
Abstract
The threat of a major coronavirus pandemic urges the development of strategies to combat these pathogens. Human coronavirus NL63 (HCoV-NL63) is an α-coronavirus that can cause severe lower-respiratory-tract infections requiring hospitalization. We report here the 3.4-Å-resolution cryo-EM reconstruction of the HCoV-NL63 coronavirus spike glycoprotein trimer, which mediates entry into host cells and is the main target of neutralizing antibodies during infection. The map resolves the extensive glycan shield obstructing the protein surface and, in combination with mass spectrometry, provides a structural framework to understand the accessibility to antibodies. The structure reveals the complete architecture of the fusion machinery including the triggering loop and the C-terminal domains, which contribute to anchoring the trimer to the viral membrane. Our data further suggest that HCoV-NL63 and other coronaviruses use molecular trickery, based on epitope masking with glycans and activating conformational changes, to evade the immune system of infected hosts.
Similar content being viewed by others

Main
Coronaviruses are enveloped viruses with large single-stranded positive-sense RNA genomes, classified in four genera (α, β, γ and δ). In humans, coronaviruses are responsible for 30% of respiratory-tract infections1. In addition, coronaviruses have received substantial attention in the past decade, owing to the emergence of two deadly viruses with tremendous pandemic potential: severe acute respiratory syndrome coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV)2. To date, there are no approved antiviral treatments or vaccines for any human coronavirus.
Coronaviruses are zoonotic viruses, and surveillance studies have suggested that both SARS-CoV and MERS-CoV originated from bats and that camels are also likely hosts for MERS-CoV3,4. Moreover, sequencing data have demonstrated that bats serve as a reservoir of coronaviruses that have the potential to cross the species barrier and infect humans. This phenomenon is illustrated by the observation that substitution of three amino acid residues in the spike (S) glycoprotein receptor-binding domain of the bat-infecting HKU4-CoV enhances its affinity for human DPP4 (the MERS-CoV receptor) by two orders of magnitude5,6. In addition, substitution of two other residues enables processing by human proteases and allows the HKU4-CoV S protein to mediate entry into human cells7. As a result, cross-species transmission of coronaviruses poses an imminent and long-term threat to human health. Recombination with coronaviruses frequently involved in mild respiratory infections may potentially lead to the emergence of highly pathogenic viruses4. Understanding the pathogenesis, cross-species transmission and recombination of coronaviruses is crucial to prevent or control their spread in humans and to evaluate the potential for long-term emerging diseases.
To date, α- and β-coronavirus genera have been implicated in human diseases and zoonoses. The human coronavirus NL63 (HCoV-NL63) is an α-coronavirus that is genetically distinct from the β-coronaviruses mouse hepatitis virus (MHV, the prototypical coronavirus), MERS-CoV and SARS-CoV, and was first isolated from a 7-month-old patient with a respiratory-tract infection8,9. Further studies have revealed that HCoV-NL63 infections appear to be common in childhood, and most adult sera contain antibodies that neutralize the virus8,10. HCoV-NL63 is a major cause of bronchiolitis and pneumonia in newborns worldwide and can cause severe lower-respiratory-tract infections that require hospitalization, especially among young children, the elderly and immunocompromised adults11. HCoV-NL63 infections have been reported in countries across Europe, Asia and North America, thus indicating its circulation among the human population worldwide. Other α-coronaviruses related to the human respiratory pathogen HCoV-229E have recently been identified in camels co-infected with MERS-CoV4, an observation further underscoring the importance of characterizing this coronavirus genus. Additionally, the emergence of the highly lethal porcine epidemic diarrhea coronavirus (PEDV, α-genus) has recently had devastating consequences for the US swine industry12.
Coronaviruses use S homotrimers to promote cell attachment and fusion of the viral and host membranes. Because it is virtually the only antigen present at the virus surface, S is the main target of neutralizing antibodies during infection and a focus of vaccine design13. S is a class I viral fusion protein that is synthesized as a single-chain precursor of ∼1,300 amino acids and trimerizes after folding14. It is composed of an N-terminal S1 subunit, containing the receptor-binding domain, and a C-terminal S2 subunit, driving membrane fusion. After virion uptake by target host cells, cleavage at the S2′ site (next to the putative fusion peptide) is required for fusion activation of all coronavirus S proteins, so that they can subsequently transition to the postfusion conformation15,16,17.
Our previously reported cryo-EM reconstruction of the MHV S glycoprotein at 4.0-Å resolution reveals the prefusion architecture of the machinery mediating entry of β-coronaviruses into cells18. It also demonstrates that coronavirus S and paramyxovirus F proteins share a common evolutionary origin. Here, we set out to characterize the conservation of the 3D organization of spike proteins among coronaviruses belonging to different genera. We report the atomic-resolution structure of the pathogenic HCoV-NL63 S-glycoprotein trimer, which belongs to the α-coronavirus genus. The substantial resolution improvement as compared with earlier studies allows visualization of the S glycoprotein at an unprecedented level of detail, which is a prerequisite for guiding drug and vaccine design, and reveals both shared and unique features of the α-genus of human pathogens. Our results suggest that HCoV-NL63 and other coronaviruses use molecular trickery, based on epitope masking with glycans and activating conformational changes, to evade the immune system of infected hosts, in a manner similar that described for HIV-1.
Results
Structure determination
We used Drosophila S2 cells to produce the HCoV-NL63 S ectodomain N-terminally fused to a GCN4 trimerization motif downstream from the heptad-repeat 2 (HR2) helix. We imaged frozen-hydrated HCoV-NL63 spike ectodomain particles with an FEI Titan Krios electron microscope equipped with a Gatan Quantum GIF energy filter operated in zero-loss mode, with a slit width of 20 eV, and a Gatan K2 Summit electron-counting camera19 (Online Methods).
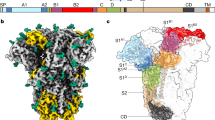
We determined a 3D reconstruction of the HCoV-NL63 spike at 3.4-Å resolution, using the gold-standard Fourier shell correlation (FSC) criterion of 0.143 (refs. 20,21) (Fig. 1 and Supplementary Fig. 1). The final model, which we built and refined with Coot22 and Rosetta23,24,25, includes residues 23 to 1224, with internal breaks between residues 110–121, 882–890 and 992–1001 (Supplementary Fig. 1 and Table 1). The HCoV-NL63 S ectodomain is a 160-Å-long trimer with a triangular cross-section.

(a) Representative micrograph of frozen-hydrated HCoV-NL63 S particles (defocus 3.4 μm). Scale bar, 355 Å. (b) Five selected class averages showing the particles along different orientations. Scale bar, 60 Å. (c,d) 3D map filtered at 3.4-Å resolution and colored by protomer. Two orthogonal views of the S trimer from the side (c) and from the top, facing toward the viral membrane, (d) are shown. (e,f) Ribbon diagrams showing the HCoV-NL63 S atomic model, oriented as in c and d, respectively.
The ordered glycan shield
A notable feature of this structure is the extraordinary number of N-linked oligosaccharides that cover the spike trimer. In the cryo-EM reconstruction, we observed density for 31 N-linked glycans extending tangentially relative to the protein surface (Fig. 2a,b, Supplementary Fig. 1 and Supplementary Table 1). At least the two core N-acetylglucosamine moieties are visible for the majority of glycosylation sites.

(a,b) Ribbon diagrams showing two orthogonal views of the S trimer, from the side (a) and from the top (b), facing toward the viral membrane. Glycans are shown as dark-blue spheres. (c) Residue-level schematic of N-linked glycans. The most extensive glycan structure detected by MS at each site is represented except for glycans observed only by cryo-EM, for which the resolved sugar moieties are shown. FP, fusion peptide, HR1, heptad-repeat 1 region; HR2, heptad-repeat 2 region (shown with a dashed line because it is not resolved in the map); TM, transmembrane domain (the striated texture indicates regions that are not part of the construct); GlcNac, N-acetylglucosamine; Man, mannose; Fuc, fucose.
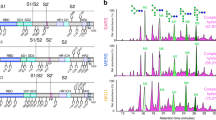
Using on-line reversed-phase liquid chromatography with electron transfer/high-energy collision-dissociation tandem MS26, we detected 25 N-linked glycosylation sites overlapping with those observed in the cryo-EM map and identified three additional sites (Fig. 2c, Supplementary Fig. 2 and Supplementary Table 1). We identified these sites from both intact glycopeptides and peptides with the glycan trimmed down to the N-linked core N-acetylglucosamine moiety. The cryo-EM and MS data together provide evidence for glycosylation at 34 out of 39 possible NXS/T glycosylation sequons. The intact glycopeptides detected by MS/MS for HCoV-NL63 S expressed in Drosophila S2 cells corresponded to either paucimannosidic glycans containing three mannose residues (with or without core fucosylation) or high-mannose glycans containing four to nine mannose residues. Previous reports have suggested that several coronavirus S glycans are of the high-mannose type, as a result of direct budding from the endoplasmic reticulum–Golgi intermediate compartment27,28, thus supporting the biological relevance of the potential glycan structures identified.
In the refined model, N-linked glycans cover a substantial amount of the accessible surface of the trimer (Fig. 2a,b). The higher glycan density per accessible surface area detected for the S2 subunits (831 Å2/glycan) compared with the S1 subunits (1,386 Å2/glycan) may explain why most coronavirus neutralizing antibodies isolated to date target the latter region. Because many of the observed glycosylation sites are topologically conserved among coronavirus S proteins, we suggest that the glycan footprint observed here may be representative of those of other S proteins. Besides potentially contributing to immune evasion, as discussed below, S glycans have been proposed to play a role in host-cell entry29 by using L-SIGN lectin, which is an alternative receptor for SARS-CoV30 and HCoV-229E27.
Structure of the S2′ trigger loop
The HCoV-NL63 and MHV S2 fusion machineries are structurally similar and can be superimposed with excellent agreement (Fig. 3a and Supplementary Fig. 3; DALI31 Z score 29.6, r.m.s. deviation 2.2 Å over 315 residues). In contrast to our previous MHV S structure18, most of the HCoV-NL63 S2′ trigger loop, which connects the upstream helix to the fusion peptide and participates in fusion activation, is resolved in the reconstruction (Fig. 3b). The trigger loop runs almost perpendicularly to the long axis of the S2 subunit and forms three helical segments before looping back to connect to the fusion peptide. Multiple arginine residues, forming two putative furin-cleavage sites, are present in the C-terminal region of the S2′ loop (863-RNIRSSR-870), which is characterized by weaker density, as would be expected from a protease-sensitive polypeptide segment. These observations are consistent with results of previous studies suggesting that fusion activation of the HCoV-NL63 S glycoprotein occurs after S2′ proteolytic processing at the plasma membrane (by trypsin-like proteases such as TMPRS2) or in the endosomal pathway (by furin or cysteine proteases)15,32.

(a) Ribbon diagram of the S2 trimer, colored by protomer with glycans rendered as dark-blue spheres. (b) Zoomed-in view of the S2′ trigger-loop region comprising the central helix and the fusion peptide (light blue). N-linked glycans are shown as dark-blue spheres. The polypeptide segment corresponding to the putative cleavage site is poorly resolved in the density, and this part of the model should be considered to be hypothetical. (c,d) Ribbon diagrams showing two orthogonal views of the S2′ C-terminal region, which is assembled from the connector domains and stem helices. (e,f) Ribbon diagrams of the HCoV-NL63 S2 subunit (e) and of the RSV F protein (f). Conserved structural elements are colored identically to highlight the similar 3D organization of these two fusion machineries, whereas nonconserved regions are colored gray. The topology diagrams underscore the similar topology of the HCoV-NL63 S connector domain and the equivalent RSV F domain, although the tertiary structures of these domains are different, and several structural motifs have been added to the latter domain throughout evolution. The RSV F secondary-structural elements are annotated according to ref. 34. The N- and C-terminal extremities of the polypeptide segments are indicated.
The lack of strict amino acid sequence conservation at the S2′ cleavage site among coronavirus S proteins reflects the usage of different proteases found in distinct cellular compartments for fusion activation15,17. Similarly to the additional cleavage site present between the S1 and S2 subunits of MERS-CoV7, the multiple glycans present in the vicinity of the S2′ loop probably further influence protease sensitivity (Fig. 3b). However, we emphasize that S2′ processing occurs at topologically equivalent positions for HCoV-NL63 S, MERS-CoV S, MHV S and probably most coronavirus S glycoproteins.
Anchoring of the fusion machinery to the viral membrane
The HCoV-NL63 S reconstruction (Fig. 3a) resolves a large part of the S2 C-terminal region that has not been observed in previous studies18,33. We were able to build an atomic model for the connector domain, which links the HR2 region and the stem helix. The connector folds as a β-rich domain decorated with one short α-helix. At its C-terminal end, the polypeptide chain folds as an α-helix (stem helix, Fig. 3a,c,d) aligned along the three-fold molecular axis, which turns into the HR2 domain, corresponding to 71 additional residues not resolved in our map. In the trimer, the connector domains assemble as a cup flanking the viral membrane-proximal side of the ectodomain, and the stem helices form a bundle stabilized by hydrophobic interactions.
The coronavirus S connector domain and the equivalent paramyxovirus F domain share a related topology, although their tertiary structures are different, and several structural motifs have been added to the latter domain throughout evolution34,35 (Fig. 3e,f). Moreover, the trimer of stem helices assembles as a helical bundle starting with the HR2 domain in a manner reminiscent of the heptad repeat B (HRB) region of paramyxovirus prefusion F structures34,35. These observations lend further support to the evolutionary connection that we have previously proposed for the fusion machineries of these two viral families18.
Comparison of the prefusion HCoV-NL63 S2 subunit with the structure of the postfusion core suggests that the C-terminal region of the connector domain and the stem helix must refold and/or change conformation to yield the canonical 'trimer of hairpin' conformation that mediates fusion of the host and viral membrane in all class I fusion proteins18,36,37.
Duplication of the N-terminal domain in α-coronaviruses
The HCoV-NL63 S structure shows the presence of an additional N-terminal domain not present in β-coronaviruses. Phylogenetic analyses suggest that this is a canonical feature of most α-coronavirus S glycoproteins (Fig. 4a–c). This domain, which we named domain 0, adopts a galectin-like β-sandwich fold supplemented with a three-stranded β-sheet, similarly to domain A (Fig. 4d–f, DALI Z score 6.9, r.m.s. deviation 4.0 Å over 149 residues), thus suggesting a gene-duplication event. Domain 0 interacts with the viral-membrane-proximal side of domain A and with domain D.

(a) Schematic representation of several α-coronavirus S-glycoprotein S1 subunits, highlighting the presence of one or several domains 0 (blue), as compared with β-coronaviruses. HCoV-NL63 (GenBank YP_003767.1), 229-rel. CoV 1 (GenBank ALK28775.1), 229-rel. CoV 2 (GenBank ALK28765.1), HCoV-229E (GenBank NP_073551.1), porcine epidemic diarrhea virus (PEDV; GenBank AAK38656.1), transmissible gastroenteritis virus strain Purdue P115 (TGEV; GenBank ABG89325.1), porcine respiratory coronavirus strain ISU-1 (PRCV; GenBank ABG89317.1), feline enteric coronavirus strain UU23 (FECV-UU23; GenBank ADC35472.1) and feline infectious peritonitis coronavirus strain UU21 (FIPV-UU21; GenBank ADL71466.1). The β-coronavirus MHV S1 subunit is shown for comparison. Domains A–D are indicated for MHV and HCoV-NL63. (b) Ribbon diagram of the HCoV-NL63 S1 subunit. (c) Ribbon diagram of the MHV S1 subunit. (d–g) Ribbon diagrams of HCoV-NL63 domain 0 (d), domain A (e), MHV domain A (f) and rotavirus VP8* (g), showing their structural similarity, which suggests common ancestry. HCoV-NL63 domain 0 and A probably arose from a duplication event.
We determined that domain 0 is also structurally similar to the VP8* sialic acid–binding domain of the rotavirus VP4 spike protein38 (Fig. 4g; PDB 1KQR, DALI Z score 8.9, r.m.s. deviation 3.1 Å over 112 residues). In line with this finding, domain 0 of transmissible gastroenteritis coronavirus (TGEV) and of PEDV bind to sialic acid, and deletion of this domain in α-coronavirus S appears to correlate with a loss of enteric tropism39. We detected no sialic acid binding activity for the HCoV-NL63 S1 subunit (Supplementary Fig. 4), thus possibly explaining the strict respiratory tropism of this virus. Instead, host-cell heparan sulfate proteoglycans have been shown to participate in HCoV-NL63 anchoring and infection40, and we detected binding of heparan sulfate to the HCoV-NL63 S protein by using surface plasmon resonance (SPR) (Supplementary Fig. 5a). We hypothesize that these interactions may be mediated either by domain 0, which exhibits several positively charged patches on its surface (Supplementary Fig. 5b), or domain A, which has been reported to bind carbohydrates in the case of a bovine coronavirus41.
A putative immune-evasion strategy
Domain B, which is the HCoV-NL63 receptor-binding domain, exhibits a structure distinct from those of β-coronavirus B domains, although a topological relatedness has been detected among these β-rich domains42. Superimposition of the HCoV-NL63 and MHV S1 subunits highlights that their B domains feature opposite orientations related by an ∼180° rotation (Fig. 5a,b). As a result, many of the HCoV-NL63 receptor-binding residues are buried through interaction with domain A of the same protomer, are masked by the glycan at residue Asn358 and are not available to engage the host-cell receptor (human angiotensin-converting enzyme 2, ACE2). Comparison of the HCoV-NL63 domain-B structure in our cryo-EM-derived model with the crystal structure of the same domain in complex with ACE2 (ref. 43) revealed that the receptor-binding loop containing residues 531–539 undergoes substantial conformational changes after binding (and is defined by weak density; Fig. 5c). These findings explain the markedly higher ACE2 binding affinity of HCoV-NL63 domain B, compared with that of the full-length S1 domain (Fig. 5d).

(a) Ribbon diagram of the HCoV-NL63 S trimer, highlighting the conformation of the S1 subunit. Domains 0, A, B, C and D are colored for one protomer. (b) The HCoV-NL63 receptor-binding loops are buried via interactions with domain A of the same protomer (including the glycan moiety at Asn358) and are not available to engage host-cell receptors. Superimposition of the HCoV-NL63 (purple) and MHV (light gray) S1 subunits via their C domains highlights that their B domains feature opposite orientations related by an ∼180° rotation, thus suggesting a putative trajectory for the conformational changes that must occur to engage the host-cell receptor. Only domain B is shown for MHV S. (c) Comparison of the HCoV-NL63 domain-B structure in our cryo-EM-derived model (purple) with the crystal structure of the same domain in complex with ACE2 (green and dark gray), showing that the receptor-binding loop containing residues 531–539 substantially changes its conformation after binding. (d) ACE2 binding ELISA showing that isolated HCoV-NL63 domain B (HCoV-NL63 S1-B-mFc) binds ACE2 with higher affinity than does the full-length S1 domain (HCoV-NL63 S1-mFc). SARS-CoV S1 (HCoV-NL63 S1-mFc) is a positive control. HCoV-NL63 S1 domain 0 (HCoV-NL63 S1-0-mFc) and PEDV S1 (PEDV S1-mFc), which do not bind ACE2, are negative controls. Mean values and s.d. of three independent experiments are shown.
Because the receptor-binding loops elicit potent neutralizing antibodies in the case of TGEV44, MERS-CoV45 and SARS-CoV46,47,48,49, we speculate that HCoV-NL63 has evolved to limit exposure of this vulnerable site to B-cell receptors via protein-protein interactions and glycan masking. This mechanism is reminiscent of the HIV-1 immune evasion strategy, which relies on a glycan shield and conformational changes that are triggered by binding of CD4 and expose the chemokine-receptor-interacting motif (V3 loop)50,51.
Discussion
Viruses have evolved several immune-evasion strategies including rapid antigenic evolution, masking of epitopes and exposure of non-neutralizing immune-dominant 'decoy' epitopes. For example, HIV-1 (ref. 52), Lassa virus53, hepatitis C virus54 and Epstein–Barr virus55 exhibit extensive N-linked glycosylation, covering exposed protein surfaces, as well as glycan masses that may exceed that of the protein component. The HCoV-NL63 S trimer is covered by an extensive glycan shield consisting of 102 N-linked oligosaccharides obstructing the protein surface. This observation is reminiscent of descriptions of the HIV-1 envelope trimer52, although the glycan density is 30% higher in the latter case. Furthermore, our data suggest that, similarly to HIV-1, coronavirus S glycans mask the protein surface and consequently limit access to neutralizing antibodies and thwart the humoral immune response. This strategy is illustrated by the presence of a glycan linked to Asn358 in the HCoV-NL63 structure reported here. This glycan, along with the proteinaceous moiety of domain A, contributes to masking the receptor-binding loops, which have been shown to elicit potent neutralizing antibodies for other coronaviruses44,45,46,47,48,49 and appear to represent a potential 'Achilles' heel' of these viruses. This hypothesis is further supported by the observation of three additional glycans directly protruding from the viral-membrane-distal side of domain B. As a result, conformational changes are required for the HCoV-NL63 S glycoprotein to be able to interact with ACE2 (ref. 43). These rearrangements and/or receptor binding are likely to participate in initiating the fusion reaction by disrupting the interactions formed between domain B and the HR1 C-terminal region. Interactions with heparan sulfate proteoglycans present at the host-cell surface might potentially contribute to activating HCoV-NL63 S and promote subsequent interactions with ACE2. A common theme arising from the analysis of α- and β-coronavirus S-glycoprotein structures is that domain-B-mediated host anchoring involves major structural rearrangements that expose the binding motifs18,33.
Visualization of the glycan shield obstructing access to the S surface and deciphering the molecular trickery used by some coronaviruses provide a rational basis for understanding the accessibility to neutralizing antibodies and may pave the way for guiding future design of immunogens therapeutics. We have previously suggested that targeting the fusion machinery bears the promise of finding broadly neutralizing inhibitors of coronavirus infection18, and the high density of glycans decorating this region will need to be taken into consideration to increase the likelihood of success.
Methods
Plasmids.
A gene fragment encoding the HCoV-NL63 S ectodomain (residues 16–1291, UniProt Q6Q1S2) was PCR-amplified from a plasmid containing the full-length S gene. The PCR product was ligated to a gene fragment encoding a GCN4 trimerization motif (LIKRMKQIEDKIEEIESKQKKIENEIARIKKIK)18,35,56, a thrombin-cleavage site (LVPRGSLE), an eight-residue-long Strep-Tag (WSHPQFEK) and a stop codon. Subsequent cloning was performed in the pMT-BiP-V5-His expression vector (Invitrogen) in frame with the Drosophila BiP secretion signal downstream the metallothionein promoter.
Production of recombinant HCoV-NL63 S ectodomain in Drosophila S2 cells.
To generate a stable Drosophila S2 cell line expressing the recombinant HCoV-NL63 S ectodomain, we used Effectene (Qiagen) and 2 μg of plasmid. Puromycin N-acetyltransferase was cotransfected and used as a dominant selectable marker. Stable HCoV-NL63 S–expressing cell lines were selected by addition of 7 μg/ml puromycin (Invivogen) to the culture medium 48 h after transfection. For large-scale production, the cells were cultured in spinner flasks and induced by 5 μM of CdCl2 at a density of approximately 107 cells/mL. After one week at 28 °C, clarified cell supernatants were concentrated 40-fold with Vivaflow tangential filtration cassettes (Sartorius, 10-kDa cutoff) and adjusted to pH 8.0, before affinity purification with a StrepTactin Superflow column (IBA) followed by gel-filtration chromatography with a Superose 6 10/300 GL column (GE Life Sciences) equilibrated in 20 mM Tris-HCl, pH 7.5, and 100 mM NaCl. The purified protein was quantified according to absorption at 280 nm and concentrated to approximately 3 mg/mL.
Cryo-EM specimen preparation and data collection.
2 μl of purified HCoV-NL63 spike at 1.0 mg/mL was applied to a 1.2/1.3 C-flat grid (Protochips), which had been glow-discharged for 30 s at 20 mA. Grids were then plunge-frozen in liquid ethane with an FEI Mark I Vitrobot with 7.5-s blot time and an offset of −3 mm at 100% humidity and 25 °C. Data were collected with Leginon automatic data-collection software57 on an FEI Titan Krios operated at 300 kV and equipped with a Gatan Quantum GIF energy filter, operated in zero-loss mode with a slit width of 20 eV, and a Gatan K2 Summit direct electron detector camera. The dose rate was adjusted to 8 counts/pixel/s, and each movie was acquired in counting mode fractionated in 50 frames of 200 ms. 1,400 micrographs were collected in a single session with a defocus range between 2.0 and 4.0 μm.
Cryo-EM data processing.
Whole-frame alignment was carried out with DOSEFGPU DRIFTCORR19. The parameters of the microscope contrast-transfer function were initially estimated with CTFFIND4 (ref. 58) and then with GCTF59. Micrographs were manually masked with Appion60 to exclude the visible carbon edge from images. Particles were automatically picked with DoGPicker61. Particle images were extracted and processed with Relion 1.4 (ref. 62) with a box size of 320 pixels2 and a pixel size of 1.36 Å. After reference-free 2D classification, we retained 180,000 out of 474,000 particles to run 3D classification with C1 symmetry62. We used the initial model previously generated for MHV18 with Optimod63 and low-pass-filtered the data to 60 Å as a starting reference for 3D classification. 118,000 particles were selected and used to run gold-standard 3D refinement with Relion20, thus yielding a map at 3.95-Å resolution. After particle-motion and radiation-damage correction with Relion particle polishing64, another round of 3D classification with C3 symmetry was performed to select 79,667 particles. After gold-standard 3D refinement with this subset of particles, we obtained a reconstruction at 3.76-Å resolution. Per-particle defocus parameters were estimated with GCTF and used to run an identical round of 3D refinement that yielded the final 3.4-Å-resolution map. Post processing was performed with Relion to apply an automatically generated B factor of −129 Å2. Reported resolutions were based on the gold-standard FSC = 0.143 criterion20,21, and FSC curves were corrected for the effects of soft masking by high-resolution noise substitution65. The soft mask used for FSC calculation had a 10-pixel cosine-edge fall-off.
Model building and analysis.
UCSF Chimera66 and Coot22,67 were used to fit atomic models into the cryo-EM map. The MHV S2 subunit was fit into the density and rebuilt manually in Coot. The crystal structure of HCoV-NL63 domain B was then fit into the density, and the rest of the S1 subunit was built with a combination of manual building in Coot and de novo building with Rosetta23,24,25. Glycan density coming after an NXS/T motif was initially manually built into the density, and glycan geometry was then refined with Rosetta, optimizing the fit-to-density as well as the energetics of protein-glycan contacts. The glycans were not as well defined as the protein region in the reconstruction, owing to flexibility and compositional heterogeneity. The final model was refined by application of strict noncrystallographic symmetry constraints with Rosetta, with a training map corresponding to one of the two maps generated by the gold-standard refinement procedure in Relion. The second map (testing map) was used only for calculation of the FSC compared with the atomic model and preventing overfitting68. The quality of the final model was analyzed with MolProbity69 and Privateer70. Structure analysis was performed with the DALI server31 and areaimol71. Electrostatic-potential calculations were performed with PDB2PQR72 and APBS73. All figures were generated with UCSF Chimera66. Local resolution estimation was performed with Resmap74.
Mass spectrometry.
HCoV-NL63 S was prepared for MS analysis unaltered or subjected to Endo H (NEB), subjected to Endo F3 (Millipore) or subjected to combined Endo H and Endo F3 deglycosylation treatment. 2 μl of the relevant endoglycosidases was incubated with 20 μg of HCoV-NL63 S for 14 h overnight in 50 mM sodium acetate, pH 4.4, at 37 °C in a 20-μL reaction. 6 μg of HCoV-NL63 S was then incubated in a freshly prepared solution containing 100 mM Tris, pH 8.5, 2% sodium deoxycholate, 10 mM Tris(2-carboxyethyl)phosphine and 40 mM iodoacetamide at 95 °C for 5 min; this was followed by an incubation at 25 °C for 30 min in the dark. 1.6 μg of denatured, reduced and alkylated HCoV-NL63 S was then diluted into freshly prepared 50 mM ammonium bicarbonate and incubated for 14 h at 37 °C with 0.032 μg of either trypsin (Sigma Aldrich) or chymotrypsin (Sigma Aldrich). Formic acid was then added to a final concentration of 2% to precipitate the sodium deoxycholate in the samples. Samples were then centrifuged at 14,000 r.p.m. for 20 min. The supernatant containing the (glyco)peptides was collected and spun again at 14,000 r.p.m. for 5 min immediately before sample analysis. Between 4 and 7 μL was run on a Thermo Scientific Orbitrap Fusion Tribrid mass spectrometer. A 35-cm analytical column and a 3-cm trap column filled with ReproSil-Pur C18AQ 5 μm (Dr. Maisch) beads were used. Nanospray LC-MS/MS was used to separate peptides over a 110-min gradient from 5% to 30% acetonitrile with 0.1% formic acid. A positive spray voltage of 2,100 was used with an ion-transfer-tube temperature of 350 °C. An electron-transfer/higher-energy collision dissociation ion-fragmentation scheme26 was used with calibrated charge-dependent ETD parameters and a supplemental higher-energy collision dissociation energy of 0.15 for the samples with intact glycopeptides and 0.2 for the samples treated with endoglycosidases. A resolution setting of 120,000 with an AGC target of 2 × 105 was used for MS1, and a resolution setting of 30,000 with an AGC target of 1 × 105 was used for MS2. The data were searched against a custom database including recombinant coronavirus S-glycoprotein sequences, a list of common contaminant proteins including trypsin, chymotrypsin and the endoglycosidases, as well as 998 decoy reverse yeast sequences, with trypsin or chymotrypsin as the protease, allowing up to two missed cleavages. All searches included carbamidomethylation of cysteine as a fixed modification and oxidation of methionine as a variable modification. An initial comprehensive search for glycosylation revealed that (core-fucosylated) paucimannose and high-mannose structures were the only identified glycan species in the samples. On the basis of these findings, a final search was performed with COMET75 on the same data with the following list of variable modifications of asparagine residues: +HexNAc(2)Hex(3), +HexNAc(2)Hex(3)dHex(1), +HexNAc(2)Hex(3)dHex(2), +HexNAc(2)Hex(4), +HexNAc(2)Hex(5), +HexNAc(2)Hex(6), +HexNAc(2)Hex(7), +HexNAc(2)Hex(8) and +HexNAc(2)Hex(9). The samples treated with endoglycosidases were searched with +HexNAc, +HexNAc(1)dHex(1) and +HexNAc(1)dHex(2) as variable modifications of asparagine. We used a precursor mass tolerance of 20 p.p.m., 0.02 fragment bin size, including b/c/y/z fragments, with monoisotopic masses for both precursor and fragment ions. The search results were filtered for modification of asparagine residues and the presence of an NX(S/T) sequon at the protein level. All appropriate peptide spectrum matches (PSMs) were manually inspected, and only those with reasonable peptide sequence coverage were kept. In addition, the spectra were inspected for the presence of glycan fragment ions. All glycosylation sites identified by MS listed in Supplementary Table 1 are based on multiple PSMs, often with multiple different glycans and additional confirmation from overlap between the trypsin- and chymotrypsin-treated samples. The greatest number of glycopeptide identifications was made in the chymotrypsin-digested samples.
Hemagglutination assay.
The S1 subunit of HCoV-NL63 C-terminally tagged with the Fc portion of human IgG (S1-Fc) was tested alone or premixed with 1 μl of Protein A–coupled, 200-nm-sized nanoparticles (nano-screenMAG-Protein A beads; Chemicell, cat.no. 4503-1) to increase the avidity of S1-Fc proteins for sialic acids on the erythrocyte surface. The sialic acid–binding S1 subunit of PEDV (strain GDU, GenBank AFP81695.1) C-terminally fused to the human Fc portion was used as a positive control. 'Mock' indicates the conditions in which no S1 subunit was used (negative control). The initial concentration of S1-Fc was 5 μg, and two-fold serial dilutions of S1-Fc-nanoparticle mixtures were made in 50 μl phosphate-buffered saline supplemented with 0.1% bovine serum albumin. 50 μl erythrocyte suspension (0.5%) was mixed with 50 μl of S1-Fc-nanoparticle dilution in V-shaped 96-well plates and incubated for 2 h on ice, after which the wells were photographed.
Protein expression of S1 variants and ACE2.
Different S1 variants of HCoV-NL63 S protein, including S1 (residues 1–718), S1 domain 0 (S1-0, residues 1–209) and S1 domain B (S1-B, residues 481–616), were C-terminally fused to the Fc region of mouse IgG (mFc), expressed in HEK-293T cells and affinity purified as previously described76. Likewise, an S1-mFc expression plasmid was made for the SARS-CoV S1 domain (isolate CUHK-W1, residues 1–676) and the PEDV S1 domain (strain GDU; residues 1–728). Expression of the human angiotensin-converting enzyme ectodomain (ACE2; residues 1–614) fused to the Fc portion of human IgG (hFc) was performed as previously described76.
ACE2 binding ELISA.
The ability of the HCoV-NL63 S1-mFc and S1-B-mFc chimeric proteins to bind the ACE2-hFc receptor was evaluated with an ELISA-based assay. 100 μl of hACE2-hFc (20 μg/ml, diluted in PBS) was coated on a 96-well MaxiSorb plate overnight at 4 °C. Nonspecific binding sites were subsequently blocked with a 3% (w/v) solution of bovine serum albumin in PBS. Plates were washed with washing buffer (PBS with 0.05% Tween 20) and subsequently incubated with serially diluted S1-mFc proteins (starting with equimolar concentrations) for 1 h at room temperature, after which plates were washed three times with washing buffer. mFc-tagged S1 proteins were detected with HRP-conjugated polyclonal rabbit-anti-mouse immunoglobulins (1:2,000 dilution in PBS with 0.1% BSA; DAKO, P0260), and a colorimetric reaction was produced after incubation with tetramethylbenzidine substrate (BioFX). The optical density (OD) was subsequently measured at 450 nm with an ELISA reader (EL-808, BioTEK). Background (signal from HRP-conjugated anti-mFc antibody alone) was subtracted from the OD450nm values. The mFc-tagged SARS-CoV S1 subunit was used as a positive control, whereas the mFc-tagged HCoV-NL63 S1 domain 0 (HCoV-NL63 S1-0-mFc) and PEDV S1 subunit (PEDV S1-mFc), both of which do not bind ACE2, were used as negative controls.
Surface plasmon resonance (SPR).
SPR was performed on a GE Healthcare Biacore T200 with a running buffer containing 20 mM HEPES, pH 7.5, 100 mM NaCl and 0.5% Tween-20, with a flow rate of 30 μL/min at 25 °C. A carboxymethylated dextran (CM5) chip (GE Healthcare) was activated with N-hydroxysulfosuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC). We then either quenched the CM5 surface with ethanolamine (yielding a blank flow cell) or immobilized HCoV-NL63 S before quenching. 10 μg of HCoV-NL63 S was diluted into 10 mM sodium acetate, pH 5.5 and was directly immobilized for 700 s, thus yielding 28,000 RUs. After immobilization quenching, running buffer was flowed for 10 min to ensure a steady baseline before experimental binding. Heparan sulfate (Sigma Aldrich) was reconstituted in running buffer at 5.0 mg/mL. Two concentrations of heparan sulfate, 5.0 mg/mL and 2.5 mg/mL, were injected for 80 s with a dissociation time of 400 s. All data were subtracted from the blank flow cell, to account for any nonspecific interactions of heparan sulfate with the CM5 chip, and the baseline was normalized to 0.
Accession codes.
The cryo-EM map has been deposited in the Electron Microscopy Data Bank under accession code EMD-8331; the corresponding atomic model has been deposited into the Protein Data Bank under accession code PDB 5SZS. The MS data (including the raw data, COMET search results and annotated tandem MS spectra of all accepted glycopeptide identifications) have been deposited in the proteomics identifications (PRIDE) database under dataset PXD004557.
Accession codes
Primary accessions
Electron Microscopy Data Bank
Protein Data Bank
Proteomics Identifications Database
Referenced accessions
GenBank/EMBL/DDBJ
NCBI Reference Sequence
Protein Data Bank
Swiss-Prot
References
Zumla, A., Chan, J.F., Azhar, E.I., Hui, D.S. & Yuen, K.Y. Coronaviruses: drug discovery and therapeutic options. Nat. Rev. Drug Discov. 15, 327–347 (2016).
Vijay, R. & Perlman, S. Middle East respiratory syndrome and severe acute respiratory syndrome. Curr. Opin. Virol. 16, 70–76 (2016).
Ge, X.Y. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013).
Sabir, J.S. et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 351, 81–84 (2016).
Yang, Y. et al. Receptor usage and cell entry of bat coronavirus HKU4 provide insight into bat-to-human transmission of MERS coronavirus. Proc. Natl. Acad. Sci. USA 111, 12516–12521 (2014).
Wang, Q. et al. Bat origins of MERS-CoV supported by bat coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe 16, 328–337 (2014).
Yang, Y. et al. Two mutations were critical for bat-to-human transmission of Middle East respiratory syndrome coronavirus. J. Virol. 89, 9119–9123 (2015).
van der Hoek, L. et al. Identification of a new human coronavirus. Nat. Med. 10, 368–373 (2004).
Fouchier, R.A. et al. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl. Acad. Sci. USA 101, 6212–6216 (2004).
Hofmann, H. et al. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl. Acad. Sci. USA 102, 7988–7993 (2005).
Chiu, S.S. et al. Human coronavirus NL63 infection and other coronavirus infections in children hospitalized with acute respiratory disease in Hong Kong, China. Clin. Infect. Dis. 40, 1721–1729 (2005).
Mole, B. Deadly pig virus slips through US borders. Nature 499, 388 (2013).
Du, L. et al. The spike protein of SARS-CoV: a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 7, 226–236 (2009).
Bosch, B.J., van der Zee, R., de Haan, C.A. & Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77, 8801–8811 (2003).
Burkard, C. et al. Coronavirus cell entry occurs through the endo-/lysosomal pathway in a proteolysis-dependent manner. PLoS Pathog. 10, e1004502 (2014).
Millet, J.K. & Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 111, 15214–15219 (2014).
Millet, J.K. & Whittaker, G.R. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 202, 120–134 (2015).
Walls, A.C. et al. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531, 114–117 (2016).
Li, X. et al. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013).
Scheres, S.H. & Chen, S. Prevention of overfitting in cryo-EM structure determination. Nat. Methods 9, 853–854 (2012).
Rosenthal, P.B. & Henderson, R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 333, 721–745 (2003).
Brown, A. et al. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr. D Biol. Crystallogr. 71, 136–153 (2015).
DiMaio, F. et al. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nat. Methods 12, 361–365 (2015).
Wang, R.Y. et al. De novo protein structure determination from near-atomic-resolution cryo-EM maps. Nat. Methods 12, 335–338 (2015).
Song, Y. et al. High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742 (2013).
Frese, C.K. et al. Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD). J. Proteome Res. 12, 1520–1525 (2013).
Jeffers, S.A., Hemmila, E.M. & Holmes, K.V. Human coronavirus 229E can use CD209L (L-SIGN) to enter cells. Adv. Exp. Med. Biol. 581, 265–269 (2006).
Ritchie, G. et al. Identification of N-linked carbohydrates from severe acute respiratory syndrome (SARS) spike glycoprotein. Virology 399, 257–269 (2010).
Zhou, Y. et al. A single asparagine-linked glycosylation site of the severe acute respiratory syndrome coronavirus spike glycoprotein facilitates inhibition by mannose-binding lectin through multiple mechanisms. J. Virol. 84, 8753–8764 (2010).
Jeffers, S.A. et al. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. USA 101, 15748–15753 (2004).
Holm, L. & Rosenström, P. Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 (2010).
Kawase, M., Shirato, K., van der Hoek, L., Taguchi, F. & Matsuyama, S. Simultaneous treatment of human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 86, 6537–6545 (2012).
Kirchdoerfer, R.N. et al. Pre-fusion structure of a human coronavirus spike protein. Nature 531, 118–121 (2016).
McLellan, J.S. et al. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340, 1113–1117 (2013).
Yin, H.S., Wen, X., Paterson, R.G., Lamb, R.A. & Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439, 38–44 (2006).
Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 15, 690–698 (2008).
Zheng, Q. et al. Core structure of S2 from the human coronavirus NL63 spike glycoprotein. Biochemistry 45, 15205–15215 (2006).
Dormitzer, P.R., Sun, Z.Y., Wagner, G. & Harrison, S.C. The rhesus rotavirus VP4 sialic acid binding domain has a galectin fold with a novel carbohydrate binding site. EMBO J. 21, 885–897 (2002).
Krempl, C., Schultze, B., Laude, H. & Herrler, G. Point mutations in the S protein connect the sialic acid binding activity with the enteropathogenicity of transmissible gastroenteritis coronavirus. J. Virol. 71, 3285–3287 (1997).
Milewska, A. et al. Human coronavirus NL63 utilizes heparan sulfate proteoglycans for attachment to target cells. J. Virol. 88, 13221–13230 (2014).
Peng, G. et al. Crystal structure of bovine coronavirus spike protein lectin domain. J. Biol. Chem. 287, 41931–41938 (2012).
Li, F. Evidence for a common evolutionary origin of coronavirus spike protein receptor-binding subunits. J. Virol. 86, 2856–2858 (2012).
Wu, K., Li, W., Peng, G. & Li, F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. USA 106, 19970–19974 (2009).
Reguera, J. et al. Structural bases of coronavirus attachment to host aminopeptidase N and its inhibition by neutralizing antibodies. PLoS Pathog. 8, e1002859 (2012).
Ying, T. et al. Junctional and allele-specific residues are critical for MERS-CoV neutralization by an exceptionally potent germline-like antibody. Nat. Commun. 6, 8223 (2015).
Prabakaran, P. et al. Structure of severe acute respiratory syndrome coronavirus receptor-binding domain complexed with neutralizing antibody. J. Biol. Chem. 281, 15829–15836 (2006).
Hwang, W.C. et al. Structural basis of neutralization by a human anti-severe acute respiratory syndrome spike protein antibody, 80R. J. Biol. Chem. 281, 34610–34616 (2006).
Sui, J. et al. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc. Natl. Acad. Sci. USA 101, 2536–2541 (2004).
Zhu, Z. et al. Potent cross-reactive neutralization of SARS coronavirus isolates by human monoclonal antibodies. Proc. Natl. Acad. Sci. USA 104, 12123–12128 (2007).
Chen, B. et al. Structure of an unliganded simian immunodeficiency virus gp120 core. Nature 433, 834–841 (2005).
Huang, C.C. et al. Structure of a V3-containing HIV-1 gp120 core. Science 310, 1025–1028 (2005).
Stewart-Jones, G.B. et al. Trimeric HIV-1-Env structures define glycan shields from clades A, B, and G. Cell 165, 813–826 (2016).
Sommerstein, R. et al. Arenavirus glycan shield promotes neutralizing antibody evasion and protracted infection. PLoS Pathog. 11, e1005276 (2015).
Falkowska, E., Kajumo, F., Garcia, E., Reinus, J. & Dragic, T. Hepatitis C virus envelope glycoprotein E2 glycans modulate entry, CD81 binding, and neutralization. J. Virol. 81, 8072–8079 (2007).
Szakonyi, G. et al. Structure of the Epstein–Barr virus major envelope glycoprotein. Nat. Struct. Mol. Biol. 13, 996–1001 (2006).
Eckert, D.M., Malashkevich, V.N. & Kim, P.S. Crystal structure of GCN4-pIQI, a trimeric coiled coil with buried polar residues. J. Mol. Biol. 284, 859–865 (1998).
Suloway, C. et al. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 (2005).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Zhang, K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016).
Lander, G.C. et al. Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 (2009).
Voss, N.R., Yoshioka, C.K., Radermacher, M., Potter, C.S. & Carragher, B. DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213 (2009).
Scheres, S.H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Lyumkis, D., Vinterbo, S., Potter, C.S. & Carragher, B. Optimod: an automated approach for constructing and optimizing initial models for single-particle electron microscopy. J. Struct. Biol. 184, 417–426 (2013).
Scheres, S.H. Beam-induced motion correction for sub-megadalton cryo-EM particles. eLife 3, e03665 (2014).
Chen, S. et al. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy 135, 24–35 (2013).
Goddard, T.D., Huang, C.C. & Ferrin, T.E. Visualizing density maps with UCSF Chimera. J. Struct. Biol. 157, 281–287 (2007).
Emsley, P., Lohkamp, B., Scott, W.G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
DiMaio, F., Zhang, J., Chiu, W. & Baker, D. Cryo-EM model validation using independent map reconstructions. Protein Sci. 22, 865–868 (2013).
Chen, V.B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Agirre, J. et al. Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834 (2015).
Lee, B. & Richards, F.M. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 55, 379–400 (1971).
Dolinsky, T.J., Nielsen, J.E., McCammon, J.A. & Baker, N.A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32, W665–W667 (2004).
Baker, N.A., Sept, D., Joseph, S., Holst, M.J. & McCammon, J.A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 98, 10037–10041 (2001).
Kucukelbir, A., Sigworth, F.J. & Tagare, H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014).
Eng, J.K., Jahan, T.A. & Hoopmann, M.R. Comet: an open-source MS/MS sequence database search tool. Proteomics 13, 22–24 (2013).
Raj, V.S. et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495, 251–254 (2013).
Acknowledgements
Research reported in this publication was supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under award number 1R01GM120553-01 (D.V.) and T32GM008268 (A.C.W.). J.S. acknowledges support from the Netherlands Organization for Scientific Research (NWO, Rubicon 019.2015.2.310.006) and the European Molecular Biology Organisation (EMBO, ALTF 933-2015). M.A.T. and F.A.R. acknowledge support from the Institute Pasteur and the CNRS. The authors acknowledge the use of instruments at the Electron Imaging Center for NanoMachines supported by the NIH (1S10RR23057 and 1S10OD018111), NSF (DBI-1338135) and CNSI at UCLA. The authors are grateful to H. Choe (The Scripps Research Institute) for providing the HCoV-NL63 S gene, J. Labonte (Johns Hopkins School of Medicine) for providing the Rosetta code handling glycans and P.J.M. Rottier (Utrecht University) for scientific advice. This work was partly supported by the University of Washington's Proteomics Resource (UWPR95794), and the authors thank P.D. von Haller and J.K. Eng for their assistance and expertise. SPR experiments were performed with the help of J. Sumida at the University of Washington School of Pharmacy Analytical Biopharmacy Core. Part of this research was facilitated by the Hyak supercomputer system at the University of Washington.
Author information
Authors and Affiliations
Contributions
B.-J.B. designed and cloned the protein construct. M.A.T. carried out protein expression and purification under the supervision of F.A.R. A.C.W. performed cryo-EM sample preparation, data collection and processing under the supervision of D.V. A.C.W., B.F., F.D. and D.V. built the atomic model. A.C.W. and J.S. performed the MS experiments. W.L. performed the hemagglutination assays and ELISAs under the supervision of B.-J.B. A.C.W. performed the SPR experiments under the supervision of D.V. All authors analyzed the data. A.C.W., J.S., B.-J.B. and D.V. prepared the manuscript with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Cryo-EM analysis of the HCoV-NL63 S trimer.
a, Gold-standard (blue) and model/map (red) Fourier shell correlation (FSC) curves. The resolution was determined to 3.4 Å. The 0.143 and 0.5 cut-off values are indicated by horizontal grey bars. b, The glycan linked to Asn 240 is rendered as ball and sticks and the corresponding region of the cryoEM map is shown as a blue mesh. c, The glycan linked to Asn 426 is rendered as ball and sticks and the corresponding region of the cryoEM map is shown as a blue mesh. In panels (b-c), carbon, nitrogen and oxygen atoms are colored grey, blue and red, respectively. d, HCoV-NL63 S cryoEM map colored according to local resolution. e, HCoV-NL63 S atomic model colored according to refined B factors.
Supplementary Figure 2 Characterization of the HCoV-NL63 S glycans by using mass spectrometry.
Tandem MS EThcD spectrum of a 3+ glycopeptide with HexNAc(2)Hex(6) attached to Asn 699 of the HCoV-NL63 S glycoprotein digested with chymotrypsin. The relative intensity normalized to the most intense ion is plotted against mass-to-charge ratio. The peaks under the horizontal dashed line are multiplied by 3 for visualization. "M" denotes the molecular ion. The charge state of the fragment ions is indicated in brackets. Ions relating to the glycopeptide, glycan and peptide fragments are colored red, green and blue, respectively. In this example of a glycopeptide identification the matched fragment ions define a large part of the peptide sequence and also provide detailed information about the glycan composition.
Supplementary Figure 3 Structural similarity of coronavirus fusion machineries.
Ribbon diagram of the HCoV-NL63 (blue) and MHV (tan) S2 fusion subunits. The dashed box highlights the two extra helical turns present in the S protein HR1 region of α-coronaviruses but not β-coronaviruses.
Supplementary Figure 4 The HCoV-NL63 S1 subunit does not bind sialic acid.
Binding of sialic acid by the HCoV-NL63 S1 subunit (N-terminally fused to human IgG Fc) was assessed by probing the hemagglutination of human eryhthrocytes. The porcine epidemic diarrhea coronavirus S1 subunit was used as a positive control. Mock indicates the absence of coronavirus S1 subunit (negative control). The assays were performed using either free S1-Fc or nanoparticle-displaying S1-Fc to increase the avidity for sialic acid on the erythrocyte surface. Wells showing hemagglutination are circled.
Supplementary Figure 5 HCoV-NL63 binds heparan sulfate.
a, Surface plasmon resonance sensorgram showing binding of heparan sulfate to HCoV NL63 S. The right panel shows a blow-up view of the sensorgram corresponding to 2.5 mg/mL heparan sulfate. b, Ribbon diagram of the HCoV-NL63 S atomic model colored by protomer. Domain 0 is shown in surface representation colored according to its electrostatic surface potential for one protomer. The positively charged patch on its surface could putatively mediate binding to heparan sulfate.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–5 (PDF 1503 kb)
Supplementary Table 1
Characterization of the HCoV-NL63 S glycans by using mass spectrometry and cryo-EM (XLSX 11 kb)
Rights and permissions
About this article

Cite this article
Walls, A., Tortorici, M., Frenz, B. et al. Glycan shield and epitope masking of a coronavirus spike protein observed by cryo-electron microscopy. Nat Struct Mol Biol 23, 899–905 (2016). https://doi.org/10.1038/nsmb.3293
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.3293
This article is cited by
-
Neutralization, effector function and immune imprinting of Omicron variants
Nature (2023)
-
Antigenic mapping reveals sites of vulnerability on α-HCoV spike protein
Communications Biology (2022)
-
Challenges and developments in universal vaccine design against SARS-CoV-2 variants
npj Vaccines (2022)
-
Leveraging metabolic modeling to identify functional metabolic alterations associated with COVID-19 disease severity
Metabolomics (2022)
-
Stable trimer formation of spike protein from porcine epidemic diarrhea virus improves the efficiency of secretory production in silkworms and induces neutralizing antibodies in mice
Veterinary Research (2021)
