
Abstract
Recent findings on the role of transforming growth factor (TGF)-β/Smad3 signaling in the pathogenesis of obesity and type 2 diabetes have underscored its importance in metabolism and adiposity. Indeed, elevated TGF-β has been previously reported in human adipose tissue during morbid obesity and diabetic neuropathy. In this review, we discuss the pleiotropic effects of TGF-β/Smad3 signaling on metabolism and energy homeostasis, all of which has an important part in the etiology and progression of obesity-linked diabetes; these include adipocyte differentiation, white to brown fat phenotypic transition, glucose and lipid metabolism, pancreatic function, insulin signaling, adipocytokine secretion, inflammation and reactive oxygen species production. We summarize the recent in vivo findings on the role of TGF-β/Smad3 signaling in metabolism based on the studies using Smad3−/− mice. Based on the presence of a dual regulatory effect of Smad3 on peroxisome proliferator-activated receptor (PPAR)β/δ and PPARγ2 promoters, we propose a unifying mechanism by which this signaling pathway contributes to obesity and its associated diabetes. We also discuss how the inhibition of this signaling pathway has been implicated in the amelioration of many facets of metabolic syndromes, thereby offering novel therapeutic avenues for these metabolic conditions.
Similar content being viewed by others


Introduction
Obesity has become a global epidemic over the past decades. Obesity is associated with an array of secondary metabolic abnormalities, including insulin resistance, type 2 diabetes and hyperlipidemia, which are collectively termed metabolic syndrome.1 Type 2 diabetes has afflicted a staggering 180 million people worldwide, and its associated complications, including diabetic nephropathy, extremity amputation and heart failure, have become the principal contributors of morbidity and mortality in the western world.2 The mechanisms by which obesity causes insulin resistance and type 2 diabetes, as well as the pathological events that occur during this process, have been vigorously studied.3 Dynamic interactions among different metabolic organs, such as the pancreas, skeletal muscle, liver and adipose tissue, have a key role in the pathogenesis of obesity and diabetes. The hallmark events involved in the development of obesity-linked diabetes include elevation of plasma free fatty acid (FFA), ectopic lipid accumulation in metabolic organs, chronic low-grade inflammation in adipose tissue, peripheral insulin resistance, chronic hyperglycemia and eventual pancreatic β-cell dysfunction.3 These seemingly independent events are actually interconnected in terms of their etiology and downstream effects. Indeed, recent studies have investigated the molecular and genetic basis that governs the whole-body response to this disease. However, many questions remain unsolved. Recent novel findings on the involvement of transforming growth factor (TGF)-β/Smad3 signaling in the pathogenesis of obesity and type 2 diabetes have shed some light on its roles in adipose physiology and metabolism.4, 5, 6 Several groups of researchers have recently demonstrated that TGF-β/Smad3 signaling regulates insulin gene transcription in the pancreatic islet β-cells,7 whereas Smad3 deficiency in mice protects against insulin resistance and type 2 diabetes during high-fat diet-induced obesity.4, 5 These Smad3-knockout (Smad3−/−) mice exhibited diminished adiposity with improved glucose tolerance and insulin sensitivity.4, 5 These mutant mice also displayed increased β-oxidation in the adipose tissue upon administration of a high-fat diet, thus ameliorating gluco- and lipotoxicity in the pancreas, skeletal muscle and liver by preventing ectopic fat accumulation.4 In addition, Smad3 gene deletion in mice triggered a brown phenotypic transformation and elevated mitochondrial biogenesis in the white adipose tissue (WAT), thereby allowing for the dissipation of the excess energy stored in fat by thermogenesis.5 Notably, when TGF-β/Smad3 signaling was blunted by treatment with a TGF-β neutralizing antibody, it protected the mice from obesity and type 2 diabetes.5 These findings indicate that Smad3, the canonical intracellular mediator of TGF-β, serves as a multifaceted regulator of metabolic homeostasis, thus identifying Smad3 as a potential target in the treatment of obesity and its associated disorders.
In this review, we discuss the molecular mechanisms by which TGF-β/Smad3 signaling contributes to the pathogenesis of obesity and diabetes, including systemic glucose and lipid metabolism, pancreatic β-cell function, adipocyte differentiation, white-to-brown fat transformation, reactive oxygen species (ROS) production, adipocytokine secretion and inflammation. Here, we review current findings regarding the role of TGF-β/Smad3 in metabolism and discuss how the inhibition of this signaling pathway has been proposed to ameliorate many facets of metabolic syndromes. These findings discussed here support that TGF-β/Smad3 signaling has a crucial role in the events leading to obesity-linked diabetes by acting as the molecular intersection point where a number of pathological events converge. We attempt to summarize the systemic actions of TGF-β/Smad3, thereby identifying the plausible therapeutic avenues for the treatment of these diseases.
TGF-β/Smad3 signaling in obesity and diabetes
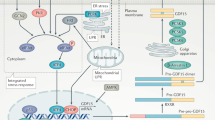
TGF-β1 is the prototype of a superfamily of highly conserved and structurally related secreted cytokines, which also includes the bone morphogenetic proteins, activins/inhibins and myostatin. These pleiotropic cytokines regulate a plethora of biological processes during embryogenesis and adult tissue homeostasis, such as cell proliferation, differentiation and specification of developmental fate.8 Particularly, TGF-β1 has a central role in fibrosis, contributing to the infiltration and activation of inflammatory cells, fibroblasts and matrix remodeling.8 TGF-β1 signals through a complex of two membrane-bound receptor serine/threonine kinases, the TGF-β type I and type II receptors, which recruit and phosphorylate receptor-activated Smads, including Smad2 and Smad3, the canonical intracellular mediators of TGF-β (Figure 1). Smads are modular proteins with two well-conserved domains, the N-terminal Mad-homology-1 domain and the C-terminal Mad-homology-2 domain, joined together by a proline-rich linker region (Figure 1).9 A Ser-Ser-X-Ser motif is located at the extreme C-terminal end of the Smad3.9 In cell signaling pathways, various transcription factors are phosphorylated at multiple sites by upstream kinases. TGF-β type I receptors, activin type IB and IC receptors phosphorylate the C-terminal Ser-Ser-X-Ser motif of Smad2 and Smad3, whereas mitogenic signals alternatively cause the phosphorylation of receptor-activated Smads at specific sites in their linker regions.10 Once phosphorylated, Smad2 and Smad3 oligomerize with Smad4 before their translocation into the nucleus, where they bind to DNA to modulate transcriptional events.9 Smad2 and Smad3 have apparently distinct functions, although they share >90% homology in their amino-acid sequences.11 The essential role of Smad2 in embryonic development was initially revealed by embryonic lethality in Smad2-knockout mice at embryonic days 7.5–12.5.12 In contrast, Smad3−/− mice are viable and exhibit postnatal growth retardation, immune defects and accelerated wound healing.13, 14 Smad3−/− mice are also resistant to radiation- and chemical-induced fibrosis and other fibrotic disorders, such as scleroderma and cystic fibrosis.15

TGF-β/Smad canonical signaling pathway. The TGF-β1 signals through a complex of TGF-β type I (TGFβRI) and type II receptors that recruit and activate intracellular mediator Smads, such as Smad2 and Smad3, which translocate into the nucleus transcriptionally regulate gene expression.
The systemic impact of TGF-β/Smad3 deficiency in the etiology of metabolic syndrome has recently been uncovered. Emerging evidence has implicated TGF-β1 in obesity and diabetes. For instance, an increased activation of the TGF-β/Smad3 signaling pathway was detected in the WAT of Lepob/ob mice.6 Elevated TGF-β1 in humans have been shown to positively correlate with increased adiposity, a poor metabolic profile and negatively correlate with fitness.5 TGF-β1 level in adipose tissue and body mass index are closely associated in individuals with morbid obesity.16 Further, plasma TGF-β are also augmented in hypertension and other cardiovascular diseases, indicative of its role in metabolic syndrome.17 Elevated TGF-β1 have been associated with a higher risk for type 2 diabetes in a prospective case-cohort study.18 Moreover, the Smad3 gene was identified in a type 2 diabetes genome-wide association study.19 Treatment with anti-TGF-β antibodies protects genetically and diet-induced obese mice from obesity and type 2 diabetes.5 TGF-β signaling has also been implicated in glucose-induced cell hypertrophy upon prolonged exposure to glucose, which may result in complications associated with pancreatic β-cell dysfunction and insulin resistance.20 Polymorphisms in TGF-β1, for example, T29C polymorphism that results in a Leu-Pro substitution at the signal peptide, as well as elevated circulating TGF-β are correlated with increased body mass index, elevated fasting insulin and glucose, insulin resistance, as well as predisposition to cardiovascular disorders.21, 22 The Leu-Pro substitution disrupts the hydrophobicity and overall structure of core signal sequence to affect production of TGF-β1.23 Renal expression of TGF-β1 was upregulated in diabetic humans,24 whereas early manifestations of diabetic renal disease in mice can be prevented by systemic treatment with anti-TGF-β antibodies and antisense TGF-β1 oligodeoxynucleotides.25, 26
Adipocyte differentiation and lipid metabolism
Obesity is medically defined as a state of excessive body fat accumulation to an extent that produces adverse health consequences. In times of nutrient abundance, a positive energy balance occurs when the energy intake exceeds the energy expenditure, leading to a net storage of surplus energy in the form of triglycerides in adipose tissue. Adipose tissue mass is dependent on both the size and number of its constituent adipocytes. Expansion of adipose tissue mass occurs through two distinct processes, namely hypertrophy (increase in cell size) and hyperplasia (increase in cell number). Adipocyte hypertrophy is achieved in mature adipocytes via an increase in lipid accumulation or lipogenesis (i.e., triglyceride synthesis), which are synergetic with a decrease in lipolysis (i.e., triglyceride breakdown). In contrast, adipocyte hyperplasia relies on a complicated process called adipogenesis that involves preadipocyte differentiation. Among the many transcriptional cascades controlling adipogenesis, peroxisome proliferator-activated receptor (PPAR)γ2 is well characterized as the master regulator of the adipogenic program.27 Upon exposure to adipogenic hormones, such as glucocorticoids, cyclic AMP and insulin, the expression of transcription factors CCAAT/enhancer-binding protein (C/EBP)β and C/EBPδ is transiently increased during adipocyte differentiation. Together they induce PPARγ2 expression in the preadipocytes, subsequently triggering full-blown adipocyte differentiation.27 PPARγ2, a nuclear hormone receptor, heterodimerizes with the retinoid X receptor and induces the expression of C/EBPα, which subsequently takes over the function of C/EBPβ and C/EBPδ in maintaining the transcription of PPARγ2 via a positive feedback mechanism.27 Activated C/EBPα cooperates with PPARγ2 to induce the expression of other adipogenic genes involved in the differentiation program.
Previous in vitro studies showed that TGF-β1 inhibits adipogenesis independently of the Wnt/β-catenin signaling pathway and through the inhibitory effects of its downstream mediator Smad3 on the transactivational potential of C/EBPs, thus abolishing the subsequent activation of PPARγ2.6, 28 Retinoic acid (RA) inhibits adipogenesis and its action appears to block C/EBPβ transcriptional potential early during differentiation. This RA-mediated effect has also been found to be dependent on Smad3.29 A recent finding also revealed an important role of cellular RA-binding protein-II in regulating the transcriptional activity of RA through RA receptor. The early and sustained repression of cellular RA-binding protein-II by glucocorticoid receptor and C/EBPα, respectively, is critical for adipogenesis.30 However, recent in vivo and in vitro studies using Smad3−/− mice have demonstrated a paradoxical role of TGF-β/Smad3 as an activator of adipocyte differentiation.4, 5 Primary mouse embryonic fibroblasts extracted from Smad3−/− mice display marked impairment in their ability to differentiate into white adipocytes and show reduced expression of WAT-specific genes.5 This is consistent with the observations that Smad3−/− mice exhibit dramatic reduction in adiposity as a result of decreased adipocyte number and size,4 suggesting the presence of defective adipogenesis and lipid accumulation in these mice. Gene expression profiling of the Smad3−/− WAT revealed decreased expression of PPARγ2 transcripts, which coincided with an increased expression of the preadipocyte-specific marker preadipocyte factor-1.4 Lipid accumulation relies on the process of lipogenesis, which involves de novo synthesis of fatty acids (FAs) and glycerol, FA uptake and triglyceride synthesis. In fact, PPARγ2 not only participates in adipocyte differentiation and survival, but it also promotes lipid accumulation by activating target genes involved in lipogenesis, such as FA synthase, adipose FA-binding protein and phosphoenolpyruvate carboxykinase.31 Except for FA uptake, the expression of the rate-limiting enzymes involved in lipogenesis (e.g., FA synthase, acetyl-CoA carboxylase-1 and stearoyl-CoA desaturase-1) and triglyceride synthesis (e.g., diacylglycerol O-acyltransferase-1 and -2) was concomitantly downregulated in the Smad3−/− WAT as evidence by decrease in ex vivo triglyceride synthesis.4 In contrast with the diminished PPARγ2 expression, a marked augmentation in the expression level of PPARβ/δ and its regulated genes, such as uncoupling protein (UCP)-2, UCP-3 and acyl-CoA oxidase (ACOX)-1, has also been found in the Smad3−/− WAT.4 This is in agreement with the finding that ex vivo FA β-oxidation was elevated in the Smad3−/− WAT.4 The activation of PPARβ/δ in obese mice has been shown to selectively induce the expression of genes required for FA β-oxidation and energy dissipation, such as UCPs and ACOX-1, but not genes involved in lipogenesis and fat storage, which are controlled by PPARγ2.32 The underlying mechanism behind this disparate effect of Smad3 on fat storage and burning involves the dual effects of Smad3 deficiency on the PPARγ2 and PPARβ/δ promoters. Chromatin immunoprecipitation and re-chromatin immunoprecipitation assays showed that C/EBPβ was associated with a specific C/EBP binding site at 494–485 bp upstream from the PPARβ/δ promoter, thereby resulting in the recruitment to this element of a transcriptional repressor complex containing histone deacetylase 1 and the repression of PPARβ/δ.4 Besides, C/EBPβ can freely bind to the PPARγ2 promoter to exert its adipogenic and lipogenic effects in normal WAT.4 In contrast, Smad3 deficiency leads to upregulation of an anti-adipogenic factor C/EBP homologous protein-10, which directly binds to C/EBPβ to liberate PPARβ/δ transcription while impeding PPARγ2 activation (Figure 2).4 This finding is further validated by the fact that C/EBP homologous protein-10 acts as a dominant-negative inhibitor of C/EBP by preventing its binding to DNA.33

TGF-β/Smad3 signaling promotes adipocyte differentiation but inhibits FA β-oxidation and thermogenesis.
It is important to recognize that reductions in fat storage could have catastrophic consequences for Smad3−/− mice if not for the increased FA uptake and β-oxidation. This concern over the adverse effects of fat storage depletion stems from the fat-storing functions of adipose tissue that prevent the ectopic lipid accumulation and lipotoxicity in metabolic organs, for example, hepatic steatosis. Indeed, ablation of PPARγ2 in obese mice results in reduced fat mass but leads to severe insulin resistance, pancreatic β-cell failure and dyslipidemia due to the cumulative deposition of toxic lipid reactive species in the peripheral organs.34 Moreover, intramuscular triglyceride content has previously been reported to be elevated in diabetic patients and inversely correlated with the degree of insulin resistance.35 Nonetheless, these abnormalities were not observed in Smad3−/− mice probably because of the decreased lipolysis and increased FA uptake from the circulation, ultimately targeting FAs for β-oxidation and energy dissipation in WAT (Figure 3).4 This hypothesis is substantiated by the findings that Smad3−/− mice exhibit lower plasma FFA and reduced ectopic accumulation of FAs and triglyceride in the peripheral organs upon high-fat diet treatment.4 Despite the fact that skeletal muscle is the organ responsible for most of the body energy expenditure and ablation of PPARβ/δ in skeletal muscle has been shown to induce a muscle fiber-type switching toward a lower oxidative capacity that precedes the development of obesity and diabetes.36, 37 It is unlikely that increased FA β-oxidation in skeletal muscle contributes to the antidiabetic phenotypes in Smad3−/− mice because of the presence of muscular atrophy and constant PPARβ/δ mRNA expression level in skeletal muscle of these animals.4, 38 This is consistent with an unaltered metabolic rate in Smad3−/− mice, reflecting a balance between their reduced physical activity and increased adipose lipid oxidation.4 In conjunction, a role for TGF-β/Smad3 in β2-adrenergic-induced muscle hypertrophy, satellite cell function and myogenic differentiation has also been reported.38, 39 Although WAT is not a major energy consumer compared with skeletal muscle, increased FA β-oxidation in WAT can have a profound impact on adiposity and insulin sensitivity.40 Clearly, adipose tissue is the primary tissue responsible for the antiobesity effects of Smad3 deficiency because FA oxidation is not altered in the liver of Smad3−/− mice.4 Interestingly, a WAT-to-brown adipose tissue (BAT) phenotypic switch has been reported recently in Smad3−/− mice and in adipocytes transfected with silencing short-hairpin RNA targeting Smad3.5 The mRNA expression level of PPARγ2 is barely detectable whereas that of PPARγ1 is expressed at a low level in rodent and human skeletal muscle.41, 42, 43, 44 Hence, the effect of Smad3 on PPARγ2 and PPARβ/δ promoters is likely to be present predominantly in WAT where PPARγ2 is abundantly expressed. To date, no data has indicated a similar regulatory effect of Smad3 on the PPARγ1 promoters in the skeletal muscle.

(a) Systemic role of TGF-β/Smad3 signaling in the pathogenesis of type 2 diabetes. (b) Amelioration of type 2 diabetes upon blockade of TGF-β/Smad3 signaling.
WAT-to-BAT phenotypic transition
There are two major types of adipose tissues in mammals, white and brown. WAT is pale white in color and is comprised predominantly of large unilocular adipocytes that are specialized for fat storage. Conversely, BAT possesses a dark-red color because of its high-mitochondrial content, and it consists of small-multilocular adipocytes that are highly adapted for oxidizing chemical energy for heat production through a process called thermogenesis. Brown adipocytes express UCP-1, which uncouples respiration from ATP synthesis, allowing for heat production. PR domain containing 16 (PRDM16) is a transcriptional coregulator that governs the development of brown adipocytes in classic BAT depots such as peri-renal and interscapular brown fats.45 White and brown adipocytes were previously believed to originate from a common lineage, however, several studies have countered this assumption.46, 47 A common lineage for muscle and BAT has been proposed based on the findings that specific cells derived from the dermomyotome give rise to muscle and brown adipocytes but not white fat cells.47 Further evidence stems from the expression of myogenic factor (Myf)-5, which links muscle and brown fat cells and excludes white adipocytes.46, 48 In addition, in vivo lineage tracing has identified muscle and brown adipocytes as the only cell types in which Myf5 promoter is induced.46 This conclusion was confirmed by the conversion of cultured myoblasts into brown adipocytes upon ectopic expression of PRDM16 and the reversal of primary brown adipocytes to myocytes with the knockdown of PRDM16.46 Despite the evidence supporting the theory of the common lineage of muscle/brown adipocytes, it is also clear that a population of distinctive, recruitable Myf5-negative cells is interspersed in the WAT. These cells are probably derived from the activation of dormant precursor cells residing within the WAT in response to environmental cues, such as adrenergic stimulation or cold exposure.46 In humans, depots of WAT are found in different anatomical locations in the body, with subcutaneous and visceral fat depots representing the major compartments for fat storage. The accumulation of intra-abdominal or visceral WAT, in particular, has been reported to be the most strongly correlated with an elevated risk for metabolic dysfunction and cardiovascular disease.49 A transformation from white to brown adipocytes in WAT has been reported in several mouse knockout strains that exhibit resistance against diet-induced obesity and type 2 diabetes.50, 51
Gross examination of the Smad3−/− WAT reveals a beige appearance resembling an intermediate tinge of white and brown. The presence of smaller and multilocular adipocytes with characteristics of brown adipocytes is evident within the milieu of larger unilocular white adipocytes.5 Gene expression profiling confirmed an increase in the transcript levels of brown adipogenic markers, including PPARγ coactivator-1α, UCP-1 and PRDM16 in Smad3−/− WAT.5 Indeed, Smad3 was found to regulate PPARγ coactivator-1α promoter and PRDM16 target genes.5 Ablation of TGF-β/Smad3 signaling in normal WAT consistently induces the mitochondrial, BAT and skeletal muscle-specific transcripts in vitro and in vivo; whereas administration of exogenous TGF-β1 in 3T3-L1 preadipocytes results in elevation in WAT-specific transcripts and a concurrent suppression of BAT/mitochondrial genes.5 Smad3−/− WAT also contained increased mitochondrial DNA copy number, ATP content and mitochondria with densely packed cristae, indicative of enhanced mitochondrial biogenesis and function.5 The physiological relevance of thermogenic activation in Smad3−/− mice is illustrated by their ability to maintain a higher core temperature upon cold exposure and increased oxygen consumption.5 This may provide supports for the conclusion that TGF-β/Smad3 has a crucial role in the recruitment and activation of brown adipocytes in the white fat depots. However, the origin and characteristics of these recruited brown adipocytes have yet to be examined despite microarray analyses of Smad3−/− WAT and anti-TGF-β antibody-treated wild-type WAT have identified genes involved in skeletal muscle biology, such as tropomyosin, troponin and myosin.5 This suggests the recruitment of a myogenic lineage of brown adipocytes that expresses the Myf5 protein. Interestingly, Myf5 protein in the Smad3−/− skeletal muscle was found to be downregulated.38 However, it should be considered that the differential role of TGF-β/Smad3 may be dependent on the tissue-specific context. Moreover, the detailed molecular mechanism by which TGF-β/Smad3 signaling activates the thermogenic program in WAT still remains elusive. The dual effects of Smad3 on both PPARγ2 and PPARβ/δ promoters may provide an explanation for the anti-adipogenic and pro-thermogenic effects of the TGF-β/Smad3 signaling pathway (Figure 4). This is substantiated by the findings that PPARβ/δ controls PPARγ coactivator-1α expression in the skeletal muscle to eradicate obesity and diabetes by inducing a muscle fiber-type switch that favors FA oxidation,36 although the direct regulation of PPARγ coactivator-1α promoter by PPARβ/δ has not been examined in WAT. Another approach that TGF-β/Smad3 may adopt to induce browning in WAT is likely mediated through its interaction with the β-adrenergic system, as indicated by a crosstalk between TGF-β1 and β-adrenergic signaling pathways in cardiac hypertrophy.52 Lastly, the WAT-to-BAT phenotypic transition observed in Smad3−/− mice may be accountable for the increased fat burning capacity, thus providing an alternative therapeutic approach for tackling obesity and diabetes.

Proposed model for WAT-to-BAT phenotypic conversion upon loss of TGF-β/Smad3 signaling in adipocytes. This phenotypic transformation is mediated, at least in part, through the dual effects of Smad3 on both the PPARβ/δ and PPARγ2 promoters. Smad3 deficiency promotes a brown phenotype in the WAT milieu.
Pancreatic function, insulin signaling and glucose metabolism
Pancreatic β-cells are remarkably plastic in their ability to regulate insulin release in response to fluctuations in plasma glucose level. Most obese and insulin-resistant individuals do not initially develop hyperglycemia, as pancreatic β-cells could provisionally compensate for the reduced peripheral insulin sensitivity by increasing the production and release of insulin into the circulation.53 As the liver progressively develops insulin resistance due to hepatic steatosis, hepatic gluconeogenesis is augmented and an additional workload is placed upon the pancreas to increase insulin supply to counter the hyperglycemia. For obesity and insulin resistance to be linked with type 2 diabetes, pancreatic β-cells have to become dysfunctional and decompensated because of the chronic and synergistic effects of gluco- and lipotoxicity on the β cells.54 Although plasma FFA level is critical for normal insulin secretion, chronic exposure of pancreatic β-cells to FFAs in vivo and in vitro is associated with marked reductions in glucose-stimulated insulin secretion and insulin biosynthesis.55, 56 Excessive intramuscular FFA and triglyceride accumulation have also been shown to cause insulin resistance resulting from the adverse effects of chronic lipotoxicity.57 Recent studies have indicated that FFAs induce insulin resistance in skeletal muscle by blocking insulin activation of insulin receptor substrate (IRS)-1-associated phosphatidylinositol 3-kinase via the increased activation of protein kinase C-θ and the downstream phosphorylation of IRS-1 at Ser (307), thereby decreasing the IRS-1 tyrosine phosphorylation and the subsequent IRS-1-associated phosphatidylinositol 3-kinase activity.58 This in turn decreases glucose transporter-4 translocation, followed by decreased insulin-stimulated glucose uptake. Furthermore, the liver is a key regulator of glucose homeostasis because of its ability to store glucose as glycogen postprandially or release glucose via gluconeogenesis and glycogenolysis during starvation. Excessive intraperitoneal fat has been proposed to induce hepatic steatosis and hepatic insulin resistance via an increased influx of FFA from the portal vein directly to the liver.59 In overnight-fasted rats, FFAs were shown to increase basal endogenous glucose production and induce hepatic insulin resistance through an increase in the in vivo activity of glucose-6-phosphatase, an enzyme involved in the rate-limiting step of gluconeogenesis and glycogenolysis, and also through an impairment of in vivo glucokinase (a glycolytic enzyme) activity, which is presumably due to an inhibitory allosteric effect of fatty acyl-CoA (an activated form of FA) on the enzyme activity.60
The loss of Smad3 in mice causes a mild hypoglycemic phenotype in these animals. These mice also exhibit enhanced glucose tolerance and insulin hypersensitivity upon glucose and insulin challenge.4, 5 Hyperinsulinemic-euglycemic clamp studies and ex vivo tissue-specific glucose uptake assays further confirmed that glucose infusion rate required to maintain euglycemia was higher in Smad3−/− mice;4 this was in parallel with an elevated whole-body glucose uptake and an enhanced insulin-mediated suppression of hepatic glucose output, suggesting that Smad3 deficiency results in whole-body enhanced insulin responsiveness in WAT, skeletal muscle and liver (Figure 3).4, 5 Upon administration of a high-fat diet, Smad3−/− mice are resistant to the development of obesity and type 2 diabetes, whereas wild-type mice become obese and develop hyperinsulinemia, hepatic steatosis, hyperglycemia and insulin resistance as detected by glucose tolerance and insulin sensitivity tests.4, 5 These results particularly indicate the role of TGF-β/Smad3 signaling in the pathogenesis of obesity and type 2 diabetes. Consistently, an elevation in protein expression levels of the key mediators of insulin-signaling cascades has also been found in the Smad3−/− WAT and skeletal muscle.4 Previous studies indicate that insulin-mediated activation of protein kinase B/Akt can inhibit TGF-β1 signaling by the formation of a protein kinase B/Smad3 complex.61 This interaction causes the inhibition of Smad3-mediated gene regulation, thereby placing protein kinase B-Smad3 at a point of convergence in the crosstalk between TGF-β/Smad3 and insulin signaling pathways.61 Chronic hyperglycemia can cause tissue injury and one pathological response to tissue injury is fibrosis. Smad3 abrogation is likely to prevent tissue injury and fibrosis through its combined actions against the development of hyperglycemia and fibrotic response in the face of chronic glucose exposure. The recently described role of TGF-β/Smad3 signaling in glucose-induced cell hypertrophy and damage may have ramifications for conditions associated with insulin resistance and diabetes.20 Fibrosis is characterized by extracellular matrix deposition within the injured tissues. The development of liver fibrosis is associated with insulin resistance in hepatitis patients.62 Smad3 has been identified as the main mediator of the TGF-β pro-fibrotic response, especially in cirrhosis and other hepatic fibrotic disorders.15 It is possible that Smad3 partially ameliorates hepatic insulin resistance by preventing glucotoxicity and its associated fibrosis in the liver. Ablation of Smad3 in mice also leads to hyperinsulinemia that may explain for the hypoglycemic phenotype observed in these animals.4, 7 TGF-β/Smad3 signaling is now known to repress insulin gene transcription through direct binding of Smad3 to the insulin gene promoter.7 Therefore, the lack of Smad3-mediated repression of insulin transcription from the pancreatic β-cells results in elevated plasma insulin and mild hypoglycemia. Consistently, the expression level of genes involved in insulin biosynthesis, pro-insulin processing, glucose metabolism, insulin exocytosis and glucose-stimulated insulin secretion was concomitantly upregulated in the Smad3−/− β-cell islets, as supported by enhanced glucose-stimulated insulin secretion ex vivo and in vivo.7 Moreover, TGF-β signaling has been implicated in pancreatic diseases and development, as illustrated by the transgenic mice overexpressing TGF-β1 in the β cells that display abnormal islet formation.63 These findings demonstrate the pivotal role of TGF-β/Smad3 in the regulation of β-cell function and development, especially in the setting of increased insulin demand, such as insulin resistance, diabetes and during β-cell injury. Pharmaceutical inactivation of TGF-β/Smad3 signaling might be useful for promoting β-cell differentiation and ameliorating β-cell dysfunction during diabetes.
Adipocytokine secretion and inflammation
The WAT has traditionally been regarded solely as an energy storage organ that accumulates triglycerides during times of energy abundance and releases FFAs when energy expenditure exceeds energy intake. However, over the last decades, a novel role of adipose tissue as a dynamic endocrine organ has emerged.40 Adipose tissue is now known to secrete a myriad of bioactive molecules that are collectively termed adipocytokines, some of which have been shown to act in either an autocrine, paracrine or endocrine fashion to affect glucose and lipid homeostasis as well as insulin sensitivity.64, 65, 66 Among these adipocytokines, tumor necrosis factor (TNF)-α, interleukin (IL)-6, resistin, visfatin, leptin, monocyte chemoattractant protein (MCP)-1, plasminogen activator inhibitor (PAI)-1, retinol-binding protein-4 and adiponectin have been documented as active modulators of peripheral insulin sensitivity and implicated in the development of insulin resistance.67 Adipocytokine secretion from adipose tissue is perturbed during obesity, which in turn is associated with diseases such as metabolic syndrome, insulin resistance and cardiovascular disease.67, 68, 69 The mechanism by which adipocytokines influence insulin resistance in obesity is still not fully understood. However, accumulating evidence suggests that obesity alters the amount and spectrum of adipocytokines that are secreted from adipose tissue.69 Indeed, a differential expression of pro- and anti-inflammatory factors has been associated with the sizes of adipocytes, whereby dysregulation of hypertrophic adipocytes causes a shift toward dominance of pro-inflammatory adipocytokines.70 These pro-inflammatory adipocytokines secreted from the hypertrophic adipocytes may be involved in the development of a chronic low-grade inflammatory state, which is considered to be the ‘common soil’ for the pathogenesis of metabolic syndrome.70 Adipose tissue from the obese subjects secretes more pro-inflammatory cytokines, such as TNF-α, IL-6 and MCP-1, as evidenced by their elevated concentrations in the circulation.66, 71 Moreover, serum retinol-binding protein-4 levels are elevated in insulin-resistant mice and humans with obesity and type 2 diabetes.72 Retinol-binding protein signaling cascade also induces the expression of STAT target genes, such as suppressor of cytokine signaling-3, which inhibits insulin signaling.73 An increase in the infiltration of macrophages in WAT has also been reported in obesity, which partly contributes to the pro-inflammatory cytokines secreted from the adipose tissue.66 Obesity has been demonstrated to induce infiltration of macrophages with distinct gene expression characteristics and cytokine secretion spectra to adipose tissue, thereby inducing a phenotypic switch in the activation state of adipose tissue macrophages from an anti-inflammatory M2-polarized state.74, 75 This state protects adipocytes from progression to an M1 pro-inflammatory state, which contributes to insulin resistance.74, 75 Particularly, adipose tissue macrophages from lean mice express many genes that are characteristic of the protective M2 or ‘alternatively activated’ macrophages, including Ym1, arginase 1 and IL-10; whereas those from diet-induced obese mice express genes that are characteristic of the pro-inflammatory M1 or ‘classically activated’ macrophages, such as TNF-α and IL-6 that promote inflammation.74
Whole-body deletion of Smad3 in mice induces a lean phenotype and enhanced insulin sensitivity regardless of feeding regimen.4, 5 Smad3−/− mice exhibit less inflammatory macrophage infiltration into the WAT, with a switch in macrophage spectrum from inflammatory M1 to protective M2 macrophages.5 Smad3−/− WAT also shows augmented expression of insulin-mimetic visfatin with a concomitant reduction in the expression of resistin, PAI-1 and pro-inflammatory cytokines, such as TNF-α, IL-6 and MCP-1 in WAT as well as in the circulation.4, 5 Visfatin is predominantly produced and secreted by the visceral WAT. Visfatin regulates insulin secretion, insulin receptor signaling and mRNA expression of diabetes-related genes in mouse pancreatic β-cells.76 Human resistin, on the contrary, has been found to impair insulin-mediated glucose transport by reducing the intrinsic activity of cell-surface glucose transporters and stimulating the production and secretion of the pro-inflammatory cytokines TNF-α and IL-12 in macrophages via an NF-κB-dependent pathway.77, 78 Furthermore, increased TNF-α production has been observed in adipose tissue isolated from obese rodents and human subjects.79 Multiple mechanisms have been proposed to account for the adverse effects of TNF-α on insulin sensitivity; these include its direct inhibitory action on insulin signaling, induction of elevated FFA via stimulation of lipolysis and negative regulation of adipocyte differentiation.79 IL-6 is another adipocytokine similar to TNF-α with regards to its deleterious effects on insulin signaling in the liver, skeletal muscles and adipocytes via ubiquitin-mediated degradation of IRS through suppressor of cytokine signaling-1 and suppressor of cytokine signaling-3.80, 81
As discussed previously, infiltration of macrophages into adipose tissue is an important determinant of the increased inflammatory process in obesity. Adipocytes secrete an array of chemoattractants that draw monocytes from the circulation into the adipose tissue. MCP-1, also known as chemokine (C–C motif) ligand (CCL)-2, is one of the chemoattractants secreted by adipocytes that has a pivotal role in macrophage recruitment. Indeed, elevated plasma MCP-1 and its overexpression in adipose tissue have been found in obese humans and subjects with insulin resistance.71, 82 Mice lacking MCP-1 or its receptor (CCR-2) exhibit reduced adipose tissue macrophage infiltration and ameliorated insulin resistance and hepatic steatosis.66, 83 Although there is no evidence supporting the notion that TGF-β/Smad3 signaling directly modulates MCP-1 expression in adipose tissue, MCP-1 has been previously shown to be a direct gene target of TGF-β by Smad3/4 in endothelial cells.84 PAI-1 gene expression, which is governed by TGF-β through the binding of phosphorylated Smad2/3 to its promoter, is also highly induced in subjects with metabolic complications of obesity.85, 86 PAI-1 acts as an inhibitor of fibrinolysis by inhibiting both tissue-type and urokinase-type plasminogen activators through its serine protease inhibitor function. Its aberrant expression in obesity has a detrimental effect on cardiovascular function, leading to thrombosis and ischemic diseases. The expression of leptin in WAT of Smad3−/− mice was also found to be downregulated when compared with their wild-type counterparts despite similar caloric intake between these mice.5 Leptin has a key role in energy homeostasis by binding to receptors in the hypothalamus of the brain where it inhibits appetite and induces the feeling of satiety.87 Further, genetic abnormalities in leptin or its receptors in rodents revealed obesity-related deficits in macrophage phagocytosis and the expression of pro-inflammatory cytokines both in vivo and in vitro, whereas treatment with exogenous leptin rescued these deficits in inflammation.88 The mechanism by which TGF-β/Smad3 signaling affects leptin promoter activity has not been elucidated in vivo, although an in vitro study reported an opposite and inhibitory action of TGF-β/Smad3 on leptin promoter mediated by sequestration of C/EBPβ.28 Moreover, it is noteworthy to highlight that adiponectin may also contribute to the antidiabetic effects of Smad3−/− mice, although its plasma concentration and expression level in Smad3−/− WAT have yet to be determined. This hypothesis is supported by the findings that FFAs impair adiponectin bioactivity in hepatocytes via TGF-β-mediated pathway and that smaller adipocytes (as seen in Smad3−/− mice) secrete more adiponectin than do larger cells.89, 90 Adiponectin improves insulin sensitivity by increasing energy expenditure and FA oxidation through the activation of AMP-activated protein kinase and by increasing the expression of PPARα target genes, such as CD36, ACOX and UCP -2.91 Indeed, the mRNA transcript and protein levels of ACOX and UCP-2 have been shown to be markedly upregulated in the WAT of Smad3−/− mice.4 Interestingly, the TGF-β/Smad3 signaling pathway has been reported to directly suppress PPARα activity to reduce FA oxidation in cardiac myocytes.92 It is plausible that augmented FA β-oxidation in the WAT of Smad3−/− mice may act through an AMP-activated protein kinase- and/or PPARα-mediated mechanism by the autocrine effect of adiponectin. However, except for PAI-1 and leptin, no results have shown that the TGF-β/Smad3 signaling pathway directly regulates the production and secretion of these adipocytokines in adipose tissue. Moreover, it is still unclear whether the protective effects of Smad3 gene deletion or anti-TGF-β antibody treatment for insulin resistance could be an immediate effect because of a change in adipocytokine secretion as a consequence of altered infiltration of macrophages or reduced adiposity (Figure 5).

Elevated TGF-β/Smad3 signaling causes adipocyte hypertrophy, hyperplasia and elevated pro-inflammatory cytokines production. These cytokines interfere with insulin signaling and promote the recruitment of M1 pro-inflammatory macrophages to the WAT. Smad3 deficiency ameliorates the chronic low-grade inflammation and insulin resistance by recruiting M2 protective macrophages and reducing pro-inflammatory cytokines expression from the adipocytes. Smad3 deficiency also upregulates the mRNA level of insulin-mimetic visfatin while decreasing resistin (insulin-desentizer).
TGF-β/Smad3 as the link between ROS and insulin resistance
ROS are generated as byproducts of normal cellular metabolism and have important roles in cellular signaling and homeostasis.93 In times of environmental stress (e.g., exposure to ultraviolet radiation, heat exposure and hypoxia), ROS levels can be dramatically increased. ROS are highly reactive because of the presence of unpaired valence shell electrons, and this accounts for their damaging effects on cellular molecules in a situation known as oxidative stress. ROS are derived from the mitochondrial respiration and the activity of NADPH oxidases and their intracellular levels are constantly kept in check by the activity of endogenous antioxidant enzymes such as Cu/Zn superoxide dismutase (SOD), MnSOD and catalase, which catalyze the dismutation of superoxide into oxygen and H2O2 and the conversion of H2O2 to water and oxygen. ROS are among the many factors that have previously been suggested to have a deleterious effect on insulin resistance.94 It is based on the evidence that oxidative stress markers are associated with obesity and diabetes in humans,95, 96 and insulin resistance is induced in 3T3-L1 adipocytes upon treatment with H2O2 and ROS-inducing agents.97 The expression of oxidative stress markers has also been reported in Lepob/ob mice with insulin resistance and glucose intolerance.98 Additionally, chemical inhibitors of NADPH oxidase can improve glucose homeostasis in an obese mouse model.96 Metabolic studies also demonstrate that individuals with primary defects that affect the ROS balance, such as familiar amyotrophic lateral sclerosis, develop insulin resistance.99 ROS are now considered to have a causal role in the etiology of insulin resistance.100 An in vitro study showed that ROS were markedly elevated in TNFα-induced insulin resistance in 3T3-L1 adipocytes, and this increase in ROS preceded the onset of insulin resistance.100 In contrast, administration of antioxidant N-acetylcysteine revealed an insulin-sensitizing effect of this drug that leads to a dose-dependent suppression of insulin resistance as determined by insulin-stimulated glucose uptake in the cultured adipocytes.100 Moreover, cells carrying the transgenes encoding ROS-scavenging enzymes Cu/ZnSOD, MnSOD and catalase exhibited reduced TNFα-induced insulin resistance.100 Little information is available regarding the downstream pathways that translate elevated ROS into insulin resistance. Studies indicate that c-Jun amino-terminal kinase (JNK) signaling may be an attractive candidate, as JNK activity is abnormally elevated by oxidative stress, inflammatory cytokines and FFAs as well as in obesity.101, 102 JNK has been shown to interfere with insulin signaling through its direct association with IRS-1 and its subsequent phosphorylation inhibition.103 Inhibition of JNK activity through genetic knockout improves insulin sensitivity in mouse models of obesity.101
JNK and Smad3 cascades are essential components of the TGF-β signaling machinery and have been implicated in common transcriptional responses.104 JNK has also been shown to induce Smad3 phosphorylation at the site outside of its Ser-Ser-Val-Ser motif.104 This interdependent relationship between JNK and Smad3 in mediating TGF-β response suggests that JNK may be the intermediate player between TGF-β and Smad3 that links ROS to the development of insulin resistance and type 2 diabetes. Further, TGF-β has previously been shown to trigger intracellular ROS release via redox- and Smad3-dependent upregulation of NADPH oxidase 4 while inhibiting the expression of MnSOD and catalase in asthmatic and obstructive airway smooth muscle.105 TGF-β also mediates hepatocyte apoptosis through Smad3 generation of ROS.106 The preponderance of evidence indicates that TGF-β/Smad3 signaling has a major role in the regulation of ROS production and its breakdown. However, there is no direct evidence to support the claim that TGF-β/Smad3 regulates ROS production to cause insulin resistance and type 2 diabetes. Hypoxia may present another link between TGF-β and ROS in insulin resistance. The rise in adipose mass during obesity may result in hypoxia in clusters of adipocytes distant from the vasculature, triggering inflammation serving to increase blood flow via angiogenesis. The response of TGF-β to hypoxia is mediated through direct binding of Smad3 with hypoxia-inducible factor-1α.107 It is plausible that the hypoxic, overly expanded adipocytes trigger ROS production either via a direct or a TGF-β-mediated mechanism that ultimately leads to insulin resistance. The explanation behind the protection against insulin resistance and type 2 diabetes in Smad3−/− mice could partially be due to the decreased FFA and glucose, the small adipocyte size and the reduced levels of inflammation that directly affect the production of ROS. Indeed, increased FFA and glucose can increase ROS production and oxidative stress.96, 97 ROS are likely to influence insulin signaling in WAT, skeletal muscle and liver. Further research will be needed to test this possibility.
TGF-β/Smad3 signaling as antidiabetic drug target
There are currently four different classes of antidiabetic drugs available in the market: sulfonylureas, biguanides, acarbose and thiazolidinediones (TZD). These drugs adopt several distinctive approaches to deploy their antidiabetic effects, including stimulation of insulin production from the pancreatic β-cells (sulfonylureas), inhibition of hepatic gluconeogenesis (biguanides), reduction of intestinal glucose digestion and absorption (acarbose) and elimination of ectopic lipid build up in muscle and liver by enhancing fat storage (TZD). Acarbose inhibits intestinal α-glucosidase and pancreatic α-amylase to prevent digestion of complex carbohydrates, thereby disrupting their subsequent absorption in the small intestine. Meanwhile, sulfonylureas bind to the ATP-depedent K+ channels located on the plasma membrane of pancreatic β-cells to induce insulin secretion from the pancreatic β-cells. However, sulfonylureas are ineffective in patients who have deficits in insulin production, such as in type 1 diabetes and post-pancreatectomy. The side effects of acarbose and sulfonylureas preclude their wide use in the treatment of diabetes; these include hepatitis, cardiovascular risk and potential teratogenicity. For these reasons, the biguanide class of antidiabetic drugs, such as metformin, has been the first-line drug of choice and the most widely prescribed drug for the treatment of type 2 diabetes despite its mild side effects. Metformin improves hyperglycemia primarily through suppression of hepatic gluconeogenesis mediated by AMP-activated protein kinase activation.108 TZD, which include troglitazone, rosiglitazone and pioglitazone, have been shown to lower blood glucose level and improve insulin sensitivity by promoting lipid storage in adipose tissue through the activation of PPARγ.109 Nonetheless, treatment with TZD in diabetic individuals often results in weight gain, although plasma FFA and peripheral ectopic lipid accumulation are reduced.110 An increased risk of bladder cancer risk among diabetic patients treated with pioglitazone has also been reported in a longitudinal cohort study.111 Furthermore, the withdrawal of troglitazone and rosiglitazone from the market in United States and Europe, respectively, due to an increased incidence of hepatitis and cardiovascular events has raised concerns over the use of other TZDs.
Although most of the current obesity therapies aim to reduce caloric intake, much attention is now focused on an alternative strategy of increasing cellular energy expenditure. Because of the presence of a host of undesirable effects associated with the use of currently available antidiabetic drugs, there is a demand for a better therapeutic target for the treatment of type 2 diabetes. Targeting TGF-β/Smad3 signaling may represent a more holistic approach in tackling these conditions (Figure 6). Indeed, Lepob/ob and diet-induced obese mice treated with anti-TGF-β neutralizing antibody exhibit weight loss, enhanced glucose tolerance and insulin sensitivity, suppression of hepatic steatosis and reduced blood glucose level, ultimately resulting in protection against obesity and type 2 diabetes.5 The beneficial effects of TGF-β antibody treatment rely on its ability to transform energy-storing WAT into energy-dissipating BAT in obese and diabetic mouse models.5 Clinical use of TGF-β antagonist is now being evaluated for the treatment of fibrosis, scarring, cancer and diabetic nephropathy where increased TGF-β are implicated. It is likely that attention will be focused on their application in diabetes treatment. Notably, a specific inhibitor of Smad3 (SIS3) has recently been characterized as a potent and selective inhibitor of Smad3 function in vitro.112 Immunoprecipitation revealed that SIS3 specifically attenuated the TGF-β1-induced phosphorylation of Smad3 and its interaction with Smad4 in a dose-dependent manner whereas Smad2 phosphorylation remained unaltered. The biological relevance of SIS3 in Smad3 inhibition was further confirmed by evaluation of its ability to suppress the TGF-β1-induced type I pro-collagen upregulation and myofibroblast differentiation in human dermal and scleroderma fibroblasts.113 Although SIS3 may seem to be a promising candidate for drug development for diabetes treatment, its effects on glucose and lipid metabolism remain elusive in vivo. Further effort should also be focused on target specification, as WAT is the primary target responsible for the antidiabetic effects of TGF-β/Smad3 antagonism. Particularly, Smad3 deletion in mice causes muscle atrophy and defective immune response,13, 38 which may represent as off-target effects if specification of the TGF-β/Smad3 antagonist is compromised. Gene therapy using short-hairpin RNA to specifically target WAT may represent an attractive approach to circumvent this problem. In support of this possibility, suppression of Smad3 expression using lentivirus-based short-hairpin RNA technology induces a WAT-to-BAT phenotypic switch and thermogenic program in adipocyte cell lines.5 Losartan, an angiotensin II receptor type 1 receptor antagonist, is another interesting candidate for antidiabetic drug development targeting TGF-β. Losartan has been shown to decrease plasma TGF-β1 in transplant patients with chronic allograft nephropathy and attenuate the development of obesity and insulin resistance in diet-induced obese mice probably mediated through downregulation of PAI-1 (a target gene of TGF-β/Smad3 signaling).113, 114, 115, 116, 117, 118, 119, 120

Summary of the whole-body effects of TGF-β/Smad3 signaling in obesity-linked diabetes and how blockage of TGF-β/Smad3 signaling ameliorates obesity and type 2 diabetes.
Conclusion
In summary, TGF-β/Smad3 signaling regulates a wide range of events involved in metabolism and the pathogenesis of obesity and type 2 diabetes. Smad3−/− mice provide an invaluable tool for the elucidation of molecular mechanisms behind the diverse roles of TGF-β1 in adipocyte differentiation, lipid and glucose metabolism, white to brown fat transformation, pancreatic insulin production, adipocytokine secretion, inflammatory cell recruitment and ROS production (Figure 7). All of these processes have profound impacts on the etiology of obesity and its associated complications. Our knowledge on the mechanistic actions of TGF-β/Smad3 signaling will certainly allow us to dissect and understand the complicated processes involved during the development of obesity and diabetes. The main theme behind the recent findings suggests that TGF-β/Smad3 signaling may represent a common site of intersection between the various pathologies that comprise metabolic syndrome. Turning the ‘bad’ fat (fat-storing white adipocytes) into ‘good’ fat (fat-burning brown adipocytes) will inevitably present a novel unifying approach for the treatment of all facets of obesity and diabetes. The availability of Smad3-specific inhibitors most certainly holds the promise of lessening the burden of global epidemic of obesity and diabetes.

Proposed unifying molecular mechanisms by which TGF-β/Smad3 signaling regulates adipogenesis, FA β-oxidation, thermogenesis and white-to-brown fat transformation by its pleiotropic effects on the expression of PPARs. Numbers in bracket indicate the relevant references.
References
Kopelman PG, Albon L . Obesity, non-insulin-dependent diabetes mellitus and the metabolic syndrome. Br Med Bull 1997; 53: 322–340.
Wild S, Roglic G, Green A, Sicree R, King H . Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 2004; 27: 1047–1053.
Kahn SE, Hull RL, Utzschneider KM . Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006; 444: 840–846.
Tan CK, Leuenberger N, Tan MJ, Yan YW, Chen Y, Kambadur R et al. Smad3 deficiency in mice protects against insulin resistance and obesity induced by a high-fat diet. Diabetes 2011; 60: 464–476.
Yadav H, Quijano C, Kamaraju AK, Gavrilova O, Malek R, Chen W et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab 2011; 14: 67–79.
Tsurutani Y, Fujimoto M, Takemoto M, Irisuna H, Koshizaka M, Onishi S et al. The roles of transforming growth factor-β and Smad3 signaling in adipocyte differentiation and obesity. Biochem Biophys Res Commum 2011; 407: 68–73.
Lin HM, Lee JH, Yadav H, Kamaraju AK, Liu E, Zhigang D et al. Transforming growth factor-beta/Smad3 signaling regulates insulin gene transcription and pancreatic islet beta-cell function. J Biol Chem 2009; 284: 12246–12257.
Massagué J, Blain SW, Lo RS . TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 2000; 103: 295–309.
Shi Y, Massagué J . Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 2003; 113: 685–700.
Mori S, Matsuzaki K, Yoshida K, Furukawa F, Tahashi Y, Yamagata H et al. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene 2004; 23: 7416–7429.
Brown KA, Pietenpol JA, Moses HL . A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-beta signaling. J Cell Biochem 2007; 101: 9–33.
Heyer J, Escalante-Alcalde D, Lia M, Boettinger E, Edelmann W, Stewart CL et al. Postgastrulation Smad2-deficient embryos show defects in embryo turning and anterior morphogenesis. Proc Natl Acad Sci USA 1999; 96: 12595–12600.
Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J 1999; 18: 1280–1291.
Ashcroft GS, Mills SJ, Flanders KC, Lyakh LA, Anzano MA, Gilliver SC et al. Role of Smad3 in the hormonal modulation of in vivo wound healing responses. Wound Repair Regen 2003; 11: 468–473.
Flanders KC . Smad3 as a mediator of the fibrotic response. Int J Exp Pathol 2004; 85: 47–64.
Alessi MC, Bastelica D, Morange P, Berthet B, Leduc I, Verdier M et al. Plasminogen activator inhibitor 1, transforming growth factor-beta 1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes 2000; 49: 1374–1380.
Gordon KJ, Blobe GC . Role of transforming growth factor-beta superfamily signaling pathways in human disease. Biochim Biophys Acta 2008; 1782: 197–228.
Herder C, Zierer A, Koenig W, Roden M, Meisinger C, Thorand B . Transforming growth factor-beta1 and incident type 2 diabetes: results from the MONICA/KORA case-cohort study, 1984-2002. Diabetes Care 2009; 32: 1921–1923.
Perry JR, McCarthy MI, Hattersley AT, Zeggini E, Weedon MN, Frayling TM . Interrogating type 2 diabetes genome-wide association data using a biological pathway-based approach. Diabetes 2009; 58: 1463–1467.
Wu L, Derynck R . Essential role of TGF-beta signaling in glucose-induced cell hypertrophy. Dev Cell 2009; 17: 35–48.
Rosmond R, Chagnon M, Bouchard C, Björntorp P . Increased abdominal obesity, insulin and glucose levels in nondiabetic subjects with a T29C polymorphism of the transforming growth factor-beta1 gene. Horm Res 2003; 59: 191–194.
Grainger DJ, Heathcote K, Chiano M, Snieder H, Kemp PR, Metcalfe JC et al. Genetic control of the circulating concentration of transforming growth factor type beta1. Hum Mol Genet 1999; 8: 93–97.
Yu SK, Kwon OS, Jung HS, Bae KS, Kwon KA, Kim YK et al. Influence of transforming growth factor-beta1 gene polymorphism at codon 10 on the development of cirrhosis in chronic hepatitis B virus carriers. J Korean Med Sci 2010; 25: 564–569.
Sharma K, Ziyadeh FN, Alzahabi B, McGowan TA, Kapoor S, Kurnik BR et al. Increased renal production of transforming growth factor-beta1 in patients with type II diabetes. Diabetes 1997; 46: 854–859.
Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci USA 2000; 97: 8015–8020.
Han DC, Hoffman BB, Hong SW, Guo J, Ziyadeh FN . Therapy with antisense TGF-beta1 oligodeoxynucleotides reduces kidney weight and matrix mRNAs in diabetic mice. Am J Physiol Renal Physiol 2000; 278: F628–F634.
Rosen ED, Hsu CH, Wang X, Sakai S, Freeman MW, Gonzalez FJ et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev 2002; 16: 22–26.
Choy L, Derynck R . Transforming growth factor-beta inhibits adipocyte differentiation by Smad3 interacting with CCAAT/enhancer-binding protein (C/EBP) and repressing C/EBP transactivation function. J Biol Chem 2003; 278: 9609–9619.
Marchildon F, St-Louis C, Akter R, Roodman V, Wiper-Bergeron NL . Transcription factor Smad3 is required for the inhibition of adipogenesis by retinoic acid. J Biol Chem 2010; 285: 13274–13284.
Berry DC, Soltanian H, Noy N . Repression of cellular retinoic acid-binding protein II during adipocyte differentiation. J Biol Chem 2010; 285: 15324–15332.
Imai T, Takakuwa R, Marchand S, Dentz E, Bornert JM, Messaddeq N et al. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc Natl Acad Sci USA 2004; 101: 4543–4547.
Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H et al. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell 2003; 113: 159–170.
Ron D, Habener JF . CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev 1992; 6: 439–453.
Medina-Gomez G, Gray SL, Yetukuri L, Shimomura K, Virtue S, Campbell M et al. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet 2007; 3: e64.
Jacob S, Machann J, Rett K, Brechtel K, Volk A, Renn W et al. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes 1999; 48: 1113–1119.
Schuler M, Ali F, Chambon C, Duteil D, Bornert JM, Tardivel A et al. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab 2006; 4: 407–414.
Luquet S, Lopez-Soriano J, Holst D, Fredenrich A, Melki J, Rassoulzadegan M et al. Peroxisome proliferator-activated receptor delta controls muscle development and oxidative capability. FASEB J 2003; 17: 2299–2301.
Ge X, McFarlane C, Vajjala A, Lokireddy S, Ng ZH, Tan CK et al. Smad3 signaling is required for satellite cell function and myogenic differentiation of myoblasts. Cell Res 2011; 21: 1591–1604.
Pearen MA, Ryall JG, Lynch GS, Muscat GE . Expression profiling of skeletal muscle following acute and chronic beta2-adrenergic stimulation: implications for hypertrophy, metabolism and circadian rhythm. BMC Genomics 2009; 10: 448.
Ahima RS, Flier JS . Adipose tissue as an endocrine organ. Trends Endocrinol Metab 2000; 11: 327–332.
Escher P, Braissant O, Basu-Modak S, Michalik L, Wahli W, Desvergne B . Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology 2001; 142: 4195–4202.
Vidal-Puig A, Jimenez-Liñan M, Lowell BB, Hamann A, Hu E, Spiegelman B et al. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest 1996; 97: 2553–2561.
Kruszynska YT, Mukherjee R, Jow L, Dana S, Paterniti JR, Olefsky JM . Skeletal muscle peroxisome proliferator-activated receptor-gamma expression in obesity and non-insulin-dependent diabetes mellitus. J Clin Invest 1998; 101: 543–548.
Vidal-Puig AJ, Considine RV, Jimenez-Liñan M, Werman A, Pories WJ, Caro JF et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest 1997; 99: 2416–2422.
Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M et al. Transcriptional control of brown fat determination by PRDM16. Cell Metab 2007; 6: 38–54.
Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008; 454: 961–967.
Atit R, Sgaier SK, Mohamed OA, Taketo MM, Dufort D, Joyner AL et al. Beta-catenin activation is necessary and sufficient to specify the dorsal dermal fate in the mouse. Dev Biol 2006; 296: 164–176.
Enerbäck S . The origins of brown adipose tissue. N Engl J Med 2009; 360: 2021–2023.
Wang Y, Rimm EB, Stampfer MJ, Willett WC, Hu FB . Comparison of abdominal adiposity and overall obesity in predicting risk of type 2 diabetes among men. Am J Clin Nutr 2005; 81: 555–563.
Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest 2011; 121: 96–105.
Scim A, Grenier G, Huh MS, Gillespie MA, Bevilacqua L, Harper ME et al. Rb and p107 regulate preadipocyte differentiation into white versus brown fat through repression of PGC-1alpha. Cell Metab 2005; 2: 283–295.
Rosenkranz S, Flesch M, Amann K, Haeuseler C, Kilter H, Seeland U et al. Alterations of beta-adrenergic signaling and cardiac hypertrophy in transgenic mice overexpressing TGF-beta(1). Am J Physiol Heart Circ Physiol 2002; 283: H1253–H1262.
Kahn SE, Prigeon RL, McCulloch DK, Boyko EJ, Bergman RN, Schwartz MW et al. Quantification of the relationship between insulin sensitivity and beta-cell function in human subjects. Evidence for a hyperbolic function. Diabetes 1993; 42: 1663–1672.
Tripathy D, Eriksson KF, Orho-Melander M, Fredriksson J, Ahlqvist G, Groop L . Parallel manifestation of insulin resistance and beta cell decompensation is compatible with a common defect in Type 2 diabetes. Diabetologia 2004; 47: 782–793.
Sako Y, Grill VE . A 48-hour lipid infusion in the rat time-dependently inhibits glucose-induced insulin secretion and B cell oxidation through a process likely coupled to fatty acid oxidation. Endocrinology 1990; 127: 1580–1589.
Zhou YP, Grill VE . Long-term exposure of rat pancreatic islets to fatty acids inhibits glucose-induced insulin secretion and biosynthesis through a glucose fatty acid cycle. J Clin Invest 1994; 93: 870–876.
Petersen KF, Shulman GI . Pathogenesis of skeletal muscle insulin resistance in type 2 diabetes mellitus. Am J Cardiol 2002; 90: 11G–18G.
Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem 2002; 277: 50230–50236.
Björntorp P . ‘Portal’ adipose tissue as a generator of risk factors for cardiovascular disease and diabetes. Arteriosclerosis 1990; 10: 493–496.
Lam TK, van de Werve G, Giacca A . Free fatty acids increase basal hepatic glucose production and induce hepatic insulin resistance at different sites. Am J Physiol Endocrinol Metab 2003; 284: E281–E290.
Conery AR, Cao Y, Thompson EA, Townsend Jr CM, Ko TC, Luo K . Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol 2004; 6: 366–372.
Taura N, Ichikawa T, Hamasaki K, Nakao K, Nishimura D, Goto T et al. Association between liver fibrosis and insulin sensitivity in chronic hepatitis C patients. Am J Gastroenterol 2006; 101: 2752–2759.
Lee MS, Gu D, Feng L, Curriden S, Arnush M, Krahl T et al. Accumulation of extracellular matrix and developmental dysregulation in the pancreas by transgenic production of transforming growth factor-beta 1. Am J Pathol 1995; 147: 42–52.
Kim S, Moustaid-Moussa N . Secretory, endocrine and autocrine/paracrine function of the adipocyte. J Nutr 2000; 130: 3110S–3115S.
Hotamisligil GS, Shargill NS, Spiegelman BM . Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 1993; 259: 87–91.
Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante Jr AW . Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 2003; 112: 1796–1808.
Rabe K, Lehrke M, Parhofer KG, Broedl UC . Adipokines and insulin resistance. Mol Med 2008; 14: 741–751.
Guzik TJ, Mangalat D, Korbut R . Adipocytokines - novel link between inflammation and vascular function? J Physiol Pharmacol 2006; 57: 505–528.
Rasouli N, Kern PA . Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab 2008; 93: S64–S73.
Skurk T, Alberti-Huber C, Herder C, Hauner H . Relationship between adipocyte size and adipokine expression and secretion. J Clin Endocrinol Metab 2007; 92: 1023–1033.
Di Gregorio GB, Yao-Borengasser A, Rasouli N, Varma V, Lu T, Miles LM et al. Expression of CD68 and macrophage chemoattractant protein-1 genes in human adipose and muscle tissues: association with cytokine expression, insulin resistance, and reduction by pioglitazone. Diabetes 2005; 54: 2305–2313.
Yang Q, Graham TE, Mody N, Preitner F, Peroni OD, Zabolotny JM et al. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005; 436: 356–362.
Berry DC, Jin H, Majumdar A, Noy N . Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc Natl Acad Sci USA 2011; 108: 4340–4345.
Lumeng CN, Bodzin JL, Saltiel AR . Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 2007; 117: 175–184.
Patsouris D, Li PP, Thapar D, Chapman J, Olefsky JM, Neels JG . Ablation of CD11c-positive cells normalizes insulin sensitivity in obese insulin resistant animals. Cell Metab 2008; 8: 301–309.
Brown JE, Onyango DJ, Ramanjaneva M, Conner AC, Patel ST, Dunmore SJ et al. Visfatin regulates insulin secretion, insulin receptor signaling and mRNA expression of diabetes-related genes in mouse pancreatic beta-cells. J Mol Endocrinol 2010; 44: 171–178.
Kusminski CM, McTernan PG, Kumar S . Role of resistin in obesity, insulin resistance and Type II diabetes. Clin Sci (Lond) 2005; 109: 243–256.
Silswal N, Singh AK, Aruna B, Mukhopadhyay S, Ghosh S, Ehtesham NZ . Human resistin stimulates the pro-inflammatory cytokines TNF-alpha and IL-12 in macrophages by NF-kappaB-dependent pathway. Biochem Biophys Res Commun 2005; 334: 1092–1101.
Moller DE . Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab 2000; 11: 212–217.
Emanuelli B, Peraldi P, Filloux C, Sawka-Verhelle D, Hilton D, Van Obberghen E . SOCS-3 is an insulin-induced negative regulator of insulin signaling. J Biol Chem 2000; 275: 15985–15991.
Kristiansen OP, Mandrup-Poulsen T . Interleukin-6 and diabetes: the good, the bad, or the indifferent? Diabetes 2005; 54 (Suppl 2): S114–S124.
Sartipy P, Loskutoff DJ . Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci USA 2003; 100: 7265–7270.
Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 2006; 116: 1494–1505.
Ma J, Wang Q, Fei T, Han JD, Chen YG . MCP-1 mediates TGF-beta-induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood 2007; 109: 987–994.
Alessi MC, Peiretti F, Morange P, Henry M, Nalbone G, Juhan-Vague I . Production of plasminogen activator inhibitor 1 by human adipose tissue: possible link between visceral fat accumulation and vascular disease. Diabetes 1997; 46: 860–867.
Song CZ, Siok TE, Gelehrter TD . Smad4/DPC4 and Smad3 mediate transforming growth factor-beta (TGF-beta) signaling through direct binding to a novel TGF-beta-responsive element in the human plasminogen activator inhibitor-1 promoter. J Biol Chem 1998; 273: 29287–29290.
Ahima RS, Prabakaran D, Mantzoros C, Qu D, Lowell B, Maratos-Flier E et al. Role of leptin in the neuroendocrine response to fasting. Nature 1996; 382: 250–252.
Fernández-Riejos P, Najib S, Santos-Alvarez J, Martín-Romero C, Pérez-Pérez A, González-Yanes C et al. Role of leptin in the activation of immune cells. Mediators Inflamm 2010; 2010: 568343.
Wanninger J, Neumeier M, Hellerbrand C, Schacherer D, Bauer S, Weiss TS et al. Lipid accumulation impairs adiponectin-mediated induction of activin A by increasing TGFbeta in primary human hepatocytes. Biochim Biophys Acta 2011; 1811: 626–633.
Bahceci M, Gokalp D, Bahceci S, Tuzcu A, Atmaca S, Arikan S . The correlation between adiposity and adiponectin, tumor necrosis factor alpha, interleukin-6 and high sensitivity C-reactive protein levels. Is adipocyte size associated with inflammation in adults? J Endocrinol Invest 2007; 30: 210–214.
Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K . Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest 2006; 116: 1784–1792.
Sekiguchi K, Tian Q, Ishiyama M, Burchfield J, Gao F, Mann DL et al. Inhibition of PPAR-alpha activity in mice with cardiac-restricted expression of tumor necrosis factor: potential role of TGF-beta/Smad3. Am J Physiol Heart Circ Physiol 2007; 292: H1443–H1451.
Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J . Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 2007; 39: 44–84.
Afanas’ev I . Signaling of reactive oxygen and nitrogen species in Diabetes mellitus. Oxid Med Cell Longev 2010; 3: 361–373.
Urakawa H, Katsuki A, Sumida Y, Gabazza EC, Murashima S, Morioka K et al. Oxidative stress is associated with adiposity and insulin resistance in men. J Clin Endocrinol Metab 2003; 88: 4673–4676.
Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest 2004; 114: 1752–1761.
Lin Y, Berg AH, Iyengar P, Lam TK, Giacca A, Combs TP et al. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species. J Biol Chem 2005; 280: 4617–4626.
Yang S, Zhu H, Li Y, Lin H, Gabrielson K, Trush MA et al. Mitochondrial adaptations to obesity-related oxidant stress. Arch Biochem Biophys 2000; 378: 259–268.
Ferrante RJ, Browne SE, Shinobu LA, Bowling AC, Baik MJ, MacGarvey U et al. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem 1997; 69: 2064–2074.
Houstis N, Rosen ED, Lander ES . Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006; 440: 944–948.
Hirosumi J, Tuncman G, Chang L, Görgün CZ, Uysal KT, Maeda K et al. A central role for JNK in obesity and insulin resistance. Nature 2002; 420: 333–336.
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M . Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005; 120: 649–661.
Lee YH, Giraud J, Davis RJ, White MF . c-Jun N-terminal kinase (JNK) mediates feedback inhibition of the insulin signaling cascade. J Biol Chem 2003; 278: 2896–2902.
Engel ME, McDonnell MA, Law BK, Moses HL . Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem 1999; 274: 37413–37420.
Michaeloudes C, Sukkar MB, Khorasani NM, Bhavsar PK, Chung KF . TGF-β regulates Nox4, MnSOD and catalase expression, and IL-6 release in airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2011; 300: L295–L304.
Black D, Lyman S, Qian T, Lemasters JJ, Rippe RA, Nitta T et al. Transforming growth factor beta mediates hepatocyte apoptosis through Smad3 generation of reactive oxygen species. Biochimie 2007; 89: 1464–1473.
Zhang H, Akman HO, Smith EL, Zhao J, Murphy-Ullrich JE, Batuman OA . Cellular response to hypoxia involves signaling via Smad proteins. Blood 2003; 101: 2253–2260.
Collier CA, Bruce CR, Smith AC, Lopaschuck G, Dyck DJ . Metformin counters the insulin-induced suppression of fatty acid oxidation and stimulation of triacylglycerol storage in rodent skeletal muscle. Am J Physiol Endocrinol Metab 2006; 291: E182–E189.
Chao L, Marcus-Samuels B, Mason MM, Moitra J, Vinson C, Arioglu E et al. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J Clin Invest 2000; 106: 1221–1228.
Fonseca V . Effect of thiazolidinediones on body weight in patients with diabetes mellitus. Am J Med 2003; 115 (Suppl 8A): 42S–48S.
Lewis JD, Ferrara A, Peng T, Hedderson M, Bilker WB, Quesenberry Jr CP et al. Risk of bladder cancer among diabetic patients treated with pioglitazone: interim report of a longitudinal cohort study. Diabetes Care 2011; 34: 916–922.
Jinnin M, Ihn H, Tamaki K . Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol Pharmacol 2006; 69: 597–607.
Campistol JM, Iñigo P, Jimenez W, Lario S, Clesca PH, Oppenheimer F et al. Losartan decreases plasma levels of TGF-beta1 in transplant patients with chronic allograft nephropathy. Kidney Int 1999; 56: 714–719.
Hondares E, Rosell M, Díaz-Delfín J, Olmos Y, Monsalve M, Iglesias R et al. Peroxisome proliferator-activated receptor α (PPARα) induces PPARγ coactivator 1α (PGC-1α) gene expression and contributes to thermogenic activation of brown fat: involvement of PRDM16. J Biol Chem 2011; 286: 43112–43122.
Barbera MJ, Schluter A, Pedraza N, Iglesias R, Villarroya F, Giralt M . Peroxisome proliferator-activated receptor alpha activates transcription of the brown fat uncoupling protein-1 gene. A link between regulation of the thermogenic and lipid oxidation pathways in the brown fat cell. J Biol Chem 2001; 276: 1486–1493.
Lendoye E, Sibille B, Rousseau AS, Murdaca J, Grimaldi PA, Lopez P . PPARbeta activation induces rapid changes of both AMPK subunit expression and AMPK activation in mouse skeletal muscle. Mol Endocrinol 2011; 25: 1487–1498.
Barroso E, Rodríguez-Calvo R, Serrano-Marco L, Astudillo AM, Balsinde J, Palomer X et al. The PPARβ/δ activator GW501516 prevents the down-regulation of AMPK caused by a high-fat diet in liver and amplifies the PGC-1α-Lipin 1-PPARα pathway leading to increased fatty acid oxidation. Endocrinology 2011; 152: 1848–1859.
Lim JY, Oh MA, Kim WH, Sohn HY, Park SI . AMP-activated protein kinase inhibits TGF-β-induced fibrogenic response of hepatic stellate cells by targeting transcriptional coactivator p300. J Cell Physiol 2011; e-pub ahead of print 12 May 2011; doi: 10.1002/jcp.22824.
Kelly LJ, Vicario PP, Thompson GM, Candelore MR, Doebber TW, Ventre J et al. Peroxisome proliferator-activated receptors gamma and alpha mediate in vivo regulation of uncoupling protein (UCP-1, UCP-2, UCP-3) gene expression. Endocrinology 1998; 139: 4920–4927.
Lee WJ, Kim M, Park HS, Kin HS, Jeon MJ, Oh KS et al. AMPK activation increases fatty acid oxidation in skeletal muscle by activating PPARalpha and PGC-1. Biochem Biophys Res Commun 2006; 340: 291–295.
Acknowledgements
This work was funded by National Medical Research Council of Singapore (RG10MAY017) to NST.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Tan, C., Chong, H., Tan, E. et al. Getting ‘Smad’ about obesity and diabetes. Nutr & Diabetes 2, e29 (2012). https://doi.org/10.1038/nutd.2012.1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2012.1
Keywords
This article is cited by
-
Hyperglycemic conditions induce rapid cell dysfunction-promoting transcriptional alterations in human aortic endothelial cells
Scientific Reports (2022)
-
Transforming growth factor beta 1 (TGFβ1) plasmatic levels and haplotype structures in obesity: a role for TGFβ1 in steatosis development
Molecular Biology Reports (2021)
-
Smad proteins differentially regulate obesity-induced glucose and lipid abnormalities and inflammation via class-specific control of AMPK-related kinase MPK38/MELK activity
Cell Death & Disease (2018)
-
Zinc finger protein ZPR9 functions as an activator of AMPK-related serine/threonine kinase MPK38/MELK involved in ASK1/TGF-β/p53 signaling pathways
Scientific Reports (2017)
-
High fat diet-induced TGF-β/Gbb signaling provokes insulin resistance through the tribbles expression
Scientific Reports (2016)
