
Abstract
Binding of the gp120 envelope (Env) glycoprotein to the CD4 receptor is the first step in the HIV-1 infectious cycle. Although the CD4-binding site has been extensively characterized, the initial receptor interaction has been difficult to study because of major CD4-induced structural rearrangements. Here we used cryogenic electron microscopy (cryo-EM) to visualize the initial contact of CD4 with the HIV-1 Env trimer at 6.8-Å resolution. A single CD4 molecule is embraced by a quaternary HIV-1–Env surface formed by coalescence of the previously defined CD4-contact region with a second CD4-binding site (CD4-BS2) in the inner domain of a neighboring gp120 protomer. Disruption of CD4-BS2 destabilized CD4-trimer interaction and abrogated HIV-1 infectivity by preventing the acquisition of coreceptor-binding competence. A corresponding reduction in HIV-1 infectivity occurred after the mutation of CD4 residues that interact with CD4-BS2. Our results document the critical role of quaternary interactions in the initial HIV-Env-receptor contact, with implications for treatment and vaccine design.
Similar content being viewed by others

Main
The native Hiv-1 Env spike on the surface of mature virions mediates viral attachment and entry1, and it thus represents the major target for neutralizing antibodies and a central focus for vaccine development. The native spike is a trimer of gp120–gp41 heterodimers constrained into a metastable structure that is able to evade immunologic control by a variety of mechanisms, including antigenic variability, N-linked glycosylation, and conformational masking of the conserved receptor-binding sites2,3. Upon binding to the primary cellular receptor, CD4, the external gp120 Env glycoprotein undergoes major conformational changes, transitioning to a state that is competent for interaction with the CCR5 or CXCR4 coreceptor1. The CD4-binding site (CD4-BS) of gp120, which is a critical target for broad-spectrum neutralizing antibodies, has been finely mapped by both mutagenesis4,5,6,7 and cocrystallization in complex with soluble CD4 (sCD4)8,9,10,11. However, most of those studies were performed with monomeric gp120 subunits, which hindered evaluation of the role of quaternary elements assembled in the pre-fusion trimeric structure that may be involved in the initial CD4 interaction. Quaternary interactions with an adjacent gp120 protomer have indeed been documented for two monoclonal antibodies (mAbs) directed to the CD4 supersite, VRC-PG04 (ref. 12) and VRC01 (ref. 13), by cryo-EM and crystallography, respectively. In addition, Lyumkis and colleagues12 used in silico docking to model quaternary trimer interactions for additional anti-CD4-BS mAbs and for sCD4 itself, thereby predicting contacts of VRC01 and sCD4 with a limited surface of the α0 helix of an adjacent protomer, as well as more extensive contacts of other mAbs with basic residues in C2 and the V3 loop; however, they did not investigate the functional significance of these interactions.
Considerable progress has recently been made in the elucidation of the molecular anatomy of the HIV-1 Env spike, particularly with the design and crystallization of soluble truncated trimers (SOSIP.664) stabilized in a near-native configuration13,14,15,16,17,18 and with the increasing resolution of cryo-EM imaging of native, membrane-bound Env spikes19,20,21. Nevertheless, it has remained challenging to obtain structural information on the initial contact of CD4 with the pre-fusion trimer, primarily because of the rapid transition of the trimer toward an open configuration after interaction with CD4 (ref. 22). Accordingly, cryo-EM studies have visualized the adoption of an open conformation by both soluble and membrane-bound Env trimers upon CD4 binding, regardless of the presence of bound antibodies directed to CD4-induced epitopes21,23 or to epitopes compatible with the prefusion trimer state24.
Here we present the cryo-EM structure of a conformationally constrained HIV-1 Env soluble trimer complexed with sCD4 and a trimer-specific broadly neutralizing antibody, PGT145, and document the quaternary configuration of the initial CD4-contact site in the closed, pre-fusion HIV-1 Env trimer. We show that the quaternary interactive site stabilizes CD4-Env binding and plays an essential role in HIV-1 entry into host cells, as its disruption prevents the acquisition of coreceptor-binding competence. Finally, we refine the boundaries of the CD4 antigenic supersite, and show that the quaternary CD4-interactive surface is immunogenic in vivo. These results have implications for HIV-1 treatment and vaccine design.
Results
Cryo-EM structure of initial CD4 contact with the HIV-1 Env trimer
To gain structural insights into the initial interaction of CD4 with the HIV-1 Env trimer, we used a conformationally constrained soluble trimer, DS-SOSIP.664 (ref. 16), which remains in a pre-fusion, closed conformation even after interaction with CD4, as evidenced by retention of binding to trimer-specific antibodies, including the V1V2 loop-directed mAb PGT145 (ref. 25). Notably, the DS-SOSIP.664 trimer binds sCD4 with an asymmetric 1:1 (CD4:trimer) stoichiometry16, which probably reflects the earliest Env-receptor interaction. Thus, we formed complexes between DS-SOSIP.664, four-domain sCD4, and the antigen-binding fragment (Fab) of PGT145, and collected cryo-EM data, which yielded a 6.8-Å-resolution reconstruction (Fig. 1a,b, Table 1, and Supplementary Figs. 1 and 2). We used known structures of the Env trimer, sCD4, and PGT145 to position each molecule in the cryo-EM map (Supplementary Tables 1 and 2, and Supplementary Fig. 2), and we then minimized the Env-interactive interfaces with PGT145 and CD4. Because 6.8-Å resolution does not allow for experimental determination of residue side chains, we deposited coordinates with all residues truncated down to their Cβ atoms (PDB 5U1F).

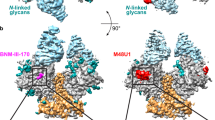
(a) Side view of the cryo-EM structure of a ternary complex comprising DS-SOSIP HIV-1 Env trimer, four-domain sCD4 (yellow), and Fab from the broadly neutralizing mAb PGT145 (green), all shown in ribbon representation, with a 6.8-Å-resolution electron-microscopy-reconstructed density shown as a gray mesh. Each gp120 protomer is displayed in a different color, with gp41 in gray. (b) View from the viral membrane of the complex in a rotated 90°. CD4 contacts two gp120s, indicated as primary and quaternary CD4-contacting protomers, but not the third gp120. (c) Zoomed-in view of the PGT145 interaction. The images at the bottom show a 90° view (middle) and electrostatic surface display for HIV-1 Env (left) and for PGT145 (right). (d) Orientation of Env protomers in the prefusion, closed Env conformation recognized by a single CD4, displayed on the Env trimer in ribbon representation (left). Trimeric Env structures were aligned to the noncontacting protomer of the single CD4-bound trimer, and the resulting orientations of the primary and quaternary CD4-contacting protomers were plotted (right). The distance and angular measurements are absolute values measured from the single CD4-bound trimer. With asymmetric trimers, such as the PGT151-bound trimer (PDB 5FUU), each protomer was aligned to the noncontacting protomer of the single CD4-bound trimer, and the alignment displayed is the orientation with median positional displacement relative to the primary CD4-contacting protomer.
The reconstructed map confirmed that the trimer was in a pre-fusion, closed conformation and was bound to a single CD4 molecule. The integrity of the pre-fusion trimer structure was reinforced by the presence of the quaternary-specific mAb PGT145, with its extended third heavy chain complementarity-determining region (CDR H3) loop penetrating through the glycan shield and reaching into a hole at the trimer apex formed by the junction of the V1V2 loops, making contacts with all three gp120 protomers, including the apex glycans, and essentially sealing the pre-fusion trimer at its apex (Fig. 1a,c and Supplementary Fig. 2). The highly electronegative CDR H3 loop of PGT145 lodged within an electropositive pocket formed by residues from all three protomers, with a sulfated tyrosine at its tip reaching into a space often occupied by SO4−2 anions in previously reported structures of the pre-fusion closed trimer13,15,16,18 (Fig. 1c).
Initial contact of CD4 with a quaternary Env surface
The reconstructed cryo-EM structure of the DS-SOSIP.664 trimer showed domain 1 of sCD4 inserted between two gp120 protomers to establish contacts not only with the previously defined CD4-BS in the outer domain of one gp120 protomer (hereinafter referred to as the primary CD4-contacting protomer), but also with CD4-BS2 in the inner domain of a neighboring protomer (quaternary CD4-contacting protomer), with no interactions with the third (non-CD4-contacting) protomer (Fig. 1b).
To define the orientations of the three Env protomers in the single CD4-bound closed trimer structure, we compared the orientation of each protomer in our reconstructed electron microscopy map with those in previously determined structures of the pre-fusion closed Env trimer (Supplementary Table 1). We superimposed the structures by aligning each previously defined protomer onto the coordinate model of the non-CD4-contacting protomer in our cryo-EM reconstruction, and compared the resulting orientations of the other two protomers (Fig. 1d). We found the position of the primary CD4-contacting protomer in our structure to be shifted by an average translation of 3.4 Å and a rotation of 2.4° relative to the mean coordinate position of other pre-fusion soluble trimer structures, whereas the quaternary CD4-contacting protomer shifted by a translation of 3.1 Å and a rotation of 1.9°, with both protomers moving outward from the trimer axis toward the CD4 molecule (Fig. 1d). The most distal orientation for both primary (difference of 5.6 Å translation and 10.5° rotation) and quaternary (difference of 5.0 Å translation and 8.7° rotation) CD4-contacting protomers occurred with the PGT151 complex20, which also induces an asymmetric conformation, but in the opposite direction. We did not observe any major change in the secondary structure of either gp120 or gp41 in the 6.8-Å map versus prior pre-fusion closed structures, which reflects the ability of the HIV-1 Env protomers to sustain movements of up to 5 Å without undergoing substantial structural rearrangements. Although the 6.8-Å resolution of the reconstructed cryo-EM map precluded experimental definition of individual side chain positions, robust fits of the Env protomer domains to the reconstructed electron microscopy map (Fig. 1a,b and Supplementary Table 1), together with a statistically significant difference of domain orientations of 6σ or greater relative to the population of trimer structures shown in gray in Figure 1d, allowed for the positioning of an asymmetric protomer domain arrangement within the single CD4-bound trimer structure.
To obtain insights into the CD4-trimer interaction, we used a molecular dynamics (MD) simulation that led us to focus on four candidate CD4-contacting residues in the quaternary CD4-contacting protomer, namely, E62, E64, and H66 in the α1--helix, and K207 at the base of the β3-β4 loop (Fig. 2a). In the crystal structure of the unliganded trimer18, the side chains of these residues project toward the incoming CD4 receptor and are exposed to the solvent (Fig. 2a). Notably, residues E64, H66, and K207 are nearly universally conserved (>99.7%) across all group M HIV-1 isolates, whereas position 62 is occupied by an acidic residue (E or D) in more than 90% of group M isolates (Fig. 2b).

(a) Top view of the cryo-EM structure of an HIV-1 Env trimer bound to a single CD4 receptor showing the quaternary interaction of CD4 (yellow) with two neighboring gp120 protomers; gp41 was removed for clarity. The image on the right shows a close-up view of the CD4-binding site (CD4-BS2) on the quaternary contacting gp120. Residues in the C1 (α1--helix) and C2 (β3-β4 loop) regions of the quaternary CD4-contacting gp120 protomer (pink) are highlighted in stick representation (E62 and E64, negatively charged; H66 and K207, positively charged). The location of K65, which points away from CD4, is shown for reference. (b) Amino acid conservation within the CD4-BS2 region. Residues E64, H66, and K207 are nearly universally conserved (>99.7%) across all group M HIV-1 isolates; position 62 is occupied by an acidic residue (either E or D) in more than 90% of group M isolates, with nonconservative mutations essentially limited to genetic subtype D (58% E→K) and subtypes G and J (100% E→S). (c) Disruption of CD4-BS2 by charge inversion abrogates HIV-1 infectivity. Infectious pseudoparticles bearing wild-type (WT) or mutated BG505-T332N (clade A) or BaL (clade B) Env were tested in TZM-bl cells. Infectivity values for each mutant were calculated relative to the value obtained for the wild type. (d) Infectivity of viral pseudotypes carrying mutated HIV-1 Env from different genetic subtypes. The infectivity of each mutant was calculated relative to that of the respective wild-type Env. The data shown in c and d represent mean ± range of duplicate wells from a representative experiment out of three performed that yielded similar results.
Critical role of quaternary CD4 interaction in HIV-1 infection
To elucidate the functional relevance of the quaternary CD4 contact to HIV-1 infection, we mutagenized residues E62, E64, H66, and K207 in full-length gp160 from six HIV-1 isolates of different genetic subtypes—namely, BG505 (clade A), BaL and JR-FL (clade B), ZM106.9 (clade C), QD435.A4 (clade D), and CM244 (clade E)—and generated infectious viral pseudoparticles for each mutant. We introduced both individual and combined charge inversions into each Env to create a reverse-charged surface; for the control condition, we mutated by charge inversion a key residue in the primary CD4-BS, D368 (refs. 5,26). Both wild-type and mutated gp160 were efficiently expressed on the surface membrane of transfected human embryonic kidney (HEK) 293T cells and did not show alterations in their folding and trimeric structure, as determined by reactivity with a wide panel of anti-envelope mAbs, including trimer-preferring and cleavage-sensitive mAbs such as PG9 (ref. 27), 35O22 (ref. 28), and PGT151 (ref. 20) (Supplementary Table 3). Charge inversions of E62, E64, H66, and K207 virtually abrogated the infectivity of all of the HIV-1 Env types tested, regardless of their genetic clades, as did the control D368R mutation (Fig. 2c,d).
We introduced a set of additional mutations into the BaL Env to obtain a more complete topological and functional mapping of the CD4-BS2 region. Alanine substitutions of E62, E64, H66, and K207, either individually or in combination, consistently reduced HIV-1 infectivity, albeit less markedly than charge inversions; nevertheless, a triple-alanine substitution (D62A, E64A, and K207A) caused a complete loss of infectious capacity (Supplementary Fig. 3). Charge inversions or alanine substitutions of neighboring solvent-exposed residues either had no effects (K59E, E102A, or E102K) or enhanced infectivity (Y61A, presumably owing to removal of the bulky side chain, which allowed for closer approach of CD4 to CD4-BS2), thus corroborating the specificity of the effects observed upon disruption of CD4-BS2. These results suggest a critical role of the quaternary trimer interaction with CD4 for the progression of the HIV-1 fusogenic process.
Stabilization of CD4 binding to the trimer by quaternary interaction
To elucidate the mechanism underlying the complete loss of infectivity induced by disruption of CD4-BS2, we initially tested whether the mutations caused reduced gp120-gp41 association, but the levels of gp120 shedding from cell-surface-expressed gp160 were unaltered (data not shown). Next, we mutagenized the four CD4-BS2 residues (E62, E64, H66, and K207) in both the DS-variant and wild-type soluble SOSIP trimers and measured the ability of the mutants to bind sCD4. The trimers were expressed in HEK293 free-style (FS) cells and purified to homogeneity (Supplementary Fig. 4). As expected from the outward orientation and complete solvent exposure of the side chains of the CD4-BS2 residues, their mutation by simple charge inversion had no effect on the correct folding and trimeric configuration of the mutants, as shown by reactivity with a panel of mAbs that included several trimer-preferring, conformation-dependent and cleavage-sensitive mAbs (Supplementary Fig. 4a). Surface plasmon resonance (SPR) analysis showed that all of the DS-SOSIP-based CD4-BS2 mutants had markedly reduced sCD4-binding affinity (KD), which was primarily due to an accelerated off-rate, with K207E exhibiting the most pronounced decrease (KD = 262.0 nM, compared to 8.0 nM for the unmutated DS-SOSIP), followed by H66E (KD = 127.2 nM) and E62K E64K (KD = 57.7 nM); no detectable binding was seen with the control D368R mutant (Fig. 3a). Likewise, all the wild-type-based mutants showed a similarly reduced ability to bind sCD4 in both ELISA (Supplementary Fig. 5a) and SPR assays (Fig. 3b), thus excluding the possibility that the reduced CD4-binding affinity could be specific for the stabilized DS-SOSIP trimer. Because soluble SOSIP trimers display some structural and antigenic differences from native Env trimers29, we also tested CD4 binding to full-length native gp160 expressed on the surface membrane of HEK293T cells. All CD4-BS2 mutants for both BG505 and BaL Env exhibited reduced binding to CD4–immunoglobulin compared with the respective wild-type proteins (Supplementary Fig. 5b). Together, these results demonstrate that CD4-BS2 has an important role in stabilizing the interaction of the HIV-1 Env trimer with CD4.

(a) The kinetics of sCD4 binding to the DS-SOSIP-based trimers as measured by single-cycle SPR. (b) The kinetics of sCD4 binding to wild-type and mutated trimers as measured by single-cycle SPR. (c) Charge inversions in CD4-BS2 abrogated the CCR5-binding capacity of BG505 SOSIP.664 trimers after sCD4 treatment. Binding of wild-type (WT) and mutated soluble BG505 SOSIP.664 trimers to CCR5 expressed on the surface of Cf2Th/syn-CCR5 cells was assessed by flow cytometry. Mean fluorescence intensity (MFI) values were calculated relative to the positive control (sCD4-bound wild-type trimer). The data shown represent the mean ± range of two independent experiments. (d) Charge inversions in CD4-BS2 abrogate the mAb 48d-binding capacity of BG505 SOSIP.664 trimers after sCD4 treatment, as assessed by ELISA. The data shown represent the mean ± range of duplicate wells from a representative experiment out of three performed that yielded similar results.
Quaternary CD4 interaction confers coreceptor-binding competence
Next we studied the events downstream of CD4 binding, which induce gp120 to acquire coreceptor-binding competence and eventually to dissociate from gp41, thus leading to activation of the fusogenic process. After preactivation with sCD4, the BG505 SOSIP.664 soluble trimer efficiently bound to cell-surface-expressed CCR5; in contrast, none of the CD4-BS2 mutants were able to bind to CCR5 regardless of sCD4 treatment (Fig. 3c). Likewise, little, if any, induction of the 48d epitope, which extensively overlaps the CCR5-binding surface30, was detected in any of the CD4-BS2 mutants after treatment with sCD4 (Fig. 3d). These results indicate that the quaternary interaction of CD4 with the gp120 inner domain is essential for the conformational transitions that lead to coreceptor binding and progression of the fusogenic process.
Limited effects of CD4-BS2 mutagenesis on monomeric gp120
As mutation of specific residues in the gp120 inner domain, including H66, was previously reported to reduce the binding of sCD4 and coreceptors to monomeric gp120 (refs. 7,31), we investigated the effects of CD4-BS2 mutations in the context of gp120 monomers. We confirmed the correct folding of the mutated gp120 proteins on the basis of their reactivity with a panel of mAbs to gp120 (Supplementary Fig. 6). Among all mutants, only H66E showed a slight reduction in sCD4-binding activity in both BG505 and BaL gp120, whereas K207E showed reduced binding only in BG505 and not in BaL gp120 (Supplementary Fig. 6a), indicating that CD4-BS2 is not a functionally relevant receptor-binding site in the context of the monomer. Mutation of CD4-BS2 residues resulted in decreased CCR5 and 48d binding in the absence of sCD4 (Supplementary Fig. 6b), which confirmed the findings of previous mutagenesis studies that attributed this effect to reduced monomer flexibility caused by altered interlayer interactions in the inner domain7,31. In contrast, in the presence of sCD4, binding of 48d was unaffected by CD4-BS2 mutations, which shows that sCD4 was able to induce the full range of conformational changes in the context of the mutated monomer, whereas binding to CCR5 was only moderately reduced, with the exception of mutant K207E that, as expected, lost the ability to bind CCR5, as this residue is part of the coreceptor-contact surface32 (Supplementary Fig. 6b). Overall, the effects observed after mutation of monomeric gp120 were only partial and limited, unlike the complete loss of CCR5 and 48d binding observed in the context of the trimer (Fig. 3c,d).
To interpret the above results, we examined the available crystal structures of monomeric gp120, which invariably revealed a disassembled CD4-BS2 region attributable to the opposite spatial orientation of the E62, E64, H66, and K207 side chains, which in the monomer point inward to establish intramolecular contacts with residues in the underlying layer 2 of the gp120 inner domain8,9,10,11; conversely, in the structures of soluble Env trimers, the same residues point outward to form a continuous, electrostatically charged surface crest13,16,17,18 (Supplementary Fig. 7). Thus, as previously suggested7,31, the slight reduction of CD4 and CCR5 binding observed in the context of monomeric gp120, as well as the reduced binding of mAb 48d in the absence of sCD4, may have been due to reduced molecular flexibility and distant allosteric effects secondary to altered interlayer adherence in the flexible inner domain of the gp120 glycoprotein.
Identification of CD4 residues involved in quaternary interaction
To further validate the role of CD4-BS2 in the HIV-1 envelope-receptor interaction, we investigated the CD4 residues that make contact with CD4-BS2 (Fig. 4a). On CD4, two discontinuous surfaces mirror the quaternary binding site on the Env trimer, with one surface interacting with the primary CD4-contacting protomer, and the other interacting with the quaternary CD4-contacting protomer (Fig. 4b). To estimate the surface areas of interaction, we used previously determined coordinates of Env trimers and two-domain sCD4 (residues 1–176) fitted to the electron microscopy density (Fig. 4c and Supplementary Table 2). Fittings of CD4 to the reconstructed cryo-EM map revealed an ∼6-Å translation and an ∼8° rotation for the median position of CD4 in the initial contact structure relative to the position of CD4 in the monomeric postfusion structure. A total of 7 Env trimers and 29 CD4 domains produced a matrix of 203 interactive surfaces, yielding 526 ± 52 Å2 of buried surface area for the primary CD4-contacting gp120, and 159 ± 41 Å2 of buried surface area for the quaternary CD4-contacting gp120 (Supplementary Table 4). Examination of the contact site in the fitted model and in the MD simulation revealed four residues in CD4 domain 1, namely, K21CD4 and K22CD4 in the CDR1-like loop, and D63CD4 and Q64CD4 in the D-E loop, as potentially interactive with CD4-BS2 in the fitted cryo-EM structure and in the MD simulation; none of these residues had been previously reported to contact gp120 (refs. 8,9,10,11).

(a) Left, top view of the cryo-EM structure of a single-CD4-bound HIV-1 Env trimer, showing the quaternary interaction of CD4 with neighboring gp120 protomers; gp41 was removed for clarity. Right, a close-up representation of CD4 residues (highlighted by stick representation and color) at the interface with CD4-BS2 in gp120 (D63, negatively charged; K21 and K22, positively charged). (b) Open-book surface representations of the quaternary HIV-1 Env–CD4 interaction in the optimized cryo-EM structure. The CD4 footprint on the HIV-1 Env trimer is shown in yellow, and the trimer footprints on CD4 are shown in purple for the primary CD4-contacting gp120 and in pink for the quaternary CD4-contacting gp120. Zoomed-in views show the contact sites (within 5.5 Å). (c) Left, top view of the cryo-EM structure of a single-CD4-bound HIV-1 Env trimer, showing CD4 molecules fitted into the cryo-EM density map (yellow) and CD4 molecules in the monomeric gp120 (gp120m)-bound (postfusion) orientation (listed in Supplementary Table 2) (gray). CD4 fits were performed with CD4 domains 1 and 2; CD4 domains 3 and 4 were added to the CD4 molecule showing the best fit by alignment with four-domain CD4 (PDB 1WIO) and by transplantation of domains 3 and 4 onto fitted domains 1 and 2. Gp41 was removed for clarity. Right, orientation of the CD4 molecules fitted into the cryo-EM density map relative to the gp120m-bound orientation. “Best-fitted CD4” refers to the CD4 fit that was chosen for the final DS-SOSIP-CD4-PGT145 model fitted to the cryo-EM density. This fit showed the highest map-to-model correlation among all the CD4 molecules. (d) Disruption of the CD4-BS2 interactive site reduces the HIV-1 receptor function of CD4. Pseudoviruses carrying wild-type (WT) BG505-T332N or BaL envelopes were tested for infectivity in Cf2Th/syn-CCR5 cells expressing different CD4 mutants. Infectivity values for the mutants were calculated relative to the value obtained with wild-type CD4. The data shown represent the mean ± range of duplicate wells from a representative experiment out of two performed that yielded similar results.
To assess the functional role of the CD4 residues predicted to interact with CD4-BS2, we mutagenized each residue by charge inversion, expressed full-length CD4 mutants on the surface of Cf2Th/syn-CCR5 cells, and tested the mutants for HIV-1 receptor function. We verified the correct expression and folding of the mutants with a panel of mAbs to CD4 (Supplementary Fig. 8); of note, mAbs RPAT4, 13B8, and 379 showed a significant loss of reactivity with mutant K22ECD4, which suggests that the mutations directly overlap their binding footprint or, less likely, that charge inversion may have caused subtle conformational changes. Charge inversion of two CD4 residues, K22CD4 and D63CD4, caused a dramatic loss of HIV-1 receptor function, as seen with both the BG505 and BaL envelopes, whereas charge inversion of K21CD4 had more limited effects, and alanine substitution of Q64CD4 had no effects; as expected, alanine substitution of the critical F43CD4 residue caused a complete loss of receptor function (Fig. 4d). Analysis of a more extended panel of CD4 mutants showed that alanine substitutions of K22CD4 and D63CD4 also reduced HIV-1 infectivity, albeit less markedly than charge inversions (Supplementary Fig. 8a). These results confirmed that alterations on both sides of the CD4:CD4-BS2 interface disrupt the functional interaction between the HIV-1 envelope and the CD4 receptor.
Role of quaternary interaction in neutralizing antibody binding
To assess the immunogenicity of CD4-BS2 and its functional relevance to antibody-mediated neutralization and immune evasion, we examined the potential interaction of this site with a panel of anti-CD4 supersite mAbs isolated from HIV-1-infected individuals. Numerous structures of these mAbs have been determined in complex with monomeric gp120 (refs. 33, 34, 35, 36, 37) and in the context of the Env trimer12,13. Docking onto the pre-fusion closed Env trimer revealed that all of the anti-CD4 supersite mAbs reach deep into the interprotomer groove, although only some of them (most notably VRC03, VRC06, and VRC-CH31) appear to make direct contact with CD4-BS2 (Fig. 5a). Optimization of quaternary contacts appeared to occur by somatic hypermutation-induced alteration of the framework regions of these antibodies (Supplementary Fig. 9). We note that quaternary interactions were predicted for several anti-CD4 supersite antibodies, including VRC01 (ref. 12), and the crystal structure of the VRC01-bound X1193.c1 SOSIP.664 trimer, but not of the JR-FL trimer, indeed documented quaternary contacts with residues Y61 and E64 in the gp120 inner domain13. These observations may relate to strain-specific modes of interaction between Env and broadly neutralizing antibodies.

(a) Fabs for select neutralizing mAbs to CD4-BS (i.e., VRC03, VRC06, and VRC-CH31) are shown along the top, docked to a surface representation of the HIV-1 Env trimer in its pre-fusion closed conformation. The close-up views below show the putative contact area with CD4-BS2, with key residues (E62, E64, H66, and K207) highlighted by stick representation with Env colored by electrostatics. (b) Disruption of CD4-BS2 affects binding of neutralizing mAbs specific for the CD4-binding site. Binding of anti-CD4-BS mAbs to wild-type (WT) and mutated soluble cleaved HIV-1 Env trimers (BG505 SOSIP.664) as assessed by ELISA. The data shown represent the mean ± range of duplicate wells from a representative experiment out of two performed that yielded similar results. (c) A surface representation of the CD4 supersite of vulnerability, with CD4-contact sites highlighted in yellow. Glycans are shown in stick representation.
Mutation of the key residues in CD4-BS2 had diverse effects on binding of anti-CD4 supersite mAbs to the BG505 SOSIP.664 trimer (Fig. 5b). Specifically, charge inversion of K207 substantially decreased recognition by VRC03 and, to a lesser extent, VRC-CH31 and 3BNC117, but not VRC06; in contrast, mutation of E62 E64 selectively affected VRC06 binding, whereas none of these mutations significantly reduced the binding of VRC01 and VRC-PG04. We confirmed these results with a more extended panel of mAbs and envelope mutants tested on cell-surface-expressed gp160 from both BG505 and BaL (Supplementary Table 3). In line with their dependence on CD4-BS2, and consistent with previous data36, both VRC03 and VRC06 exhibited trimer-preferring reactivity, with no detectable binding to monomeric BG505 and BaL gp120 in ELISA (Supplementary Fig. 6c), even though binding and even crystallization in complex with monomeric gp120 have been reported for other strains37,38. Overall, these results demonstrate that certain neutralizing human mAbs establish quaternary contacts with the HIV-1 Env spike, and thus provide inferential evidence that the CD4-BS2 region is immunogenic in vivo.
Discussion
Our work delineates the initial contact of the HIV-1 Env with the CD4 receptor, documenting the quaternary configuration of a retrovirus receptor-binding site, a feature previously recognized for picornaviruses39,40,41, but never before for enveloped viruses. We found that the initial CD4-binding site on the pre-fusion HIV-1 Env trimer spans conserved regions on two neighboring gp120 protomers, which embrace the receptor with an extended quaternary surface complementary to discontinuous elements located on two faces of the N-terminal domain of CD4. This initial interaction with CD4 represents the first event in a multistep process during which the trimer undergoes sequential structural transitions toward downstream intermediate states (i.e., a requisite intermediate high-FRET state22, a three-CD4-bound state21,42, and coreceptor-bound states22,43) before reaching a final, low-energy postfusion state1,2,3. To prevent CD4-induced conformational changes in the trimer, which until now have impeded the study of the initial events of receptor interaction, we took advantage of a conformationally stabilized soluble trimer, DS-SOSIP, which binds CD4 with an asymmetric 1:1 (CD4:trimer) stoichiometry16. Accordingly, our cryo-EM structure showed structural asymmetry in the initial Env–CD4 complex, with an ∼3-Å outward shift of both CD4-interactive protomers toward the incoming CD4 molecule. This shift represents perhaps the earliest conformational change to take place in the HIV-1 Env trimer. Its marked asymmetry seems to be maintained in the requisite transition intermediate observed by smFRET22, before binding of two additional CD4 molecules yields the 3:1 (CD4:trimer) stoichiometry commonly observed with the CD4-bound open trimer21,23,42.
The dramatic loss of HIV-1 infectivity that we observed when we mutated key residues involved in the quaternary Env-CD4 contact both on the trimer and on the receptor sides indicates that binding to the full quaternary Env surface is critical for the functional interaction of CD4 with the HIV-1 virion. The more pronounced effect induced by charge inversion versus alanine mutagenesis suggests that CD4 interaction with CD4-BS2 is predominantly driven by electrostatic forces. The quaternary site in the inner gp120 domain appears to be essential to stabilize the Env-receptor interaction and to trigger Env activation, which in turn leads to coreceptor binding and progression of the fusogenic process. In agreement with this model, the inner domain has been implicated in regulation of the transition from the pre-fusion ligand-free to the CD4-activated conformation in the context of monomeric gp120 (ref. 7). However, the orientation of the CD4-BS2 region is dramatically different in the trimer and the monomer. In the prefusion metastable trimer, the key residues that form CD4-BS2 point outward and are well exposed to the solvent, facing the incoming CD4 receptor; in contrast, the same residues display a peculiar inward orientation and are almost completely buried in the fusion-transitioned structure of the monomeric gp120 core8,9,10,11. Thus, in the context of the monomer only, CD4-BS2 residues establish intramolecular interactions with layer-2 residues, which affect interlayer adherence in the inner domain and, consequently, monomer flexibility and spontaneous access to the coreceptor-binding-competent state. This can explain the allosteric effects on CD4 binding that have been previously reported after mutation of H66 and other inner-domain residues in the context of the monomer7,31. The cryptic configuration of the CD4-BS2 region in monomeric gp120 also suggests that the quaternary CD4-contact site may be a region of conformational masking3 that facilitates the immune evasion of this critical surface.
Major efforts aimed at developing a protective vaccine are currently focused on the design of HIV-1-Env-based immunogens that display the major epitopes recognized by broad and potent neutralizing antibodies14,16,44. The definition of the initial CD4-contact surface in the HIV-1 Env trimer allowed us to reassess the supersite of vulnerability formed by the CD4-binding site (Fig. 5c), with potential implications for the development of HIV-1 vaccines. Our observation that selected neutralizing antibodies to the CD4-BS, such as VRC03 and VRC06, which preferentially recognize the trimeric envelope, develop contact with CD4-BS2 through somatic hypermutation provides evidence that this region is immunogenic in vivo and, therefore, should be considered in the design of vaccine immunogens. Furthermore, the CD4-BS2 region may provide a new molecular target for the development of HIV-1 entry inhibitors.
Methods
Sample preparation for electron microscopy analysis.
DS-SOSIP.664, four-domain CD4, and PGT145 Fab were expressed and purified as described previously16. Purified DS-SOSIP.664 at 1 mg/ml was incubated with 20 μM of four-domain sCD4 for 16 h at room temperature; PGT145 Fab was then added at three-fold molar excess of the trimer concentration, and the sample was incubated for another 3–4 h before vitrification.
Specimen vitrification for cryo-EM.
To avoid aggregation during vitrification, the sample was incubated with a small amount of dodecyl maltoside at a final concentration of 0.085 mM (2× below the critical micelle concentration). To prepare specimens for cryo-EM we applied 3 μL of sample to a freshly plasma-cleaned holey gold grid, allowed the sample to adsorb to the grid for 30 s, and then blotted the grids with filter paper and plunge-froze them in liquid ethane using the CP3 cryo-plunger (Gatan, Inc.) (20 °C, 85–90% relative humidity).
Cryo-EM data collection and raw-frame alignment.
As summarized in Table 1, data were acquired using the Leginon system45 installed on a Krios electron microscope operating at 300 kV, with a dose of ∼86 e−/Å2 and an estimated defocus ranging from 1.0 to 4.0 μm underfocus (distributed in an approximately Gaussian manner and centered at 2.5 ± 0.8 μm). The dose was fractionated over 40 raw frames collected over a 10-s exposure time (250 ms per frame) on the Gatan K2 Summit direct detection device. A total of 1,900 such 'movies' were automatically collected over a span of four data collection sessions, recorded at a nominal magnification of 22,500×, corresponding to a pixel size of 1.1 Å at the specimen level. The individual frames were globally aligned using a GPU-enabled frame-alignment program (generously provided by Y. Cheng and X. Li)46.
Image processing and refinement.
Unless otherwise noted, all of the following image-processing steps were performed within the Appion pipeline47,48. The contrast transfer function for all micrographs was estimated with the GCTF package49. All micrographs were also manually masked with the “manual masking” tool in Appion to remove regions that were over gold and/or contained large amounts of disordered or aggregated particles. To obtain a clean stack of particles on which to perform the final refinement, we first performed an iterative stack cleaning procedure (described below). Particles were initially selected using DoG Picker50 from 800 micrographs (Supplementary Fig. 1a). A phase-flipped, contrast-inverted, and 2× binned stack of 10,000 particles was created from these picked particles with a box size of 160 pixels and pixel size of 2.2 Å, and subjected to reference-free 2D alignment and clustering by CL2D51 to obtain 256 2D classes. The classes were inspected visually, and 15 of these classes (representing different views) were used as templates to select particles over the entire set of micrographs using the template-picking algorithm in Appion52,53. After 2D alignment and clustering with CL2D and RELION48 (outside of Appion) to eliminate class averages that did not display identifiable features (Supplementary Fig. 1b), 100,500 particles were retained for all subsequent analyses, which were done outside of Appion. A map of unliganded BG505 SOSIP.664 (EMD-5782), low-pass filtered to 60 Å, was used as the starting point of 3D classification/sorting to eliminate trimer particles that were not bound to both CD4 and PGT145, as well as to exclude badly formed particles. After two rounds of 3D classification separating the particles into four classes in the first round and three classes in the second round, the class that showed both PGT145 and CD4 bound (class I) (Supplementary Fig. 1c) was selected for 3D refinement in RELION (Table 1 and Supplementary Fig. 1d,e). Automasking in RELION54 yielded an 18-Å resolution map. We truncated the CD4 density from this map using the volume eraser tool in Chimera, and used the relion_mask_create program to make masks that extended from this CD4-truncated map. We then carried out 3D refinement with these manual masks, which resulted in maps with incrementally truncated density for the bound CD4 with resolutions of 11 Å (mask 1) and 9 Å (mask 2) (Supplementary Fig. 1e). Next, we carried out dose fractionation by incrementally excluding movie frames; the best resolutions of 9 Å (mask 1) and 7.5 Å (mask 2) were achieved for the two maps that were generated with masks truncated at different lengths, by using the first 30 frames, which corresponded to a total dose of ∼51 e−/Å2. In a second masking strategy we generated masks extending from the 3D map from class I (Supplementary Fig. 1c) followed by auto-refine within RELION using these manual masks, resulting in a best resolution of 6.8 Å after post-processing within RELION (Supplementary Fig. 1e). This 6.8-Å-resolution reconstruction showed the best-resolved density for the bound four-domain CD4 molecule. This map was used for all model fitting unless otherwise noted.
Model fitting.
First, all HIV-1 Env soluble trimer models available in PDB were fitted onto the 7.5-Å cryo-EM map (Supplementary Table 1). Next, each fitted trimer was separated into its gp120-gp41 protomers, and the protomers were fitted to optimize map-to-model correlation. The trimer with the optimal fit was assembled through combination of the protomers that fit the map with highest correlation. On examination of the protomers, all three of which came from PDB 5ACO, it was found that the protomers were missing residues 59–67 in the gp120 chains. This stretch of residues was added back into each of the protomers by superposition with the trimer in PDB 5FYL13 and grafting of residues 57–70 from each protomer of PDB 5FYL to each equivalent protomer of the fitted trimer. This resulted in a small increase in map-to-model correlation from 0.84 to 0.85. Finally, the fits of the protomers were optimized within the 6.8-Å cryo-EM map. Once the fit of the trimer to the map had been optimized, the coordinates of free PGT145 (PDB 3U1S)56 were fit to the density visible in the map near the V1V2 region of the trimer. The Fab was first fit as a whole; then, fits for the variable and constant domains were optimized separately to maximize map-to-model correlation. Two orientations of the antibody Fab fragment, rotated by 180° along the longitudinal axis of the Fab, were tested, and the orientation that fit with higher correlation (0.75) was also visually found to fit better to the map (Supplementary Fig. 2) compared with the Fab orientation that fit the electron microscopy map with reduced correlation (0.66). To fit CD4, we used an approach similar to that used for the trimer, using all the PDB deposits containing CD4 (Supplementary Table 2). For gp120-bound CD4, we superposed the gp120 in the gp120–CD4 complex with the gp120 protomer in the trimer that had its outer-domain CD4-binding loop facing the CD4 density in the cryo-EM map. We designated the resulting orientation as the 'post-fusion' orientation of CD4. For CD4 structures that were not bound to gp120, CD4 residues 2–176 were superimposed on residues 2–176 of CD4 from PDB 2NY4 that had already been placed in the post-fusion orientation. From this starting position, we fit each CD4 molecule using rigid-body docking within Chimera55 (Supplementary Table 2 and Fig. 4c). Following model fitting, the PGT145-trimer and CD4-trimer interfaces were minimized using the steepest descent protocol in Chimera, and the interactive areas were optimized by varying side chain rotamer conformations. The 7.5-Å map obtained with Mask2 in RELION auto-refine (Supplementary Fig. 1e) showed better definition of the glycan density around the trimer, and was used to place the glycans.
Structural analyses.
To calculate the relative orientations of the CD4-bound protomers in the single CD4-bound Env trimer structure, we first superposed all trimer structures with the PDB entries listed in Supplementary Table 1 to the single-CD4-bound structure, by aligning a protomer of each trimer to the protomer of the single-CD4-bound Env trimer that did not contact CD4. For trimers where each protomer had been determined independently (i.e., where electron microscopy maps were refined using C1 symmetry or where the entire trimer was part of one crystallographic asymmetric unit), each protomer was fit to the protomer of the single CD4-bound Env trimer that did not contact CD4. From this starting orientation, for each pairwise comparison between the single CD4-bound trimer and an Env trimer from the PDB, the rotation and the translation needed to superpose the two CD4 contacting protomers to the respective protomers of the aligned Env trimer were calculated (Fig. 4c). All superpositions were performed using the Superpose module57 within the CCP4 suite58, and rotation angles were obtained with Superpose (MATLAB, The MathWorks Inc.) and Origin (OriginLab).
For estimating surface area buried at the interfaces between trimer and CD4, starting from the Env trimers listed in Supplementary Table 1 that had been fitted to the EM density map, trimers missing any of the following CD4 contacting residues were removed: 281, 368, 371, 280, 428, 62, 64, 65, 66, or 207. This resulted in seven Env trimer structures. Similarly, for CD4, starting from Supplementary Table 2 with CD4 structures that had been fitted to the EM density map, CD4 missing any of the following Env trimer contacting residues were removed: 20, 21, 22, 40, 43, or 64. This resulted in 29 CD4 structures. Complex structures for all pairwise Env trimer–CD4 combinations were minimized using Chimera55 (only CD4 D1 domain and Env residues that are 10 Å within CD4 were allowed to move). The side chain rotamers of the interface residues were further refined using SCAP59. Buried surface areas were calculated using PISA60.
To define the CD4 supersite that is targeted by CD4-BS antibodies on the pre-fusion closed HIV-1 Env trimer, we superposed PDB coordinates of HIV-1 gp120 in complex with CD4-BS antibodies isolated from 14 human donors35 onto the BG505.SOSIP (PDB 4TVP) by aligning the outer domain residues 255:392, 412:422 and 437:459. The CD4 supersite for antibodies was defined as a set of HIV-1 Env atoms that are within 5.5 Å of docked CD4-BS antibodies, including the VH1-2 germline-derived VRC01-class antibodies, the VH1-46 germline-derived 8ANC131 class antibodies and the CDR H3-dominated antibodies (VRC13, VRC16, CH103, and HJ16). In addition to the previously defined primary antibody contact region on gp120, the CD4 supersite for CD4-BS antibodies also extended to the neighboring gp120 protomer in the trimeric pre-fusion closed Env and overlapped with CD4-BS2. Electrostatic potential molecular surfaces for antibodies and HIV-1 gp120 were generated with the APBS and PDB2PQR server (http://nbcr-222.ucsd.edu/pdb2pqr_2.1.1/).
Molecular dynamics simulation.
A model of the structure of the HIV-1 BG505-SOSIP.664 trimer with a single CD4 molecule bound was subjected to an all-atom, isobaric-isothermal (1 atm, 310 K) MD simulation. The simulation was done with the AceMD61 program (v.3212) on the Biowulf Linux cluster at the NIH, Bethesda, MD (http://biowulf.nih.gov). The complex was explicitly solvated with TIP3P water, neutralized through the addition of counterions (Na+, Cl− to 0.15 M), with periodic boundary conditions applied. Electrostatic interactions were calculated using the particle-mesh Ewald summation with grid spacing of 1 Å along with a 9-Å cutoff. The CHARMM27 force field was used61 for protein. The system was energy minimized, warmed to 310 K, and submitted to a production run. We implemented a 4-fs integration-time step by making use of a hydrogen mass repartitioning scheme, and we collected data for 668 ns. Langevin dynamics were used to maintain temperature, and a Berendsen barostat was used to control pressure.
Sequence conservation analysis.
Sequence alignment of a segment of the C1 region (centered around the α1--helix) and of the C2 region (a segment of the β3-β4 loop) of the gp120 inner domain from the all the available group M HIV-1 and SIVcpz isolates (including both subtypes A–K and recombinant forms) was obtained from the Los Alamos HIV database (http://www.hiv.lanl.gov). The sequence logo was created with WebLogo (http://weblogo.berkeley.edu). The height of each stack indicates the degree of conservation for each residue; the relative height of each letter within an individual stack represents the frequency of the indicated amino acid at that site.
Mutagenesis, protein expression, and purification.
Site-directed mutagenesis was carried out with the QuikChange II site-directed mutagenesis kit (Agilent Technologies). The BG505-SOSIP.664 trimer was expressed by cotransfection of the relevant plasmid (a kind gift of John P. Moore) with a plasmid expressing the cellular protease furin into HEK293FS cells. Cell-free supernatants were harvested after 7 d, passed through a 0.22-μm filter, and loaded onto a Galanthus nivalis lectin column (Vector laboratories). After repeated washing with PBS, bound proteins were eluted with 1 M methyl α-D-mannopyranoside in PBS, and then subjected to dialysis toward PBS. Dialyzed samples were concentrated to 2 ml using Amicon Ultra-15 centrifugal filter units (MWCO 50,000, Millipore) and applied to a PBS pre-equilibrated Superdex 200 column. The peak corresponding to the trimeric form was collected and concentrated, after which sample underwent a second round of Superdex 200 purification. Finally, purified trimers were concentrated to 0.5–1 mg/ml and stored at −80 °C. Monomeric HIV-1 BG505-T332N gp120 and BaL gp120 (gene synthesized by GeneArt) were expressed in HEK293FS cells. After 7 d, the supernatants were harvested and passed through a 0.22-μm filter. We purified gp120 by loading filtered supernatants onto a lectin column as described above. The eluted proteins were concentrated with Amicon Ultra-15 centrifugal filter units (MWCO 10,000, Millipore) and applied to a Superdex 200 column. DS-SOSIP.664-based trimers were purified as described previously16. Briefly, cell-free supernatants were loaded onto a VRC01 or 2G12 affinity column and subjected to gel filtration. The trimeric form was collected and passed through a 447-52D affinity column for negative selection. Further cleanup was done with a column of anti-V3 (1006-15D, 2219, 2557, 2558, 3074, 50.1).
Expression of full-length HIV-1 gp160 or CD4.
The full-length CD4 gene and the gene encoding gp160 from BG505-T332N and BaL (both from Edward Berger) were mutagenized as described above for the SOSIP trimer. HEK293T cells were used to express CD4 or gp160 on the cell-surface membrane. The cells were seeded into six-well plates at 300,000 cells per well in 2 ml of DMEM containing 10% FBS (DMEM 10%). After overnight incubation at 37 °C in a humidified atmosphere with 5% CO2, the culture medium was replaced with 1.5 ml of fresh 10% DMEM. We prepared DNA-FuGENE 6 complex by mixing 2 μg of DNA with 10 μl of FuGENE 6 Transfection Reagent (Promega) in Opti-MEM (Gibco) to a final volume of 150 μl and incubated it for 15 min at room temperature (RT). The cells were transfected by dropwise addition of the DNA–FuGENE 6 complex to each well, and were harvested after 48–60 h. The same protocol was used for CD4 expression on the surface membrane of Cf2Th/syn-CCR5 cells (AIDS Reagent Program).
Flow cytometry.
HEK293T cells expressing CD4 or gp160 were harvested by mechanical shaking and pipetting, washed with PBS, and incubated with anti-CD4 or anti-gp120 (5 μg/ml) or serially diluted human Fc-chimeric CD4-immunoglobulin (produced in HEK293FS cells; plasmid provided by Michael Farzan) at RT for 30 min. After being washed with PBS twice, the cells were incubated with PE-conjugated sheep anti-mouse IgG (Sigma) or goat anti-human IgG (Southern Biotech) at RT for 15 min. The cells were then washed once with PBS, fixed in 2% paraformaldehyde and analyzed on a BD FACSCanto (BD Biosciences). Data analysis was done with the FlowJo software.
Pseudovirus preparation and infectivity assays.
Viral pseudoparticles expressing wild-type or mutated gp160 from HIV-1 BG505-T332N or BaL were produced in HEK293T cells by cotransfection of Env-expressing plasmids together with a backbone plasmid, pSG3Δenv, expressing a full-length HIV-1 clone with a defective Env gene. To produce the pseudoparticles, we mixed 2 μg of each Env-expressing plasmid and 4 μg of the backbone plasmid in Opti-MEM medium (Gibco), added 24 μl of FuGENE 6 transfection reagent, and incubated the mixture for 15 min at RT. DNA–FuGENE 6 complex was then added to the cells and incubated overnight at 37 °C. After the culture medium had been replaced with 2 ml of fresh 10% DMEM, the cells were incubated for 48 h and the supernatants containing the pseudoviruses were harvested by centrifugation. For pseudovirus infection, TZM-bl cells (AIDS Reagent Program) were harvested and seeded into 96-well flat-bottom plates at 10,000 cells per well in 100 μl 10% DMEM. After 30 min at 37 °C, the pseudovirus stock (normalized to 1 ng of p24GAG protein per mutant) was added to the cells in a total volume of 200 μl/well. Reporter gene activation was detected 2 d later with a luciferase assay kit (Promega) with a luminometer (PerkinElmer). Relative light units (RLU) were recorded, and the final values were normalized against the values obtained with the wild-type Env set at 100%. All the samples were tested in duplicate wells.
We produced pseudoviruses expressing firefly luciferase by cotransfecting Env-expressing plasmids (wild-type BaL or BG505-T332N) together with a backbone plasmid, pNL4-3.Luc.R−E− (AIDS Reagent Program), at a ratio of 1:1. Supernatants containing pseudoviurses were harvested by centrifugation after 48 h of incubation. We prepared target cells by transfecting wild-type or mutated CD4-expressing plasmids to Cf2Th/syn-CCR5 cells. After incubation for 24 h, the cells were harvested and seeded to 96-well flat-bottom plates at 10,000 cells per well in 100 μl of 10% DMEM. Pseudovirus stocks were added to the cells to a total volume of 200 μl/well. The reporter gene activity was detected 2 d later with a luciferase assay kit (Promega) and expressed as RLU. Serial dilutions of the wild-type CD4 plasmid were used to generate a reference curve, which allowed us to extrapolate accurate infectivity values for each mutant on the basis of expression levels, as measured by flow cytometry using mAb OKT4. All samples were performed in duplicate.
Enzyme immunoassays.
All the antibodies and envelope proteins used in ELISA experiments were diluted in 0.02% casein in PBS. All samples were washed three times after each step with 1× wash buffer (R&D Systems). For assessment of sCD4 binding to the BG505 SOSIP.664 trimers or gp120 monomers, 96-well ELISA plates (Corning) were coated with 2 μg/ml mAb 2G12 at 4 °C overnight. After blocking with 0.2% casein in PBS, purified trimers or monomers (2 μg/ml) were added and incubated for 2 h at RT. After washing, serially diluted four-domain sCD4 (R&D Systems) was added and incubated for 1 h at RT. After washing, the samples were incubated with biotin-labeled mAb OKT4 (eBioscience; 13-0048-82) for 1 h; after additional washing, horseradish peroxidase (HRP)-conjugated streptavidin (R&D Systems; DY998) was added for 1 h at RT. The reaction was revealed by incubation with the appropriate substrate for 10 min before addition of the stop solution. Light absorption was measured at a wavelength of 450 nm. All samples were tested in duplicate or triplicate wells. To evaluate antibody binding, we coated ELISA plates with 5 μg/ml of an anti-gp120 C-terminus hyperimmune sheep antiserum (Aalto Bio Reagents; D7324) for monomeric gp120 or with 0.5 μg/ml of rabbit anti-His tag antiserum for trimers (GenScript; A00174). Monomeric gp120s or trimers were captured on the plates, and various antibodies to gp120 (0.5–5 μg/ml or serial dilutions starting from 5–20 μg/ml) were added to the wells and incubated for 1 h, and subsequently for another hour with HRP-conjugated goat anti-human IgG (Sigma; A8419) at RT. The subsequent steps were the same as described above. For all the mutants, relative binding compared to the wild type was calculated. To evaluate antibody binding to DS-SOSIP.664, we coated the ELISA plate with 10 μg/ml lectin from Galanthus nivalis (Sigma). DS-SOSIP.664 proteins were added at 1 μg/ml for capture. Various antibodies to gp120 were tested at 1–5 μg/ml or with serial dilutions starting from 5–20 μg/ml.
Surface plasmon resonance analyses.
Single-cycle kinetics analysis of two-domain sCD4 binding to the BG505 SOSIP.664 and DS-SOSIP.664 trimers was performed at 20 °C on a Biacore T200 (GE Healthcare) as described previously16. Briefly, mAb 2G12 was immobilized onto four flow cells of a CM5 sensor to ∼2,000 response units (RU) using the amine-coupling method. Purified BG505 SOSIP.664 trimers were injected to two sample flow cells at a flow rate of 5 μl/min for 2 min, and captured to 300–400 RU. The other two flow cells were left blank as a reference. Series dilutions of sCD4 (180 nM, 90 nM, 45 nM, 22.5 nM, 11.25 nM) were injected into both the reference and sample flow cells at 50 μl/min in a single cycle, starting from the lowest concentration. Ten minutes of dissociation phase was allowed after the last sCD4 injection. We carried out the same injections with HBS-EP buffer to obtain a reference curve. Data were fitted to a 1:1 Langmuir model with Biacore T200 evaluation software. By SPR analysis, we verified that the differential CD4 binding of CD4-BS2 mutants was not due to altered binding to mAb 2G12 (data not shown).
CCR5-binding assay.
Cf2Th/syn-CCR5 cells, which express high levels of CCR5 on their surface membrane, were used to assess binding of soluble BG505-SOSIP.664 trimers or BaL gp120 monomers to CCR5. The cells were harvested at ∼80% confluence with enzyme-free cell-dissociation buffer (Gibco). His-tagged BG505-SOSIP.664 trimers or BaL gp120 monomers were pre-incubated with or without two-domain sCD4 for 1 h at 4 °C and incubated with the cells for 1 h at 4 °C, and then washed with PBS. For BG505-SOSIP.664 trimers, PE-conjugated mouse anti-His tag (Miltenyi Biotec; 120-003-741) was added to the cells for 1 h at 4 °C. For gp120 monomers, D7324 was added to the cells for 1 h at 4 °C, and then incubated for 15 min with CFTM 488A-conjugated chicken anti-goat antibody (Sigma; SAB4600028). The cells were then washed once with PBS, fixed with 2% PFA, and analyzed on a BD FACSCanto instrument. The specificity of binding was assessed by abrogation of the signal with an anti-CCR5 mAb (Becton Dickinson; 555991). Data analysis was done with the FlowJo software.
Structure alignments and figures.
All graphs were plotted using Graphpad Prism, and structural figures were displayed using Pymol (https://www.pymol.org). PDB IDs are referenced in individual figures.
Data availability.
The 7.5-Å and 6.8-Å cryo-EM maps have been deposited under accession code EMD-8427. Fitted coordinates have been deposited under accession code PDB 5U1F. Primary data are available upon request from the corresponding author.
Change history
27 April 2017
In the version of this article initially published, funding information for B.C. and C.S.P. was missing NIH grant S10 OD019994-01. In addition, there was an incorrect comma in the introduction (after "glycoprotein" in the sentence "Upon binding to the primary cellular receptor, CD4, the external gp120 Env glycoprotein undergoes major conformational changes...") that has been removed. The errors have been corrected in the HTML and PDF versions of the article.
References
Wyatt, R. & Sodroski, J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science 280, 1884–1888 (1998).
Mascola, J.R. & Haynes, B.F. HIV-1 neutralizing antibodies: understanding nature's pathways. Immunol. Rev. 254, 225–244 (2013).
Kwong, P.D. et al. HIV-1 evades antibody-mediated neutralization through conformational masking of receptor-binding sites. Nature 420, 678–682 (2002).
Lasky, L.A. et al. Delineation of a region of the human immunodeficiency virus type 1 gp120 glycoprotein critical for interaction with the CD4 receptor. Cell 50, 975–985 (1987).
Olshevsky, U. et al. Identification of individual human immunodeficiency virus type 1 gp120 amino acids important for CD4 receptor binding. J. Virol. 64, 5701–5707 (1990).
Pantophlet, R., Wilson, I.A. & Burton, D.R. Hyperglycosylated mutants of human immunodeficiency virus (HIV) type 1 monomeric gp120 as novel antigens for HIV vaccine design. J. Virol. 77, 5889–5901 (2003).
Finzi, A. et al. Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell 37, 656–667 (2010).
Kwong, P.D. et al. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 393, 648–659 (1998).
Kwong, P.D. et al. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure 8, 1329–1339 (2000).
Huang, C.C. et al. Structure of a V3-containing HIV-1 gp120 core. Science 310, 1025–1028 (2005).
Pancera, M. et al. Structure of HIV-1 gp120 with gp41-interactive region reveals layered envelope architecture and basis of conformational mobility. Proc. Natl. Acad. Sci. USA 107, 1166–1171 (2010).
Lyumkis, D. et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 342, 1484–1490 (2013).
Stewart-Jones, G.B. et al. Trimeric HIV-1-env structures define glycan shields from clades A, B, and G. Cell 165, 813–826 (2016).
Sanders, R.W. et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 9, e1003618 (2013).
Julien, J.P. et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342, 1477–1483 (2013).
Kwon, Y.D. et al. Crystal structure, conformational fixation and entry-related interactions of mature ligand-free HIV-1 Env. Nat. Struct. Mol. Biol. 22, 522–531 (2015).
Garces, F. et al. Affinity maturation of a potent family of HIV antibodies is primarily focused on accommodating or avoiding glycans. Immunity 43, 1053–1063 (2015).
Pancera, M. et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 514, 455–461 (2014).
Hu, G., Liu, J., Taylor, K.A. & Roux, K.H. Structural comparison of HIV-1 envelope spikes with and without the V1/V2 loop. J. Virol. 85, 2741–2750 (2011).
Lee, J.H., Ozorowski, G. & Ward, A.B. Cryo-EM structure of a native, fully glycosylated, cleaved HIV-1 envelope trimer. Science 351, 1043–1048 (2016).
Liu, J., Bartesaghi, A., Borgnia, M.J., Sapiro, G. & Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 455, 109–113 (2008).
Munro, J.B. et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 346, 759–763 (2014).
Harris, A. et al. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc. Natl. Acad. Sci. USA 108, 11440–11445 (2011).
Scharf, L. et al. Broadly neutralizing antibody 8ANC195 recognizes closed and open states of HIV-1 Env. Cell 162, 1379–1390 (2015).
Walker, L.M. et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature 477, 466–470 (2011).
Li, Y. et al. Broad HIV-1 neutralization mediated by CD4-binding site antibodies. Nat. Med. 13, 1032–1034 (2007).
Walker, L.M. et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science 326, 285–289 (2009).
Huang, J. et al. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature 515, 138–142 (2014).
Alsahafi, N., Debbeche, O., Sodroski, J. & Finzi, A. Effects of the I559P gp41 change on the conformation and function of the human immunodeficiency virus (HIV-1) membrane envelope glycoprotein trimer. PLoS One 10, e0122111 (2015).
Xiang, S.H., Doka, N., Choudhary, R.K., Sodroski, J. & Robinson, J.E. Characterization of CD4-induced epitopes on the HIV type 1 gp120 envelope glycoprotein recognized by neutralizing human monoclonal antibodies. AIDS Res. Hum. Retroviruses 18, 1207–1217 (2002).
Ding, S. et al. A highly conserved gp120 inner domain residue modulates Env conformation and trimer stability. J. Virol. 90, 8395–8409 (2016).
Rizzuto, C.D. et al. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 280, 1949–1953 (1998).
Zhou, T. et al. Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445, 732–737 (2007).
Zhou, T. et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science 329, 811–817 (2010).
Zhou, T. et al. Structural repertoire of HIV-1-neutralizing antibodies targeting the CD4 supersite in 14 donors. Cell 161, 1280–1292 (2015).
Li, Y. et al. HIV-1 neutralizing antibodies display dual recognition of the primary and coreceptor binding sites and preferential binding to fully cleaved envelope glycoproteins. J. Virol. 86, 11231–11241 (2012).
Wu, X. et al. Focused evolution of HIV-1 neutralizing antibodies revealed by structures and deep sequencing. Science 333, 1593–1602 (2011).
Georgiev, I.S. et al. Delineating antibody recognition in polyclonal sera from patterns of HIV-1 isolate neutralization. Science 340, 751–756 (2013).
Xing, L. et al. Distinct cellular receptor interactions in poliovirus and rhinoviruses. EMBO J. 19, 1207–1216 (2000).
Neumann, E., Moser, R., Snyers, L., Blaas, D. & Hewat, E.A. A cellular receptor of human rhinovirus type 2, the very-low-density lipoprotein receptor, binds to two neighboring proteins of the viral capsid. J. Virol. 77, 8504–8511 (2003).
Xing, L. et al. Structural and functional analysis of integrin α2I domain interaction with echovirus 1. J. Biol. Chem. 279, 11632–11638 (2004).
Wang, H. et al. Cryo-EM structure of a CD4-bound open HIV-1 envelope trimer reveals structural rearrangements of the gp120 V1V2 loop. Proc. Natl. Acad. Sci. USA 113, E7151–E7158 (2016).
Huang, C.C. et al. Structures of the CCR5 N terminus and of a tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science 317, 1930–1934 (2007).
de Taeye, S.W. et al. Immunogenicity of stabilized HIV-1 envelope trimers with reduced exposure of non-neutralizing epitopes. Cell 163, 1702–1715 (2015).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Li, X. et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013).
Lyumkis, D. et al. Automation in single-particle electron microscopy: connecting the pieces. Methods Enzymol. 483, 291–338 (2010).
Lander, G.C. et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 (2009).
Zhang, K. Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Voss, N.R., Yoshioka, C.K., Radermacher, M., Potter, C.S. & Carragher, B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213 (2009).
Sorzano, C.O.S. et al. A clustering approach to multireference alignment of single-particle projections in electron microscopy. J. Struct. Biol. 171, 197–206 (2010).
Roseman, A.M. Particle finding in electron micrographs using a fast local correlation algorithm. Ultramicroscopy 94, 225–236 (2003).
Roseman, A.M. FindEM—a fast, efficient program for automatic selection of particles from electron micrographs. J. Struct. Biol. 145, 91–99 (2004).
Scheres, S.H.W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Pettersen, E.F. et al. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
McLellan, J.S. et al. Structure of HIV-1 gp120 V1/V2 domain with broadly neutralizing antibody PG9. Nature 480, 336–343 (2011).
Krissinel, E. & Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 (2004).
Winn, M.D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011).
Xiang, Z. & Honig, B. Extending the accuracy limits of prediction for side-chain conformations. J. Mol. Biol. 311, 421–430 (2001).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Harvey, M.J., Giupponi, G. & Fabritiis, G.D. ACEMD: accelerating biomolecular dynamics in the microsecond time scale. J. Chem. Theory Comput. 5, 1632–1639 (2009).
Acknowledgements
We thank J.P. Moore and A. Cupo (Weill Medical College of Cornell University, New York, New York, USA) for providing plasmids for expression of the BG505 SOSIP.664 trimer and furin; M. Farzan (Scripps Research Institute, Jupiter, Florida, USA) for providing the plasmid to express human CD4-immunoglobulin; D.R. Burton (Scripps Research Institute, La Jolla, California, USA, and Ragon Institute of MGH, MIT and Harvard, Cambridge, Massachusetts, USA), M. Connors (Laboratory of Immunoregulation, NIAID, NIH, Bethesda, Maryland, USA), J.R. Mascola (Vaccine Research Center, NIAID, NIH, Bethesda, Maryland, USA), and J.E. Robinson (Tulane University School of Medicine, New Orleans, Louisiana, USA) for anti-gp120 mAbs; E. Berger (Laboratory of Viral Diseases, NIAID, NIG, Bethesda, Maryland, USA) for the full-length CD4 gene and the gene encoding gp160 from BG505-T332N and BaL; D. Ichikawa and Y. Lin for help in performing selected experiments; J. Arthos, E.A. Berger, and R. Cimbro for helpful discussion; P. Gangopadhyay for help with MATLAB and Origin; and the AIDS Reagent Program for providing the reagents indicated in the Online Methods. The GPU-enabled frame-alignment program we used was generously provided by Y. Cheng and X. Li. The cryo-EM work was done at the National Resource for Automated Molecular Microscopy based at the Simons Electron Microscopy Center, which is supported by grants from the NIH (GM103310 and S10 OD019994-01) and the Simons Foundation (349247) to B.C. and C.S.P. This research was supported by the Intramural Programs of the Vaccine Research Center and of the Division of Intramural Research, NIAID, NIH.
Author information
Authors and Affiliations
Contributions
Q.L. and P.L. conceived the project and designed biological and virological studies; P.A., T.Z., and P.D.K. conceived and designed structural studies; Q.L., J.L., and D.G. mutagenized gp160 from different HIV-1 isolates and produced infectious pseudoparticles; Q.L., C.G., H.M., and A.K. carried out infectivity assays and antigenicity studies of surface-expressed Envs; Q.L., D.G., and P.Z. produced and purified mutants of SOSIP trimer and gp120 monomer; Q.L. characterized the purified proteins and performed immunogenicity studies and CD4-binding and CCR5-binding assays; A.D. expressed DS-SOSIP proteins for SPR and cryo-EM studies; Q.L. and P.A. purified CD4-BS2 mutants of DS-SOSIP trimer and performed SPR analysis of CD4 binding; P.A. purified DS-SOSIP, prepared the DS-SOSIP-CD4-PGT145 complex, and performed cryo-EM experiments, with W.J.R. and C.W. providing assistance with electron microscopy data collection; P.A., T.B., G.-Y.C., and T.Z. analyzed the structure; M.A.D. performed docking experiments and MD simulations; B.C., C.S.P., and P.D.K. supervised cryo-EM experiments and structural analyses; Q.L., P.A., P.D.K., and P.L. analyzed data and wrote the paper; and all coauthors read and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Cryo-EM structure determination of the DS-SOSIP trimer–sCD4–PGT145 complex.
(a) Representative micrograph (left and middle) with red circles (middle) showing the particles picked, and its Fourier transformed image (right) showing CTF fitting. (b) Representative 2D class averages from RELION. Red circles show examples of 2D classes with views of the trimer bound to CD4 and PGT145 Fab. (c) Maps of 3D classes from a RELION 3D classification run. Class I was further refined using RELION autorefine. (d) Angular distribution of particles contributing to the final reconstruction. (e) Masks used for RELION autorefine, and corresponding gold standard FSC plots.
Supplementary Figure 2 Density shots of various regions of the DS-SOSIP trimer–sCD4–PGT145 complex.
The blue mesh represents the cryo-EM map. Zoomed-in views are shown for (a) the CDR H3 loop of PGT145 binding to a proteoglycan epitope at the interface of the three gp120 protomers in the Env trimer; (b) the gp120-gp41 interface; and (c) the trimer-CD4 interface showing continuous, well-defined CD4 electron density wedged between two gp120 protomers.
Supplementary Figure 3 Infectivity of HIV-1 BaL pseudotypes with individual and combined mutations in and around the CD4-BS2 region.
The infectivity of each mutant was calculated relative to that of the wild-type (WT). The data shown represent the mean ± range of duplicate wells from a representative experiment out of three performed which yielded similar results.
Supplementary Figure 4 Reactivity with anti-gp120 monoclonal antibodies and electrophoretic mobility of BG505-SOSIP.664 trimers and their mutants.
(a) A panel of anti-gp120 mAbs was used in ELISA to verify the correct folding of purified BG505-SOSIP.664 and DS-SOSIP.664 trimers and their mutants expressed in HEK 293FS cells. Of note, mAb F105, which reacts well with monomeric gp120 but not with the functional trimer, was virtually negative on all trimers, whereas trimer-preferring mAbs (eg, PG9, PGT145, PGT151) showed similar binding levels to the WT and the mutants, indicating the correct folding of the trimers. Optical density (OD) values are shown for the WT (top row), while binding relative to WT is shown for all mutants. (b) The correct size and cleavage of recombinant trimers were assessed by SDS-PAGE under reducing and non-reducing conditions.
Supplementary Figure 5 Effect of mutations in CD4-BS2 on CD4 binding to the HIV-1 Env trimer.
(a) Binding of WT and mutated BG505 SOSIP.664 soluble trimers to sCD4 as determined by ELISA. Purified soluble trimers were captured by MAb 2G12 immobilized to the plate and tested against serial dilutions of sCD4. The table on the right side reports area-under-the-curve (AUC) values and p values for the comparison between each mutant and WT as assessed by unpaired t-test. The data shown represent the mean ± range of duplicate wells from a representative experiment out of three performed which yielded similar results. (b) Binding of WT and mutated native, cell surface-expressed Env trimers to CD4-Ig as determined by flow cytometry. HEK 293T cells were transfected with plasmids encoding WT or mutated full-length gp160 and tested after 48-72 hours for CD4-Ig surface binding. The data shown are from a representative experiment out of two performed which yielded similar results.
Supplementary Figure 6 Effects of CD4-BS2 mutations in monomeric gp120.
(a) Binding of soluble CD4 (sCD4) to WT and mutated monomeric gp120 from BG505-T332N and BaL, as determined by ELISA. The data shown represent the mean ± range of duplicate wells from a representative experiment out of three performed which yielded similar results. (b) Binding of CCR5 (left) and mAb 48d (right) to WT and mutated monomeric BaL gp120, as determined by flow cytometry and ELISA, respectively. The data shown are from a representative experiment out of three performed which yielded similar results. (c) Reactivity of WT and mutated monomeric gp120 from BG505-T332N and BaL with a panel of anti-gp120 mAbs, as determined by ELISA.
Supplementary Figure 7 Spatial orientation of CD4-BS2 residues in the structures of trimeric versus monomeric gp120.
Upper panels: side view with the key residues in CD4-BS2 (K207, E62, E64 and H66) highlighted by stick representation and color code based on charge (blue, positive; red, negative). Lower panels: front view, surface representation. In the trimer, the side chains of CD4-BS2 residues form a continuous, electrostatically-charged surface that is totally exposed to the solvent; in contrast, in monomeric gp120, the side chains and relative charges are partially or completely buried. Of note, the spatial orientation of these residues is nearly identical in all the available monomeric gp120 structures, regardless of the presence and nature of co-crystallized ligands (see Supplementary Table 1).
Supplementary Figure 8 Mutations in the CD4-BS2 contact site of CD4 affect HIV-1 entry.
(a) Full-length human CD4 mutants were expressed in Cf2Th/syn-CCR5 cells and infected with luciferase-expressing pseudoviruses carrying WT BaL or BG505-T332N Env. Relative infectivity values for the mutants were calculated as percent of the value obtained with WT CD4. The data shown represent the mean ± range of duplicate wells from a representative experiment out of two performed which yielded similar results. (b) Reactivity of WT and mutated CD4 with anti-CD4 mAbs tested by flow cytometry. Mean fluorescence intensity (MFI) values are shown for WT (top row), while MFI relative to WT is shown for each mutant.
Supplementary Figure 9 Somatic hypermutation-evolved interaction of human anti-HIV-1 Env mAbs with CD4-BS2.
(a) Structural alignment of VRC01-class antibodies. Negatively-charged residues that interact with CD4-BS2 are highlighted in green. CD4-BS2 interaction through antibody framework 1 (b) and 3 (c). Somatic hypermutation-evolved acidic residues in both antibody framework 1 and 3 show no interaction with gp120 in complexes with monomeric core gp120 (upper row). Models in the trimer context (bottom row) with antibodies docked through gp120 outer-domain superposition indicate interaction with positively-charged CD4-BS2 residues on the neighboring protomer. Electrostatic potential surfaces for negatively-charged tips of framework 1 and 3 and the neighboring gp120 protomer are shown in the panels on the right.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–9 and Supplementary Tables 1–3 (PDF 4821 kb)
Supplementary Table 4
BSA for various trimers (XLSX 20 kb)
Rights and permissions
About this article

Cite this article
Liu, Q., Acharya, P., Dolan, M. et al. Quaternary contact in the initial interaction of CD4 with the HIV-1 envelope trimer. Nat Struct Mol Biol 24, 370–378 (2017). https://doi.org/10.1038/nsmb.3382
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.3382
This article is cited by
-
HIV-1 Env trimers asymmetrically engage CD4 receptors in membranes
Nature (2023)
-
Intermediate conformations of CD4-bound HIV-1 Env heterotrimers
Nature (2023)
-
Cryo-EM structures of prefusion SIV envelope trimer
Nature Structural & Molecular Biology (2022)
-
A site of vulnerability at V3 crown defined by HIV-1 bNAb M4008_N1
Nature Communications (2021)
-
Extended antibody-framework-to-antigen distance observed exclusively with broad HIV-1-neutralizing antibodies recognizing glycan-dense surfaces
Nature Communications (2021)
