
Abstract
Mutations in genes encoding proteins required for telomere structure, replication, repair and length maintenance are associated with several debilitating human genetic disorders. These complex telomere biology disorders (TBDs) give rise to critically short telomeres that affect the homeostasis of multiple organs. Furthermore, genome instability is often a hallmark of telomere syndromes, which are associated with increased cancer risk. Here, we summarize the molecular causes and cellular consequences of disease-causing mutations associated with telomere dysfunction.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$189.00 per year
only $15.75 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others
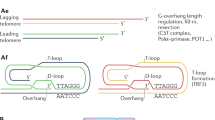
Change history
20 April 2017
The schematic of the TERT subunit in Figure 1 incorrectly identified the 'Internal RNA template': it now correctly indicates the telomerase RNA. The RNA template sequence has been corrected from AAUCCCAAU to AAUCCCAAUC, and the DNA sequence of the repeat synthesis product added to the 3′ overhang was changed from GGGTTA to GGTTAG.
References
de Lange, T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 19, 2100–2110 (2005).
Moyzis, R.K. et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc. Natl. Acad. Sci. USA 85, 6622–6626 (1988).
Wellinger, R.J., Wolf, A.J. & Zakian, V.A. Saccharomyces telomeres acquire single-strand TG1–3 tails late in S phase. Cell 72, 51–60 (1993).
Harley, C.B., Futcher, A.B. & Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 345, 458–460 (1990).
de Lange, T. How telomeres solve the end-protection problem. Science 326, 948–952 (2009).
Greider, C.W. & Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 43, 405–413 (1985).
Shippen-Lentz, D. & Blackburn, E.H. Functional evidence for an RNA template in telomerase. Science 247, 546–552 (1990).
Hao, L.Y. et al. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell 123, 1121–1131 (2005).
Hahn, W.C. et al. Creation of human tumour cells with defined genetic elements. Nature 400, 464–468 (1999).
Hiyama, E. & Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 96, 1020–1024 (2007).
Marión, R.M. & Blasco, M.A. Telomeres and telomerase in adult stem cells and pluripotent embryonic stem cells. Adv. Exp. Med. Biol. 695, 118–131 (2010).
Flores, I. et al. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 22, 654–667 (2008).
Hultdin, M. et al. Association between telomere length and V(H) gene mutation status in chronic lymphocytic leukaemia: clinical and biological implications. Br. J. Cancer 88, 593–598 (2003).
Damle, R.N. et al. Telomere length and telomerase activity delineate distinctive replicative features of the B-CLL subgroups defined by immunoglobulin V gene mutations. Blood 103, 375–382 (2004).
Palm, W. & de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 (2008).
Baumann, P. & Cech, T.R. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 292, 1171–1175 (2001).
Ye, J.Z. et al. TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J. Biol. Chem. 279, 47264–47271 (2004).
Sfeir, A. et al. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90–103 (2009).
van Steensel, B., Smogorzewska, A. & de Lange, T. TRF2 protects human telomeres from end-to-end fusions. Cell 92, 401–413 (1998).
Celli, G.B. & de Lange, T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat. Cell Biol. 7, 712–718 (2005).
Sfeir, A. & de Lange, T. Removal of shelterin reveals the telomere end-protection problem. Science 336, 593–597 (2012).
Griffith, J.D. et al. Mammalian telomeres end in a large duplex loop. Cell 97, 503–514 (1999). Demonstration by electron microscopy that TRF2 can remodel linear telomeric DNA into large telomeric duplex loops (t loops).
O'Sullivan, R.J. & Karlseder, J. Telomeres: protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 11, 171–181 (2010).
Vannier, J.B., Pavicic-Kaltenbrunner, V., Petalcorin, M.I., Ding, H. & Boulton, S.J. RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149, 795–806 (2012). Demonstration that the RTEL1 helicase promotes t-loop disassembly at telomeres and counteracts telomere fragility by unwinding G4-DNA secondary structures.
Vannier, J.B. et al. RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 342, 239–242 (2013).
Armanios, M. et al. Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc. Natl. Acad. Sci. USA 102, 15960–15964 (2005).
Heiss, N.S. et al. X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat. Genet. 19, 32–38 (1998).
Knight, S.W. et al. 1.4 Mb candidate gene region for X linked dyskeratosis congenita defined by combined haplotype and X chromosome inactivation analysis. J. Med. Genet. 35, 993–996 (1998).
Vulliamy, T. et al. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413, 432–435 (2001). Identification of telomerase RNA (hTR) as a gene mutated in dyskeratosis congenita, with an autosomal dominant inheritance pattern.
Vulliamy, T.J. et al. Mutations in the reverse transcriptase component of telomerase (TERT) in patients with bone marrow failure. Blood Cells Mol. Dis. 34, 257–263 (2005).
Yamaguchi, H. et al. Mutations in TERT, the gene for telomerase reverse transcriptase, in aplastic anemia. N. Engl. J. Med. 352, 1413–1424 (2005).
Savage, S.A. et al. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet. 82, 501–509 (2008). The first demonstration that TINF2 is mutated in a classical dyskeratosis congenita and Revesz syndrome.
Walne, A.J., Vulliamy, T., Beswick, R., Kirwan, M. & Dokal, I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood 112, 3594–3600 (2008).
Yang, D., He, Q., Kim, H., Ma, W. & Songyang, Z. TIN2 protein dyskeratosis congenita missense mutants are defective in association with telomerase. J. Biol. Chem. 286, 23022–23030 (2011).
Montanaro, L., Tazzari, P.L. & Derenzini, M. Enhanced telomere shortening in transformed lymphoblasts from patients with X linked dyskeratosis. J. Clin. Pathol. 56, 583–586 (2003).
Dokal, I. Dyskeratosis congenita. Hematology (Am Soc Hematol Educ Program) 2011, 480–486 (2011).
Alter, B.P. et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br. J. Haematol. 150, 179–188 (2010).
Alter, B.P., Giri, N., Savage, S.A. & Rosenberg, P.S. Cancer in dyskeratosis congenita. Blood 113, 6549–6557 (2009).
Murnane, J.P. Telomere dysfunction and chromosome instability. Mutat. Res. 730, 28–36 (2012).
Vulliamy, T. et al. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in TERC. Nat. Genet. 36, 447–449 (2004).
Kirwan, M. & Dokal, I. Dyskeratosis congenita, stem cells and telomeres. Biochim. Biophys. Acta 1792, 371–379 (2009).
Hoyeraal, H.M., Lamvik, J. & Moe, P.J. Congenital hypoplastic thrombocytopenia and cerebral malformations in two brothers. Acta Paediatr. Scand. 59, 185–191 (1970).
Hreidarsson, S., Kristjansson, K., Johannesson, G. & Johannsson, J.H. A syndrome of progressive pancytopenia with microcephaly, cerebellar hypoplasia and growth failure. Acta Paediatr. Scand. 77, 773–775 (1988).
Revy, P. et al. A syndrome involving intrauterine growth retardation, microcephaly, cerebellar hypoplasia, B lymphocyte deficiency, and progressive pancytopenia. Pediatrics 105, E39 (2000).
Touzot, F. et al. Heterogeneous telomere defects in patients with severe forms of dyskeratosis congenita. J. Allergy Clin. Immunol. 129, 473–82, 482 e1–3 (2012).
Ballew, B.J. et al. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum. Genet. 132, 473–480 (2013).
Le Guen, T. et al. Human RTEL1 deficiency causes Hoyeraal-Hreidarsson syndrome with short telomeres and genome instability. Hum. Mol. Genet. 22, 3239–3249 (2013).
Ding, H. et al. Regulation of murine telomere length by Rtel: an essential gene encoding a helicase-like protein. Cell 117, 873–886 (2004). Knockout of Rtel1 is embryonically lethal. Rtel1 -null embryonic stem cells display telomere loss, chromosomal breaks and fusions upon differentiation in vivo.
Barber, L.J. et al. RTEL1 maintains genomic stability by suppressing homologous recombination. Cell 135, 261–271 (2008).
Mitchell, J.R., Wood, E. & Collins, K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402, 551–555 (1999). Identification that dyskerin associates with human telomerase RNA. Fibroblasts derived from people with DC show reduced telomerase activity.
Meier, U.T. & Blobel, G. NAP57, a mammalian nucleolar protein with a putative homolog in yeast and bacteria. J. Cell Biol. 127, 1505–1514 (1994).
Mitchell, J.R., Cheng, J. & Collins, K. A box H/ACA small nucleolar RNA-like domain at the human telomerase RNA 3′ end. Mol. Cell. Biol. 19, 567–576 (1999).
Marrone, A., Walne, A. & Dokal, I. Dyskeratosis congenita: telomerase, telomeres and anticipation. Curr. Opin. Genet. Dev. 15, 249–257 (2005).
Trahan, C., Martel, C. & Dragon, F. Effects of dyskeratosis congenita mutations in dyskerin, NHP2 and NOP10 on assembly of H/ACA pre-RNPs. Hum. Mol. Genet. 19, 825–836 (2010).
Mochizuki, Y., He, J., Kulkarni, S., Bessler, M. & Mason, P.J. Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proc. Natl. Acad. Sci. USA 101, 10756–10761 (2004).
Grozdanov, P.N., Fernandez-Fuentes, N., Fiser, A. & Meier, U.T. Pathogenic NAP57 mutations decrease ribonucleoprotein assembly in dyskeratosis congenita. Hum. Mol. Genet. 18, 4546–4551 (2009).
Ruggero, D. et al. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science 299, 259–262 (2003).
Brault, M.E., Lauzon, C. & Autexier, C. Dyskeratosis congenita mutations in dyskerin SUMOylation consensus sites lead to impaired telomerase RNA accumulation and telomere defects. Hum. Mol. Genet. 22, 3498–3507 (2013).
Knight, S.W. et al. X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am. J. Hum. Genet. 65, 50–58 (1999).
Cossu, F. et al. A novel DKC1 mutation, severe combined immunodeficiency (T+B-NK- SCID) and bone marrow transplantation in an infant with Hoyeraal-Hreidarsson syndrome. Br. J. Haematol. 119, 765–768 (2002).
Batista, L.F. et al. Telomere shortening and loss of self-renewal in dyskeratosis congenita induced pluripotent stem cells. Nature 474, 399–402 (2011).
Marrone, A. et al. Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood 110, 4198–4205 (2007).
Lingner, J. et al. Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276, 561–567 (1997).
Weinrich, S.L. et al. Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat. Genet. 17, 498–502 (1997).
Canard, B., Chowdhury, K., Sarfati, R., Doublie, S. & Richardson, C.C. The motif D loop of human immunodeficiency virus type 1 reverse transcriptase is critical for nucleoside 5′-triphosphate selectivity. J. Biol. Chem. 274, 35768–35776 (1999).
Du, H.Y. et al. Complex inheritance pattern of dyskeratosis congenita in two families with 2 different mutations in the telomerase reverse transcriptase gene. Blood 111, 1128–1130 (2008).
Du, H.Y. et al. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood 113, 309–316 (2009).
Sharma, A., Myers, K., Ye, Z. & D'Orazio, J. Dyskeratosis congenita caused by a novel TERT point mutation in siblings with pancytopenia and exudative retinopathy. Pediatr. Blood Cancer 61, 2302–2304 (2014).
Peng, Y., Mian, I.S. & Lue, N.F. Analysis of telomerase processivity: mechanistic similarity to HIV-1 reverse transcriptase and role in telomere maintenance. Mol. Cell 7, 1201–1211 (2001).
Bachand, F. & Autexier, C. Functional regions of human telomerase reverse transcriptase and human telomerase RNA required for telomerase activity and RNA-protein interactions. Mol. Cell. Biol. 21, 1888–1897 (2001).
Lai, C.K., Mitchell, J.R. & Collins, K. RNA binding domain of telomerase reverse transcriptase. Mol. Cell. Biol. 21, 990–1000 (2001).
Banik, S.S. et al. C-terminal regions of the human telomerase catalytic subunit essential for in vivo enzyme activity. Mol. Cell. Biol. 22, 6234–6246 (2002).
Middleman, E.J., Choi, J., Venteicher, A.S., Cheung, P. & Artandi, S.E. Regulation of cellular immortalization and steady-state levels of the telomerase reverse transcriptase through its carboxy-terminal domain. Mol. Cell. Biol. 26, 2146–2159 (2006).
Seimiya, H. et al. Involvement of 14–3-3 proteins in nuclear localization of telomerase. EMBO J. 19, 2652–2661 (2000).
Zhong, F.L. et al. TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell 150, 481–494 (2012).
Vulliamy, T.J. et al. Differences in disease severity but similar telomere lengths in genetic subgroups of patients with telomerase and shelterin mutations. PLoS ONE 6, e24383 (2011).
Xin, Z.T. et al. Functional characterization of natural telomerase mutations found in patients with hematologic disorders. Blood 109, 524–532 (2007).
ten Dam, E., van Belkum, A. & Pleij, K. A conserved pseudoknot in telomerase RNA. Nucleic Acids Res. 19, 6951 (1991).
Comolli, L.R., Smirnov, I., Xu, L., Blackburn, E.H. & James, T.L. A molecular switch underlies a human telomerase disease. Proc. Natl. Acad. Sci. USA 99, 16998–17003 (2002).
Zhang, Q., Kim, N.K. & Feigon, J. Architecture of human telomerase RNA. Proc. Natl. Acad. Sci. USA 108, 20325–20332 (2011).
Theimer, C.A., Finger, L.D., Trantirek, L. & Feigon, J. Mutations linked to dyskeratosis congenita cause changes in the structural equilibrium in telomerase RNA. Proc. Natl. Acad. Sci. USA 100, 449–454 (2003).
Henras, A. et al. Nhp2p and Nop10p are essential for the function of H/ACA snoRNPs. EMBO J. 17, 7078–7090 (1998).
Walne, A.J. et al. Genetic heterogeneity in autosomal recessive dyskeratosis congenita with one subtype due to mutations in the telomerase-associated protein NOP10. Hum. Mol. Genet. 16, 1619–1629 (2007).
Li, L. & Ye, K. Crystal structure of an H/ACA box ribonucleoprotein particle. Nature 443, 302–307 (2006).
Vulliamy, T. et al. Mutations in the telomerase component NHP2 cause the premature ageing syndrome dyskeratosis congenita. Proc. Natl. Acad. Sci. USA 105, 8073–8078 (2008).
Venteicher, A.S. et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644–648 (2009).
Cristofari, G. et al. Human telomerase RNA accumulation in Cajal bodies facilitates telomerase recruitment to telomeres and telomere elongation. Mol. Cell 27, 882–889 (2007).
Sleeman, J.E., Ajuh, P. & Lamond, A.I. snRNP protein expression enhances the formation of Cajal bodies containing p80-coilin and SMN. J. Cell Sci. 114, 4407–4419 (2001).
Sleeman, J.E. & Lamond, A.I. Newly assembled snRNPs associate with coiled bodies before speckles, suggesting a nuclear snRNP maturation pathway. Curr. Biol. 9, 1065–1074 (1999).
Zhong, F. et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 25, 11–16 (2011).
Tycowski, K.T., Shu, M.D., Kukoyi, A. & Steitz, J.A. A conserved WD40 protein binds the Cajal body localization signal of scaRNP particles. Mol. Cell 34, 47–57 (2009).
Zhu, Y., Tomlinson, R.L., Lukowiak, A.A., Terns, R.M. & Terns, M.P. Telomerase RNA accumulates in Cajal bodies in human cancer cells. Mol. Biol. Cell 15, 81–90 (2004).
Jády, B.E., Bertrand, E. & Kiss, T. Human telomerase RNA and box H/ACA scaRNAs share a common Cajal body-specific localization signal. J. Cell Biol. 164, 647–652 (2004).
Chen, Y. et al. Human cells lacking coilin and Cajal bodies are proficient in telomerase assembly, trafficking and telomere maintenance. Nucleic Acids Res. 43, 385–395 (2015).
Freund, A. et al. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell 159, 1389–1403 (2014).
Nandakumar, J. et al. The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature 492, 285–289 (2012). Identification of the interaction between telomerase and TPP1 through a small patch of amino acids on the surface of TPP1 (TEL patch). This interaction is critical for telomerase function in vivo.
Sexton, A.N., Youmans, D.T. & Collins, K. Specificity requirements for human telomere protein interaction with telomerase holoenzyme. J. Biol. Chem. 287, 34455–34464 (2012).
Kocak, H. et al. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev. 28, 2090–2102 (2014).
Guo, Y. et al. Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood 124, 2767–2774 (2014).
Frescas, D. & de Lange, T. Binding of TPP1 protein to TIN2 protein is required for POT1a,b protein-mediated telomere protection. J. Biol. Chem. 289, 24180–24187 (2014).
Du, H.Y., Mason, P.J., Bessler, M. & Wilson, D.B. TINF2 mutations in children with severe aplastic anemia. Pediatr. Blood Cancer 52, 687 (2009).
Sasa, G.S., Ribes-Zamora, A., Nelson, N.D. & Bertuch, A.A. Three novel truncating TINF2 mutations causing severe dyskeratosis congenita in early childhood. Clin. Genet. 81, 470–478 (2012).
Chen, Y. et al. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science 319, 1092–1096 (2008).
Kim, S.H. et al. Telomere dysfunction and cell survival: roles for distinct TIN2-containing complexes. J. Cell Biol. 181, 447–460 (2008).
Ye, J.Z. & de Lange, T. TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat. Genet. 36, 618–623 (2004).
Beier, F., Foronda, M., Martinez, P. & Blasco, M.A. Conditional TRF1 knockout in the hematopoietic compartment leads to bone marrow failure and recapitulates clinical features of dyskeratosis congenita. Blood 120, 2990–3000 (2012).
Canudas, S. et al. A role for heterochromatin protein 1gamma at human telomeres. Genes Dev. 25, 1807–1819 (2011).
Houghtaling, B.R., Canudas, S. & Smith, S. A role for sister telomere cohesion in telomere elongation by telomerase. Cell Cycle 11, 19–25 (2012).
Bhanot, M. & Smith, S. TIN2 stability is regulated by the E3 ligase Siah2. Mol. Cell. Biol. 32, 376–384 (2012).
Wright, W.E., Piatyszek, M.A., Rainey, W.E., Byrd, W. & Shay, J.W. Telomerase activity in human germline and embryonic tissues and cells. Dev. Genet. 18, 173–179 (1996).
Giraud-Panis, M.J., Teixeira, M.T., Geli, V. & Gilson, E. CST meets shelterin to keep telomeres in check. Mol. Cell 39, 665–676 (2010).
Keller, R.B. et al. CTC1 mutations in a patient with dyskeratosis congenita. Pediatr. Blood Cancer 59, 311–314 (2012).
Walne, A.J. et al. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica 98, 334–338 (2013).
Anderson, B.H. et al. Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat. Genet. 44, 338–342 (2012).
Chen, L.Y., Majerska, J. & Lingner, J. Molecular basis of telomere syndrome caused by CTC1 mutations. Genes Dev. 27, 2099–2108 (2013).
Tummala, H. et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J. Clin. Invest. 125, 2151–2160 (2015).
Stuart, B.D. et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat. Genet. 47, 512–517 (2015).
Wu, M. et al. Structural basis of m7GpppG binding to poly(A)-specific ribonuclease. Structure 17, 276–286 (2009).
Wu, M. et al. Structural insight into poly(A) binding and catalytic mechanism of human PARN. EMBO J. 24, 4082–4093 (2005).
Walne, A.J., Vulliamy, T., Kirwan, M., Plagnol, V. & Dokal, I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am. J. Hum. Genet. 92, 448–453 (2013).
Ballew, B.J. et al. A recessive founder mutation in regulator of telomere elongation helicase 1, RTEL1, underlies severe immunodeficiency and features of Hoyeraal Hreidarsson syndrome. PLoS Genet. 9, e1003695 (2013).
Deng, Z. et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc. Natl. Acad. Sci. USA 110, E3408–E3416 (2013).
Khincha, P.P. & Savage, S.A. Genomic characterization of the inherited bone marrow failure syndromes. Semin. Hematol. 50, 333–347 (2013).
Fedick, A.M. et al. Carrier screening of RTEL1 mutations in the Ashkenazi Jewish population. Clin. Genet. 68, 177–181 (2015).
Vannier, J.B., Sarek, G. & Boulton, S.J. RTEL1: functions of a disease-associated helicase. Trends Cell Biol. 24, 416–425 (2014).
Acknowledgements
Research in the DNA damage–response laboratory of S.J.B. is funded by The Francis Crick Institute, a European Research Council (ERC) Advanced Investigator Grant (RecMitMei) and a Senior Investigator grant from the Wellcome Trust. S.J.B. is supported as a recipient of a Royal Society Wolfson Research Merit Award. G.S. is funded by an European Molecular Biology Organization (EMBO) Advanced Fellowship. P. Marzec and P. Margalef are funded by EMBO long-term fellowships.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Sarek, G., Marzec, P., Margalef, P. et al. Molecular basis of telomere dysfunction in human genetic diseases. Nat Struct Mol Biol 22, 867–874 (2015). https://doi.org/10.1038/nsmb.3093
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nsmb.3093
This article is cited by
-
Structure of human telomerase holoenzyme with bound telomeric DNA
Nature (2021)
-
Impact of DAXX and ATRX expression on telomere length and prognosis of breast cancer patients
Journal of the Egyptian National Cancer Institute (2020)
-
Tpz1TPP1 prevents telomerase activation and protects telomeres by modulating the Stn1-Ten1 complex in fission yeast
Communications Biology (2019)
-
Epidemiological, clinical and genetic characterization of aplastic anemia patients in Pakistan
Annals of Hematology (2019)
-
LARP7-like protein Pof8 regulates telomerase assembly and poly(A)+TERRA expression in fission yeast
Nature Communications (2018)
