 2007; 46: 1345–1352.
Dorn JM, Hovey K, Muti P, Freudenheim JL, Russell M, Nochajski TH et al. Alcohol drinking patterns differentially affect central adiposity as measured by abdominal height in women and men. J Nutr 2003; 133: 2655–2662.
Suter PM . Is alcohol consumption a risk factor for weight gain and obesity? Crit Rev Clin Lab Sci 2005; 42: 197–227.
Shelmet JJ, Reichard GA, Skutches CL, Hoeldtke RD, Owen OE, Boden G . Ethanol causes acute inhibition of carbohydrate, fat, and protein oxidation and insulin resistance. J Clin Invest 1988; 81: 1137–1145.
Bessembinders K, Wielders J, van de Wiel A . Severe hypertriglyceridemia influenced by alcohol (SHIBA). Alcohol Alcohol 2011; 46: 113–116.
Ginsberg H, Olefsky J, Farquhar JW, Reaven GM . Moderate ethanol ingestion and plasma triglyceride levels. A study in normal and hypertriglyceridemic persons. Ann Intern Med 1974; 80: 143–149.
Scherer T, O'Hare J, Diggs-Andrews K, Schweiger M, Cheng B, Lindtner C et al. Brain insulin controls adipose tissue lipolysis and lipogenesis. Cell Metab 2011; 13: 183–194.
Carpentier A, Mittelman SD, Bergman RN, Giacca A, Lewis GF . Prolonged elevation of plasma free fatty acids impairs pancreatic beta-cell function in obese nondiabetic humans but not in individuals with type 2 diabetes. Diabetes 2000; 49: 399–408.
Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH . Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci USA 1994; 91: 10878–10882.
Tushuizen ME, Bunck MC, Pouwels PJ, Bontemps S, van Waesberghe JH, Schindhelm RK et al. Pancreatic fat content and beta-cell function in men with and without type 2 diabetes. Diabetes Care 2007; 30: 2916–2921.
Kovanlikaya A, Mittelman SD, Ward A, Geffner ME, Dorey F, Gilsanz V . Obesity and fat quantification in lean tissues using three-point Dixon MR imaging. Pediatr Radiol 2005; 35: 601–607.
Baliunas DO, Taylor BJ, Irving H, Roerecke M, Patra J, Mohapatra S et al. Alcohol as a risk factor for type 2 diabetes: a systematic review and meta-analysis. Diabetes Care 2009; 32: 2123–2132.
Nguyen KH, Lee JH, Nyomba BL . Ethanol causes endoplasmic reticulum stress and impairment of insulin secretion in pancreatic beta-cells. Alcohol 2012; 46: 89–99.
Kim JY, Song EH, Lee HJ, Oh YK, Park YS, Park JW et al. Chronic ethanol consumption-induced pancreatic {beta}-cell dysfunction and apoptosis through glucokinase nitration and its down-regulation. J Biol Chem 2010; 285: 37251–37262.
Lee JH, Nguyen KH, Mishra S, Nyomba BL . Prohibitin is expressed in pancreatic beta-cells and protects against oxidative and proapoptotic effects of ethanol. FEBS J 2010; 277: 488–500.
Acknowledgements
This study was supported by grants from the Canadian Institutes of Health Research (CIHR MOP 84553 and ELA 80227). We thank Amber Knuff and Stephanie Logan for their technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Dobson, C., Mongillo, D., Brien, D. et al. Chronic prenatal ethanol exposure increases adiposity and disrupts pancreatic morphology in adult guinea pig offspring. Nutr & Diabetes 2, e57 (2012). https://doi.org/10.1038/nutd.2012.31
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2012.31
Keywords
This article is cited by
-
Prenatal ethanol exposure and changes in fetal neuroendocrine metabolic programming
Biological Research (2023)
-
From conception to infancy — early risk factors for childhood obesity
Nature Reviews Endocrinology (2019)
-
Sex-specific increase in susceptibility to metabolic syndrome in adult offspring after prenatal ethanol exposure with post-weaning high-fat diet
Scientific Reports (2015)
-
Early-Life Exposure to Substance Abuse and Risk of Type 2 Diabetes in Adulthood
Current Diabetes Reports (2015)
