
Abstract
Background:
Maternal obesity is linked with offspring obesity and type 2 diabetes. Skeletal muscle (SM) insulin resistance is central to the development of diabetes. Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is inhibited in SM of fetuses born to obese mothers.
Objective:
The aim of this study was to evaluate the effect of maternal metformin administration on AMPK activity and reversion of adverse changes in offspring SM of obese mice.
Design:
Female weanling C57BL/6J mice received either control diet (CON, 6 mice) or high-fat diet (HFD; OB, 12 mice) for 8 weeks before mating. After mating, mice continued receiving their respective CON or OB diets. In addition, 6 of those 12 mice fed with fat diet also received metformin administration (2 mg per ml in drinking water) throughout gestation and lactation (MET). After weaning at postnatal 21 days, offspring were fed a HFD to mimic a postnatal obesogenic environment until necropsy.
Results:
Mothers receiving the fat diet developed obesity. OB offspring showed higher adiposity than CON and MET offspring. AMPK phosphorylation was lower in SM of OB offspring. β-Catenin and myogenic regulatory factors, MyoD and myogenin, were downregulated in OB muscle, whereas the adipogenic marker, peroxisome proliferator-activated receptor-γ, was upregulated compared with CON muscle. Metformin administration prevented these changes in OB offspring SM. OB but not MET offspring demonstrated glucose intolerance. Mitochondrial content decreased, and activities of citrate synthase and β-hydroxyacyl-CoA dehydrogenase also decreased in OB offspring SM, whereas they were recovered in MET offspring SM.
Conclusion:
Maternal metformin administration improves SM development in OB offspring.
Similar content being viewed by others


Introduction
Obesity and type 2 diabetes are increasing at alarming rates, even in teenagers and infants.1, 2 At the same time, the prevalence of overweight and obese women of childbearing age is a growing public health concern.3 Western high-energy diets combined with maternal obesity (MO) represent a special problem that can result in harmful, persistent phenotypic outcomes in offspring, including predisposition to obesity and diabetes.4, 5, 6, 7 Although accumulating evidence from both epidemiological studies and animal studies shows that MO predisposes offspring to obesity and diabetes,6, 8, 9 the causative mechanisms have not been elucidated.
Skeletal muscle (SM) is a potential target for antiobese and antidiabetic therapies because it constitutes about 40–50% of body mass and is the main peripheral tissue responsive to insulin-stimulated uptake of glucose and fatty acids.10 Development of insulin resistance in SM is an essential step in the development of type 2 diabetes.11, 12, 13 The fetal stage is crucial for SM development, because no net increase in the number of muscle fibers occurs after birth.14
Fetal SM contains a large number of pluripotent cells. Although the majority of these cells differentiate into muscle fibers through myogenesis, a portion becomes adipocytes.14 The canonical Wnt/β-catenin signaling pathway is essential for myogenesis.15, 16 Enhancing Wnt/β-catenin signaling increases expression of myogenic regulatory factors, including Myf5 and MyoD,17, 18, 19 whereas downregulating Wnt/β-catenin signaling pathway promotes adipogenesis by enhancing the expression of peroxisome proliferator-activated receptor-γ (PPAR-γ), a key regulator of adipogenesis.20 Enhanced adipogenesis in fetal SM increases intramuscular adipocytes, an event linked to SM insulin resistance.10, 21 Thus, impairment of fetal SM development will predispose offspring to diabetes and obesity in later life.14
Adenosine monophosphate (AMP)-activated protein kinase (AMPK) is a master controller of energy metabolism in SM.22, 23 AMPK controls SM development by enhancing the insulin signaling pathway24 and inhibiting adipogenesis,10, 25, 26 both of which link to SM insulin resistance. Activation of AMPK promotes myogenesis, whereas inhibition promotes adipogenesis.9, 27 Metformin is the most common drug currently in use for treating type 2 diabetes. Metformin's activity is mainly mediated by AMPK.28, 29, 30 Using a maternal obese sheep model, we have showed that MO downregulates AMPK activity in fetal SM, which is expected to have profound impact on glucose uptake, insulin signaling and properties of fetal and offspring SM.9 Based on these findings, we hypothesized that activation of fetal SM AMPK by maternal metformin administration would prevent the changes in fetal and offspring SM properties that result from exposure to MO. The objective of the present study was to investigate the effects of metformin administration to obese mothers on offspring SM composition and to assess the importance of AMPK in the development of SM.
Materials and methods
Care and use of animals
Weaning C57BL/6J mice (Charles River Laboratories, Wilmington, MA, USA) were maintained according to the standard protocols approved by the University of Wyoming Animal Use and Care Committee. Mice were housed two per cage under a 12-h light/dark cycle (from 0630 to 1830 hours), with ad libitum access to diet and water. At 4 weeks of age, females were randomized to receive either a control diet (D12450B) with 10% energy from fat (CON) or a high-fat diet (HFD; D12451) with 45% energy from fat for 8 weeks. Control diet (D12450B, a starch, casein and sucrose-based diet containing protein 19.2%, carbohydrate 67.3%, fat 4.3% and vitamins 1.0%, 3.85 kcal g−1) and high-energy diet (D12451, a lard and sucrose-based diet containing protein 24.0%, carbohydrate 41.0%, fat 24.0% and vitamins 1.0%, 4.73 kcal g−1) were purchased from Research Diets (New Brunswick, NJ, USA). At 3 months of age, mice (6 CON mice and 12 high-fat-fed mice) were housed individually and bred. Following mating, all mice continued to receive their respective diets until their litters were weaned. The 12 high-fat-fed mice were further separated into two groups, with one group (n=6) receiving metformin (Spectrum Chemical MFG Corp., New Brunswick, NJ, USA) administration (2 mg per ml in the drinking water, about 350 mg kg−1 per day, MET) throughout pregnancy and lactation and the other group (n=6) supplied with normal drinking water (OB). The dosage of metformin is based on a previous study.31 The litter size was normalized to six mice per litter. Body weight (BW) and diet intake of dams and offspring were monitored twice per week.
At weaning, maternal mice were killed, and serum was collected for blood profile analyses. The maternal carcasses were grounded, and the fat content was analyzed using Folch extraction method.32 After weaning, all offspring were fed with the HFD (D12451) to mimic a postnatal obesogenic environment until necropsy at 60 days of age.
A separate cohort of 12 female mice were assigned to CON, OB and MET groups (n=4) and treated as described above. Offspring male mice were used for glucose tolerance test.
Sample collection
Offspring were killed on postnatal day 1 (two mice per litter were killed), day 15 (one male per litter) and day 60 (one male per litter). Each mouse was skinned and eviscerated. Owing to the small size of the offspring, the muscle from the whole hind (after removing skin, bone and visible connective tissue and fat) was collected from postnatal day-1 mice (muscles from 2 mice per litter were pooled) and day-15 male mice (1 mouse per litter, n=6 per treatment), frozen in liquid N2 and stored at −80 °C for further analyses. For postnatal day-60 mice, gastrocnemius muscle and gonadal fat from males (n=6 per group) were separated, weighed and collected. Also, a section (from first to last lumbar vertebrae) of trunk from day-1 and -15 mice was embedded in OCT compound (Sakura Fineteck USA Inc., Torrance, CA, USA), frozen in liquid N2 and stored in −80 °C for histological examination of muscle structure.
Antibodies
Antibodies against AMPKα (cat. number 2532), phospho-AMPKα at Ser172 (cat. number 2535) and β-catenin (cat. number 9587) were purchased from Cell Signaling (Danvers, MA, USA). Antibody against MyoD (cat. number 5117) was purchased from GenScript Corporation (Piscataway, NJ, USA). Antibody against myogenin was obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City). Antibody against PPAR-γ (cat. number DB134) was purchased from Delta Biolabs (Gilroy, CA, USA). Antibody against glyceraldehyde 3-phosphate dehydrogenase was purchased from Ambion Inc. (Austin, TX, USA). Secondary antibodies were purchased from LI-COR Biosciences (Lincoln, NE, USA).
Immunoblotting analysis
Frozen muscle samples (0.1 g) were homogenized in 500 μl extraction buffer containing 20 mM Tris-HCl (pH 7.4), 2% SDS, 1% Triton X-100, 5.0 mM EDTA, 5.0 mM EGTA, 1 mM DTT, 100 mM NaF, 2 mM sodium vanadate, 0.5 mM phenylmethylsufonyl fluoride, 10 μl per ml leupeptin and 10 μl per ml pepstatin. Protein concentration was determined using the Bio-Rad DC Protein Assay kit (Bio-Rad, Hercules, CA, USA) and separated by SDS-polyacrylamide gel electrophoresis Following electrophoresis, proteins on the gel were transferred to nitrocellulose membranes. After blocking in 50% Odyssey Infrared Imaging blocking buffer (LI-COR Biosciences) and 50% phosphate-buffered saline for 60 min, membranes were incubated overnight with primary antibodies at 4 °C, followed by incubation with infrared dye-conjugated secondary antibodies at room temperature for 60 min. Membranes were visualized using an Odyssey Infrared Imaging System (LI-COR Biosciences). Density of bands was quantified using the Odyssey Infrared Imaging Software Version 2.1 and using glyceraldehyde 3-phosphate dehydrogenase as the internal control.
Hematoxylin and eosin staining
Samples of the trunk (first lumbar vertebrae) were cross-sectioned at 10-μm thickness using a Reichert HistoStat (Scientific Instruments, Buffalo, NY, USA), stained with hematoxylin and eosin and examined by light microscopy.
Intraperitoneal glucose tolerance test
Glucose tolerance test was performed on one male offspring per litter at day 15 and day 60 postnatal. Before the glucose tolerance test, mice were weighed and fasted overnight with ad libitum access to water. The tail tip was nicked with a razor blade, and a small drop of blood was sampled from the tail of each mouse by gentle stroking. Baseline blood glucose was measured by the glucose oxidase method using a glucometer (Bayer Contour, Tarrytown, NY, USA). Following the baseline sampling, 2 g per kg BW of 20% D-glucose was injected into the intraperitoneal cavity, and blood glucose measured at 15, 30, 60 and 120 min after injection.
Myosin heavy-chain isoform separation
Homogenates from muscle samples were prepared, and myosin heavy-chain isoform distribution was assessed using gel electrophoresis, as previously described.33
Real-time PCR for mitochondrial DNA and genomic DNA copy number
Total DNA was obtained from muscles by an organic solvent extraction-based method, as described previously.34 DNA concentration was measured using a Nanodrop 1000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA), and subsequently diluted in water to a final concentration of 50 ng μl−1. Real-time PCR was carried out using an iQ5 RT-PCR detection system (Bio-Rad) using SYBR Green RT-PCR kit from Bio-Rad and the following cycle parameters: one cycle of 50 °C for 2 min, and 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 60 s. Melting point dissociation curves and agarose gel electrophoresis were performed to confirm that only a single product was amplified. The murine genomic DNA was analyzed by measuring the single-copy uncoupling protein 2 (UCP2) gene with primers: GCGTTCTGGGTACCATCCTAAC (forward) and GCGACCAGCCCATTGTAGA (reverse). Mitochondrial DNA was analyzed by measuring cytochrome C oxidase II (Cyt C) mtDNA gene with primers: TTTTCAGGCTTCACCCTAGATGA (forward) and GAAGAATGTTATGTTTACTCCTACGAATATG (reverse). Mitochondrial DNA copy number was calculated based on the difference in ΔCt values between Cyt C and UCP2.35
Blood profile analyses
Serum glucose concentrations were analyzed using Glucose Assay Kit (Sigma-Aldrich, Saint Louis, Missouri, USA). Serum triglyceride concentrations were analyzed using Mouse Serum Triglyceride Determination Kit (Sigma-Aldrich). Serum insulin concentrations and leptin concentrations were determined by kits (Linco Research, St Charles, MO, USA). All experimental assays were performed according to the manufacturer's instructions.
Enzyme activity assays
Muscle samples (0.03 g) were homogenized in 1:20 dilution (wt/vol) of an extraction buffer (0.1 M KH2PO4/Na2PO4 and 2 mM EDTA, pH 7.2). Citrate synthase and β-hydroxyacyl-CoA dehydrogenase (β-HAD) activities were assayed spectrophotometrically, as described elsewhere.36 CS and β-HAD activity were calculated and reported as μmol per ming wet weight of muscle.
Statistical analysis
Each litter was considered as an experimental unit. All data were expressed as mean±s.e. Data from each time point (days 1, 15 and 60) were analyzed independently using the GLM procedure of SAS (SAS Inst. Inc., Cary, NC, USA), and Tukey's studentized range test was used for multi-comparison to determine significant differences among means. Statistical significance was considered as P<0.05.
Results
BW and diet consumption of dams and offspring
There was no difference in maternal BW at the start of dietary treatments (Supplementary Figure 1). At week 8, BW of HFD dams and CON dams clearly diverged (P<0.05). By the end of treatments, BW gain in HFD dams was greater than CON dams (P<0.05, Supplementary Figure 1). At week 4, the diet intake of HFD mice was lower than CON mice. At weeks 8 and 12, the diet consumption did not differ between groups (Supplementary Figure 1).
At weaning (21 days), the OB maternal BW and fat content were higher in OB and MET groups (Table 1). The circulating glucose level was higher in OB compared with CON and MET treatments, but there was no difference in the concentrations of insulin and triglyceride in serum. The leptin content was higher for both OB and MET compared with CON mice (Table 1).
The birth weight of offspring born to OB and MET mothers were heavier than CON mothers (OB mothers, 1.37±0.02 g; MET mothers, 1.35±0.01 g; and CON mothers, 1.26±0.04 g), whereas there was no difference between OB and MET mothers (Supplementary Figure 2A). Dietary intake did not differ substantially between groups (Supplementary Figure 2D). OB offspring showed greater weight gain than CON offspring throughout the experiment (Figure 2a), whereas MET offspring was intermediate. At day 60, the gonadal fat of OB male offspring (1.05±0.17 g) was much heavier than that of CON offspring (0.57±0.06 g, P<0.05) and tended to be heavier than that of MET offspring (0.74±0.07 g, P=0.08; Supplementary Figures 2B and C).
Maternal metformin administration stimulated AMPK activity in offspring SM
AMPK was activated in the SM of MET offspring. Phosphorylation of AMPK at Thr 172 in OB offspring SM was reduced compared with CON mothers (P<0.05 at days 15 and 60, and P=0.06 at day 1, Figure 1). The phosphorylation of AMPK at Thr 172 was rescued in MET offspring SM at the three time points examined compared with OB offspring SM. Using western blotting, no significant difference in the protein content of AMPKα subunit was observed (data not shown).

AMPK phosphorylation in day-1 and -15 offspring hindleg muscle, and day-60 male offspring gastrocnemius muscle of CON, OB and MET mothers. Data are expressed as mean±s.e., n=6. #P<0.05 versus CON; (#)P<0.1 versus CON; and *P<0.05 versus OB.
Expression of β-catenin and myogenic markers in offspring SM
At day 1, β-catenin protein content tended to be lower in OB offspring muscle (P=0.07) compared with CON offspring muscle, and was increased by metformin administration (Figure 2). This difference in β-catenin was maintained in the day-60 gastrocnemius muscle. There was a trend that MyoD protein expression at days 1 (P=0.06) and 15 were decreased in OB offspring SM (P=0.09), as well as myogenin at day 1 (P=0.07), indicating that myogenesis might be impaired. Metformin administration tended to restore MyoD at days 1 and 15 (P=0.06 and 0.08, respectively), and myogenin expression at day 15 (P=0.05, Figure 2).

Contents of β-catenin, myogenic markers and PPAR-γ in day-1 and -15 offspring hindleg muscle, and day-60 male offspring gastrocnemius muscle of CON, OB and MET mothers. (a) Beta-catenin content; (b) MyoD content; (c) Myogenin content; (d) PPARgamma content. Data are expressed as mean±s.e., n=6. #P<0.05 versus CON; (#)P<0.1 versus CON; *P<0.05 versus OB; and (*)P<0.1 versus OB.
Adipogenesis in offspring SM
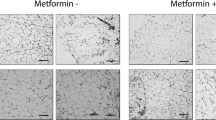
PPAR-γ protein expression was much higher in OB offspring SM compared with CON SM (P<0.05; Figure 2d). Metformin administration decreased PPAR-γ protein expression compared with OB (P<0.05). We next examined the morphology of offspring muscle. There were no obvious adipocytes visible in muscle cross-section at day 1 (Figure 3), but at day 15, mature adipocytes were visible in OB offspring SM; metformin inhibited the formation of intramuscular adipocytes in offspring SM induced by the maternal HFD (Figure 3).

Hematoxylin and eosin (H&E) staining of day-1 and -15 offspring muscle of CON, OB and MET mothers. A section of dorsum of the trunk was dissected, embedded in OCT and frozen in liquid N2. Cryofixed muscle specimens were cross-sectioned at 10-um thickness and stained with H&E, × 100 magnification. (a) CON muscle at day 1; (b) OB muscle at day 1; (c) MET muscle at day 1; (d) CON muscle at day 15; (e) OB muscle at day 15; (f) MET muscle at day 15.
Maternal metformin administration increased glucose uptake of offspring
There were no differences in the glucose concentration among treatments at a single time point (Figure 4). However, when the total glucose disposal was compared by combining the incremental area under the curve, offspring of OB dam had a lower blood glucose disposal rate compared with those of CON and MET mothers at both days 15 and 60. The improvement of glucose uptake following metformin administration was associated with enhanced Akt signaling (Supplementary Figure 3).

Glucose tolerance test (GTT) on postnatal day-15 and -60 offspring of CON, OB, and MET mothers. (a) Intraperitoneal GTT in day-15 male offspring mice. The histogram represents the incremental area under the respective glucose curve. (b) Intraperitoneal GTT in postnatal day-60 male offspring mice. The histogram represents the incremental area under the glucose curve. Data are expressed as mean±s.e., n=4 for both ages. #P<0.05 versus CON; (#)P<0.10 versus CON; *P<0.50 versus OB; and (*)P<0.50 versus OB.
Maternal HFD and metformin administration had no effects on muscle fiber type distribution in the offspring SM
Myosin heavy-chain isoforms in postnatal mice muscle were separated (Supplementary Figure 4). There are two developmental myosin heavy-chain isoforms, namely, embryonic and neonatal myosin heavy-chain isoform, present in newborn mouse gastrocnemius, which are replaced by adult isoforms, including type IIA, IIX and IIB by day 14.37 As shown in Supplementary Figure 4, no difference in muscle fiber composition was detected among different treatments at all time points. Also, type I fiber was indistinguishable at day 60 with this gel electrophoresis method.
Mitochondria DNA copy number and muscle metabolic activity in the offspring was affected by maternal HFD and metformin administration
Different muscle fibers have distinct mitochondrial density. Although we found no difference in muscle fiber composition among different treatments, mitochondrial DNA copy number was decreased in OB offspring at days 15 and 60 (Table 2). Metformin administration to OB dams increased mitochondrial copy number in the offspring at day 15. Moreover, mitochondrial function in offspring SM was affected by maternal HFD and metformin administration, as evidenced by the decrease of citrate synthase activity and β-HAD activity in OB offspring and restoration of their activity after maternal metformin administration (Table 2).
Discussion
Both obesity and diabetes are increasing at alarming rates and among a younger and younger population.1 MO predisposes offspring to obesity and diabetes, which also contributes to the increase in obesity, especially in children.9 One explanation is that MO programs hypothalamic regulation of BW and energy homeostasis.38, 39 MO in rats does slightly increase energy intake in offspring.38 Offspring of obese rat mothers are hyperphagic.40, 41 However, in our study, we only observed a trend toward a higher dietary intake in OB offspring compared with CON offspring, which could be because of our small sample size. Also, the diet used in our study differed from that in the published rat studies, and offspring may have distinctive preference for sugary and fatty items in different diets. In addition, strain differences have been noted in the response to HFDs.41 The observation that OB offspring showed more weight gain and adiposity while lacking a significant difference in diet intake suggests that hyperphagia may not be the only cause of obesity but differences in basal energy expenditure may also contribute. SM is the main tissue responsible for glucose and fatty acid oxidation. Reduction in SM oxidative capacity could lead to more offspring energy storage and BW gain. Mitochondria are the main sites of nutrient oxidation to generate ATP. Citrate synthase is a key enzyme in the tricarboxylic acid cycle, and β-HAD is a key enzyme in fatty acid oxidation. Both enzymes are good markers of SM oxidative capacity.42 Our data showed that both mitochondrial content and function were impaired in OB offspring SM, indicating the impairment of SM oxidative capacity. Maternal metformin administration rescued mitochondrial density and function. In adult muscle, activation of AMPK enhances the oxidative capacity of SM via a PPAR-γ coactivator-1α-mediated mechanism.43, 44 Mitochondrial dysfunction leads to obesity, insulin resistance and type 2 diabetes.11 Thus, our results support the notion that low oxidative capacity in muscle has a role in the etiology of obesity.
AMPK has a critical role in maintaining systemic and cellular energy status. The growing appreciation that AMPK coordinates anabolic and catabolic metabolic processes suggests that it may be an attractive target for preventing diabetes and related metabolic diseases. Physiological activation by exercise or pharmacological activation of AMPK improves blood glucose disposal, lipid profile and blood pressure in insulin-resistant humans and rodents, which indicates that AMPK is an effective drug target in the therapeutic applications in diabetes and other metabolic diseases.45 Using a sheep MO model, we detected that MO downregulates AMPK activity in fetal SM.9 This observation prompted us to investigate whether activation of AMPK in fetal and neonatal SM could have a positive effect on its development and function. In the present study, we elected to continue metformin administration throughout lactation and suckling, as SM development during this period in mice is approximately similar to the development that occurs from mid to late gestation in sheep and humans. Metformin readily crosses the placenta.46, 47, 48 Metformin is commonly used to treat type 2 diabetes, and its effect is believed to be mainly mediated by AMPK.28, 29, 30 Our data show that metformin administration during pregnancy and lactation increased AMPK activity in offspring SM of obese mothers.
β-Catenin is a key mediator of the canonical Wnt/β-catenin signaling pathway that is essential for myogenesis, and blocking the β-catenin pathway reduces the total number of myocytes.15, 16 MyoD and myogenin belong to the family of muscle-specific transcription factors that control SM differentiation;49 their expression correlates with myogenesis. We observed that β-catenin, MyoD and myogenin were downregulated in fetal OB SM, which is consistent with our previous observation in sheep SM.10 PPAR-γ is a critical regulator of adipogenesis. Increased expression of PPAR-γ leads to differentiation of pluripotent cells to adipocytes.20 Our observation of increased PPAR-γ in OB offspring SM indicates that adipogenesis was upregulated, a conclusion confirmed by the presence of increased numbers of intramuscular adipocytes in OB offspring SM, detected in OB offspring SM. Maternal metformin administration reversed these changes, indicating that metformin is effective in improving SM development in fetuses and neonatal mice born to obese mothers. Whether this beneficial effect is due to the positive impacts of metformin on maternal whole-body energy metabolism, improved insulin sensitivity and intrauterine environment or a direct action on the fetuses of metformin that has crossed the placenta remains to be clarified.
Currently, metformin administration is avoided during human pregnancy because of the lack of data about its effects on fetal development.46, 48 Our data show that metformin administration during pregnancy and lactation has no obvious negative effect on overall fetal and offspring development. Progeny from metformin-treated dams appeared healthy. In addition, our data clearly show that metformin administration during pregnancy and lactation improves the development of fetal SM of obese mothers, which may provide a promising strategy to prevent the fetal-programming effect of obesity during pregnancy on fetal and offspring SM development.
References
Petersen KF, Dufour S, Shulman GI . Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med 2005; 2: e233.
Galtier F, Raingeard I, Renard E, Boulot P, Bringer J . Optimizing the outcome of pregnancy in obese women: from pregestational to long-term management. Diabetes Metab 2008; 34: 19–25.
Siega-Riz AM, Siega-Riz AM, Laraia B . The implications of maternal overweight and obesity on the course of pregnancy and birth outcomes. Matern Child Health J 2006; 10: 153–156.
Morino K, Petersen KF, Dufour S, Befroy D, Frattini J, Shatzkes N et al. Reduced mitochondrial density and increased IRS-1 serine phosphorylation in muscle of insulin-resistant offspring of type 2 diabetic parents. J Clin Invest 2005; 115: 3587–3593.
Fowden AL, Ward JW, Wooding FP, Forhead AJ, Constancia M . Programming placental nutrient transport capacity. J Physiol 2006; 572: 5–15.
Nathanielsz PW . Animal models that elucidate basic principles of the developmental origins of adult diseases. Ilar J 2006; 47: 73–82.
Barker DJ . Fetal programming of coronary heart disease. Trends Endocrinol Metab 2002; 13: 364–368.
Armitage JA, Khan IY, Taylor PD, Nathanielsz PW, Poston L . Developmental programming of the metabolic syndrome by maternal nutritional imbalance: how strong is the evidence from experimental models in mammals? J Physiol 2004; 561: 355–377.
Zhu MJ, Han B, Tong J, Ma C, Kimzey JM, Underwood KR et al. AMP-activated protein kinase signalling pathways are down regulated and skeletal muscle development impaired in fetuses of obese, over-nourished sheep. J Physiol 2008; 586: 2651–2664.
Tong JF, Yan X, Zhu MJ, Ford SP, Nathanielsz PW, Du M . Maternal obesity downregulates myogenesis and beta-catenin signaling in fetal skeletal muscle. Am J Physiol Endocrinol Metab 2009; 296: E917–E924.
Lowell BB, Shulman GI . Mitochondrial dysfunction and type 2 diabetes. Science 2005; 307: 384–387.
Ozanne SE, Jensen CB, Tingey KJ, Storgaard H, Madsbad S, Vaag AA . Low birthweight is associated with specific changes in muscle insulin-signalling protein expression. Diabetologia 2005; 48: 547–552.
Selak MA, Storey BT, Peterside I, Simmons RA . Impaired oxidative phosphorylation in skeletal muscle of intrauterine growth-retarded rats. Am J Physiol Endocrinol Metab 2003; 285: E130–E137.
Du M, Yan X, Tong JF, Zhao J, Zhu MJ . Maternal obesity, inflammation, and fetal skeletal muscle development. Biol Reprod 2010; 82: 4–12.
Mermelstein CS, Portilho DM, Mendes FA, Costa ML, Abreu JG . Wnt/beta-catenin pathway activation and myogenic differentiation are induced by cholesterol depletion. Differentiation 2007; 75: 184–192.
Armstrong DD, Esser KA . Wnt/beta-catenin signaling activates growth-control genes during overload-induced skeletal muscle hypertrophy. Am J Physiol 2005; 289: C853–C859.
Cossu G, Borello U . Wnt signaling and the activation of myogenesis in mammals. EMBO J 1999; 18: 6867–6872.
Yamanouchi K, Hosoyama T, Murakami Y, Nishihara M . Myogenic and adipogenic properties of goat skeletal muscle stem cells. J Reprod Dev 2007; 53: 51–58.
Pan W, Jia Y, Wang J, Tao D, Gan X, Tsiokas L et al. Beta-catenin regulates myogenesis by relieving I-mfa-mediated suppression of myogenic regulatory factors in P19 cells. Proc Natl Acad Sci USA 2005; 102: 17378–17383.
Spiegelman BM, Puigserver P, Wu Z . Regulation of adipogenesis and energy balance by PPARgamma and PGC-1. Int J Obes Relat Metab Disord 2000; 24 (Suppl 4): S8–S10.
Aguiari P, Leo S, Zavan B, Vindigni V, Rimessi A, Bianchi K et al. High glucose induces adipogenic differentiation of muscle-derived stem cells. Proc Natl Acad Sci USA 2008; 105: 1226–1231.
Hardie DG . AMP-activated protein kinase: a key system mediating metabolic responses to exercise. Med Sci Sports Exerc 2004; 36: 28–34.
Hardie DG . New roles for the LKB1 → AMPK pathway. Curr Opin Cell Biol 2005; 17: 167–173.
Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE . 5′-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J Biol Chem 2001; 276: 46912–46916.
Dagon Y, Avraham Y, Berry EM . AMPK activation regulates apoptosis, adipogenesis, and lipolysis by eIF2alpha in adipocytes. Biochem Biophys Res Commun 2006; 340: 43–47.
Habinowski SA, Witters LA . The effects of AICAR on adipocyte differentiation of 3T3-L1 cells. Biochem Biophys Res Commun 2001; 286: 852–856.
Giri S, Rattan R, Haq E, Khan M, Yasmin R, Won JS et al. AICAR inhibits adipocyte differentiation in 3T3L1 and restores metabolic alterations in diet-induced obesity mice model. Nutr Metab (Lond) 2006; 3: 31.
Hawley SA, Gadalla AE, Olsen GS, Hardie DG . The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes 2002; 51: 2420–2425.
Lee JO, Lee SK, Jung JH, Kim JH, You GY, Kim SJ et al. Metformin induces Rab4 through AMPK and modulates GLUT4 translocation in skeletal muscle cells. J Cell Physiol 2010; 226: 974–981.
Zou MH, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles IV WG, Schlattner U et al. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo. Role of mitochondrial reactive nitrogen species. J Biol Chem 2004; 279: 43940–43951.
Ma TC, Buescher JL, Oatis B, Funk JA, Nash AJ, Carrier RL et al. Metformin therapy in a transgenic mouse model of Huntington's disease. Neurosci Lett 2007; 411: 98–103.
Folch J, Lees M, Sloane Stanley GH . A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem 1957; 226: 497–509.
Underwood KR, Tong J, Zhu MJ, Shen QW, Means WJ, Ford SP et al. Relationship between kinase phosphorylation, muscle fiber typing, and glycogen accumulation in longissimus muscle of beef cattle with high and low intramuscular fat. J Agric Food Chem 2007; 55: 9698–9703.
Guo W, Jiang L, Bhasin S, Khan SM, Swerdlow RH . DNA extraction procedures meaningfully influence qPCR-based mtDNA copy number determination. Mitochondrion 2009; 9: 261–265.
Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA et al. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes 2005; 54: 1926–1933.
Call JA, Voelker KA, Wolff AV, McMillan RP, Evans NP, Hulver MW et al. Endurance capacity in maturing mdx mice is markedly enhanced by combined voluntary wheel running and green tea extract. J Appl Physiol 2008; 105: 923–932.
Agbulut O, Noirez P, Beaumont F, Butler-Browne G . Myosin heavy chain isoforms in postnatal muscle development of mice. Biol Cell 2003; 95: 399–406.
Page KC, Malik RE, Ripple JA, Anday EK . Maternal and postweaning diet interaction alters hypothalamic gene expression and modulates response to a high-fat diet in male offspring. Am J Physiol Regul Integr Comp Physiol 2009; 297: R1049–R1057.
Kirk SL, Samuelsson AM, Argenton M, Dhonye H, Kalamatianos T, Poston L et al. Maternal obesity induced by diet in rats permanently influences central processes regulating food intake in offspring. PLoS One 2009; 4: e5870.
Nivoit P, Morens C, Van Assche FA, Jansen E, Poston L, Remacle C et al. Established diet-induced obesity in female rats leads to offspring hyperphagia, adiposity and insulin resistance. Diabetologia 2009; 52: 1133–1142.
Samuelsson AM, Matthews PA, Argenton M, Christie MR, McConnell JM, Jansen EH et al. Diet-induced obesity in female mice leads to offspring hyperphagia, adiposity, hypertension, and insulin resistance: a novel murine model of developmental programming. Hypertension 2008; 51: 383–392.
Bruce CR, Anderson MJ, Carey AL, Newman DG, Bonen A, Kriketos AD et al. Muscle oxidative capacity is a better predictor of insulin sensitivity than lipid status. J Clin Endocrinol Metab 2003; 88: 5444–5451.
Atherton PJ, Babraj J, Smith K, Singh J, Rennie MJ, Wackerhage H . Selective activation of AMPK-PGC-1alpha or PKB-TSC2-mTOR signaling can explain specific adaptive responses to endurance or resistance training-like electrical muscle stimulation. FASEB J 2005; 19: 786–788.
Suwa M, Egashira T, Nakano H, Sasaki H, Kumagai S . Metformin increases the PGC-1alpha protein and oxidative enzyme activities possibly via AMPK phosphorylation in skeletal muscle in vivo. J Appl Physiol 2006; 101: 1685–1692.
Hardie DG . Biochemistry. Balancing cellular energy. Science 2007; 315: 1671–1672.
Feig DS . Metformin therapy for gestational diabetes mellitus: are we there yet? Nat Clin Pract Endocrinol Metab 2008; 4: 654–655.
Kovo M, Kogman N, Ovadia O, Nakash I, Golan A, Hoffman A . Carrier-mediated transport of metformin across the human placenta determined by using the ex vivo perfusion of the placental cotyledon model. Prenat Diagn 2008; 28: 544–548.
Paglia MJ, Coustan DR . The use of oral antidiabetic medications in gestational diabetes mellitus. Curr Diab Rep 2009; 9: 287–290.
Sabourin LA, Rudnicki MA . The molecular regulation of myogenesis. Clin Genet 2000; 57: 16–25.
Acknowledgements
The research is supported by the National Institute of Child Health & Human Development: R01HD067449, and the National Center for Research Resources of NIH and the Wyoming INBRE: P20RR016474, and USDA-NRI-2008-35206-18826.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Nutrition and Diabetes website
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Tong, J., Yan, X., Zhao, J. et al. Metformin mitigates the impaired development of skeletal muscle in the offspring of obese mice. Nutr & Diabetes 1, e7 (2011). https://doi.org/10.1038/nutd.2011.3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nutd.2011.3
Keywords
This article is cited by
-
Metformin alleviates muscle wasting post-thermal injury by increasing Pax7-positive muscle progenitor cells
Stem Cell Research & Therapy (2020)
-
Gestational exposure to metformin programs improved glucose tolerance and insulin secretion in adult male mouse offspring
Scientific Reports (2018)
