
Key Points
-
Ageing-associated changes promote the development of osteoarthritis (OA), but ageing and OA are independent processes
-
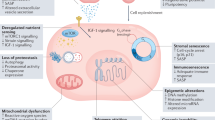
Several hallmarks of ageing could contribute to OA: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, dysregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication
-
The increase in fat mass and related metabolic changes that occur with ageing can result in ageing-related inflammation (referred to as 'inflammaging'), a chronic low-grade systemic proinflammatory state
-
Elevated levels of reactive oxygen species can contribute to OA by causing oxidative damage and disrupting normal cell signalling, leading to imbalanced anabolic and catabolic activity and ultimately cell death
-
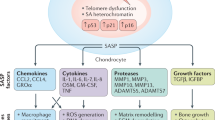
Chondrocytes can undergo cellular senescence with age and OA in response to growth signals released as a result of underlying cellular damage
-
The non-enzymatic crosslinking of collagen that occurs with ageing alters the mechanical properties of cartilage, and the resulting changes to mechanotransduction pathways reduce extracellular matrix synthesis by chondrocytes
Abstract
Ageing-associated changes that affect articular tissues promote the development of osteoarthritis (OA). Although ageing and OA are closely linked, they are independent processes. Several potential mechanisms by which ageing contributes to OA have been elucidated. This Review focuses on the contributions of the following factors: age-related inflammation (also referred to as 'inflammaging'); cellular senescence (including the senescence-associated secretory phenotype (SASP)); mitochondrial dysfunction and oxidative stress; dysfunction in energy metabolism due to reduced activity of 5′-AMP-activated protein kinase (AMPK), which is associated with reduced autophagy; and alterations in cell signalling due to age-related changes in the extracellular matrix. These various processes contribute to the development of OA by promoting a proinflammatory, catabolic state accompanied by increased susceptibility to cell death that together lead to increased joint tissue destruction and defective repair of damaged matrix. The majority of studies to date have focused on articular cartilage, and it will be important to determine whether similar mechanisms occur in other joint tissues. Improved understanding of ageing-related mechanisms that promote OA could lead to the discovery of new targets for therapies that aim to slow or stop the progression of this chronic and disabling condition.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others

References
Johnson, V. L. & Hunter, D. J. The epidemiology of osteoarthritis. Best Pract. Res. Clin. Rheumatol. 28, 5–15 (2014).
Prieto-Alhambra, D. et al. Incidence and risk factors for clinically diagnosed knee, hip and hand osteoarthritis: influences of age, gender and osteoarthritis affecting other joints. Ann. Rheum. Dis. 73, 1659–1664 (2014).
Losina, E. et al. Lifetime risk and age at diagnosis of symptomatic knee osteoarthritis in the US. Arthritis Care Res. (Hoboken) 65, 703–711 (2013).
Vos, T. et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 380, 2163–2196 (2012).
March, L. M. & Bachmeier, C. J. Economics of osteoarthritis: a global perspective. Baillieres Clin. Rheumatol. 11, 817–834 (1997).
Cram, P. et al. Total knee arthroplasty volume, utilization, and outcomes among Medicare beneficiaries, 1991–2010. JAMA 308, 1227–1236 (2012).
Loeser, R. F., Goldring, S. R., Scanzello, C. R. & Goldring, M. B. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum. 64, 1697–1707 (2012).
Pauli, C. et al. Macroscopic and histopathologic analysis of human knee menisci in aging and osteoarthritis. Osteoarthritis Cartilage 19, 1132–1141 (2011).
Hasegawa, A. et al. Anterior cruciate ligament changes in the human knee joint in aging and osteoarthritis. Arthritis Rheum. 64, 696–704 (2012).
Busse, B. et al. Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone. Aging Cell 9, 1065–1075 (2010).
Lopez-Otin, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Attur, M. et al. Low-grade inflammation in symptomatic knee osteoarthritis: prognostic value of inflammatory plasma lipids and peripheral blood leukocyte biomarkers. Arthritis Rheumatol. 67, 2905–2915 (2015).
Scanzello, C. R. & Loeser, R. F. Inflammatory activity in symptomatic knee osteoarthritis: not all inflammation is local. Arthritis Rheumatol. 67, 2797–2800 (2015).
Franceschi, C. et al. Inflamm-aging: an evolutionary perspective on immunosenescence. Ann. NY Acad. Sci. 908, 244–254 (2000).
Ershler, W. B. Interleukin-6: a cytokine for gerontologists. J. Am. Geriatr. Soc. 41, 176–181 (1993).
Morrisette-Thomas, V. et al. Inflamm-aging does not simply reflect increases in pro-inflammatory markers. Mech. Ageing Dev. 139, 49–57 (2014).
Spector, T. D. et al. Low-level increases in serum C-reactive protein are present in early osteoarthritis of the knee and predict progressive disease. Arthritis Rheum. 40, 723–727 (1997).
Livshits, G. et al. Interleukin-6 is a significant predictor of radiographic knee osteoarthritis: the Chingford study. Arthritis Rheum. 60, 2037–2045 (2009).
Smolen, J. S. et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet 371, 987–997 (2008).
de Hooge, A. S. et al. Male IL-6 gene knock out mice developed more advanced osteoarthritis upon aging. Osteoarthritis Cartilage 13, 66–73 (2005).
Delmonico, M. J. et al. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 90, 1579–1585 (2009).
Wang, X., Hunter, D., Xu, J. & Ding, C. Metabolic triggered inflammation in osteoarthritis. Osteoarthritis Cartilage 23, 22–30 (2015).
Courties, A., Gualillo, O., Berenbaum, F. & Sellam, J. Metabolic stress-induced joint inflammation and osteoarthritis. Osteoarthritis Cartilage 23, 1955–1965 (2015).
Sellam, J. & Berenbaum, F. Is osteoarthritis a metabolic disease? Joint Bone Spine 80, 568–573 (2013).
Guilak, F. Biomechanical factors in osteoarthritis. Best Pract. Res. Clin. Rheumatol. 25, 815–823 (2011).
Lee, S., Kim, T. N. & Kim, S. H. Sarcopenic obesity is more closely associated with knee osteoarthritis than is nonsarcopenic obesity: a cross-sectional study. Arthritis Rheum. 64, 3947–3954 (2012).
Scott, D., Blizzard, L., Fell, J. & Jones, G. Prospective study of self-reported pain, radiographic osteoarthritis, sarcopenia progression, and falls risk in community-dwelling older adults. Arthritis Care Res. (Hoboken) 64, 30–37 (2012).
Chuckpaiwong, B., Charles, H. C., Kraus, V. B., Guilak, F. & Nunley, J. A. Age-associated increases in the size of the infrapatellar fat pad in knee osteoarthritis as measured by 3T MRI. J. Orthop. Res. 28, 1149–1154 (2010).
Ushiyama, T., Chano, T., Inoue, K. & Matsusue, Y. Cytokine production in the infrapatellar fat pad: another source of cytokines in knee synovial fluids. Ann. Rheum. Dis. 62, 108–112 (2003).
Bastiaansen-Jenniskens, Y. M. et al. Infrapatellar fat pad of patients with end-stage osteoarthritis inhibits catabolic mediators in cartilage. Ann. Rheum. Dis. 71, 288–294 (2012).
Lotz, M. & Loeser, R. F. Effects of aging on articular cartilage homeostasis. Bone 51, 241–248 (2012).
Mobasheri, A., Matta, C., Zakany, R. & Musumeci, G. Chondrosenescence: definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 80, 237–244 (2015).
Sharpless, N. E. & Sherr, C. J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 15, 397–408 (2015).
Aigner, T. et al. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: a study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 44, 1304–1312 (2001).
Kozhemyakina, E. et al. Identification of a Prg4-positive articular cartilage progenitor cell population. Arthritis Rheumatol. 67, 1261–1273 (2015).
Munoz-Espin, D. & Serrano, M. Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 (2014).
Bovee, J. V., Hogendoorn, P. C., Wunder, J. S. & Alman, B. A. Cartilage tumours and bone development: molecular pathology and possible therapeutic targets. Nat. Rev. Cancer 10, 481–488 (2010).
Freund, A., Orjalo, A. V., Desprez, P. Y. & Campisi, J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246 (2010).
Coppe, J. P. et al. Tumor suppressor and aging biomarker p16INK4a induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 286, 36396–36403 (2011).
Freund, A. & Patil, C. K. & Campisi, J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548 (2011).
Tsuchida, A. I. et al. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res. Ther. 16, 441 (2014).
Demidenko, Z. N. & Blagosklonny, M. V. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle 7, 3355–3361 (2008).
Rose, J. et al. DNA damage, discoordinated gene expression and cellular senescence in osteoarthritic chondrocytes. Osteoarthritis Cartilage 20, 1020–1028 (2012).
Harbo, M. et al. The relationship between ultra-short telomeres, aging of articular cartilage and the development of human hip osteoarthritis. Mech. Ageing Dev. 134, 367–372 (2013).
Jurk, D. et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell 11, 996–1004 (2012).
Liu, L. & Rando, T. A. Manifestations and mechanisms of stem cell aging. J. Cell Biol. 193, 257–266 (2011).
Philipot, D. et al. p16INK4a and its regulator miR-24 link senescence and chondrocyte terminal differentiation-associated matrix remodelling in osteoarthritis. Arthritis Res. Ther. 16, R58 (2014).
Sousa-Victor, P. et al. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321 (2014).
Vincent, T., Hermansson, M., Bolton, M., Wait, R. & Saklatvala, J. Basic FGF mediates an immediate response of articular cartilage to mechanical injury. Proc. Natl Acad. Sci. USA 99, 8259–8264 (2002).
Hoshiyama, Y. et al. Chondrocyte clusters adjacent to sites of cartilage degeneration have characteristics of progenitor cells. J. Orthop. Res. 33, 548–555 (2015).
Fukui, N., Zhu, Y., Maloney, W. J., Clohisy, J. & Sandell, L. J. Stimulation of BMP-2 expression by pro-inflammatory cytokines IL-1 and TNF-α in normal and osteoarthritic chondrocytes. J. Bone Joint Surg. Am. 85-A (Suppl. 3), 59–66 (2003).
Blaney Davidson, E. N. et al. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Res. Ther. 9, R102 (2007).
Johmura, Y. et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol. Cell 55, 73–84 (2014).
Krenning, L., Feringa, F. M., Shaltiel, I. A., van den Berg, J. & Medema, R. H. Transient activation of p53 in G2 phase is sufficient to induce senescence. Mol. Cell 55, 59–72 (2014).
Ashraf, S. et al. Regulation of senescence associated signaling mechanisms in chondrocytes for cartilage tissue regeneration. Osteoarthritis Cartilage 24, 196–205 (2016).
Gossan, N. et al. The circadian clock in murine chondrocytes regulates genes controlling key aspects of cartilage homeostasis. Arthritis Rheum. 65, 2334–2345 (2013).
Khapre, R. V., Kondratova, A. A., Susova, O. & Kondratov, R. V. Circadian clock protein BMAL1 regulates cellular senescence in vivo. Cell Cycle 10, 4162–4169 (2011).
Dudek, M. et al. The chondrocyte clock gene Bmal1 controls cartilage homeostasis and integrity. J. Clin. Invest. 126, 365–376 (2016).
Baker, D. J. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011).
Baker, D. J. et al. Naturally occurring p16ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Demaria, M. et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31, 722–733 (2014).
Zhu, Y. et al. The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658 (2015).
Takayama, K. et al. Involvement of ERCC1 in the pathogenesis of osteoarthritis through the modulation of apoptosis and cellular senescence. J. Orthop. Res. 32, 1326–1332 (2014).
Chang, J. et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 22, 78–83 (2016).
Laberge, R. M. et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 17, 1049–1061 (2015).
Jones, D. P. Redox theory of aging. Redox Biol. 5, 71–79 (2015).
Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15 (2011).
Hui, W. et al. Oxidative changes and signalling pathways are pivotal in initiating age-related changes in articular cartilage. Ann. Rheum. Dis. 75, 449–458 (2016).
Finkel, T. Signal transduction by mitochondrial oxidants. J. Biol. Chem. 287, 4434–4440 (2012).
Wang, Y., Zhao, X., Lotz, M., Terkeltaub, R. & Liu-Bryan, R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor γ coactivator 1α. Arthritis Rheumatol. 67, 2141–2153 (2015).
Blanco, F. J., Rego, I. & Ruiz-Romero, C. The role of mitochondria in osteoarthritis. Nat. Rev. Rheumatol. 7, 161–169 (2011).
Loeser, R. F. Aging and osteoarthritis. Curr. Opin. Rheumatol. 23, 492–496 (2011).
Koike, M. et al. Mechanical overloading causes mitochondrial superoxide and SOD2 imbalance in chondrocytes resulting in cartilage degeneration. Sci. Rep. 5, 11722 (2015).
Aigner, T. et al. Large-scale gene expression profiling reveals major pathogenetic pathways of cartilage degeneration in osteoarthritis. Arthritis Rheum. 54, 3533–3544 (2006).
Ruiz-Romero, C. et al. Mitochondrial dysregulation of osteoarthritic human articular chondrocytes analyzed by proteomics: a decrease in mitochondrial superoxide dismutase points to a redox imbalance. Mol. Cell. Proteomics 8, 179–189 (2008).
Scott, J. L. et al. Superoxide dismutase downregulation in osteoarthritis progression and end-stage disease. Ann. Rheum. Dis. 69, 1502–1510 (2010).
Yin, W., Park, J. I. & Loeser, R. F. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-Kinase-Akt and MEK–ERK MAPK signaling pathways. J. Biol. Chem. 284, 31972–31981 (2009).
Loeser, R. F., Gandhi, U., Long, D. L., Yin, W. & Chubinskaya, S. Aging and oxidative stress reduce the response of human articular chondrocytes to insulin-like growth factor 1 and osteogenic protein 1. Arthritis Rheumatol. 66, 2201–2209 (2014).
Klomsiri, C., Karplus, P. A. & Poole, L. B. Cysteine-based redox switches in enzymes. Antioxid. Redox Signal. 14, 1065–1077 (2011).
Wood, S. T. et al. Cysteine-mediated redox regulation of cell signaling in chondrocytes stimulated with fibronectin fragments. Arthritis Rheumatol. 68, 117–126 (2016).
Collins, J. A. et al. Oxidative stress promotes peroxiredoxin hyperoxidation and attenuates pro-survival signalling in aging chondrocytes. J. Biol. Chem. 291, 6641–6654 (2016).
Hardie, D. G. & Ashford, M. L. AMPK: regulating energy balance at the cellular and whole body levels. Physiology (Bethesda) 29, 99–107 (2014).
Petursson, F. et al. Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res. Ther. 15, R77 (2013).
Terkeltaub, R., Yang, B., Lotz, M. & Liu-Bryan, R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1β and tumor necrosis factor α. Arthritis Rheum. 63, 1928–1937 (2011).
Lotz, M. K. & Carames, B. Autophagy and cartilage homeostasis mechanisms in joint health, aging and OA. Nat. Rev. Rheumatol. 7, 579–587 (2011).
Ulgherait, M., Rana, A., Rera, M., Graniel, J. & Walker, D. W. AMPK modulates tissue and organismal aging in a non-cell-autonomous manner. Cell Rep. 8, 1767–1780 (2014).
Carames, B., Olmer, M., Kiosses, W. B. & Lotz, M. K. The relationship of autophagy defects to cartilage damage during joint aging in a mouse model. Arthritis Rheumatol. 67, 1568–1576 (2015).
Carames, B., Taniguchi, N., Otsuki, S., Blanco, F. J. & Lotz, M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum. 62, 791–801 (2010).
Bohensky, J., Leshinsky, S. Srinivas, V. & Shapiro, I. M. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr. Nephrol. 25, 633–642 (2010).
Johnson, S. C. & Rabinovitch, P. S. & Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 493, 338–345 (2013).
Zhang, Y. et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann. Rheum. Dis. 74, 1432–1440 (2015).
Michan, S. & Sinclair, D. Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13 (2007).
Burnett, C. et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477, 482–485 (2011).
Kugel, S. & Mostoslavsky, R. Chromatin and beyond: the multitasking roles for SIRT6. Trends Biochem. Sci. 39, 72–81 (2014).
Mostoslavsky, R. et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 124, 315–329 (2006).
Kanfi, Y. et al. The sirtuin SIRT6 regulates lifespan in male mice. Nature 483, 218–221 (2012).
Liu-Bryan, R. & Terkeltaub, R. Emerging regulators of the inflammatory process in osteoarthritis. Nat. Rev. Rheumatol. 11, 35–44 (2015).
Nagai, K. et al. Depletion of SIRT6 causes cellular senescence, DNA damage, and telomere dysfunction in human chondrocytes. Osteoarthritis Cartilage 23, 1412–1420 (2015).
Wu, Y. et al. Overexpression of Sirtuin 6 suppresses cellular senescence and NF-κB mediated inflammatory responses in osteoarthritis development. Sci. Rep. 5, 17602 (2015).
Sanchez-Adams, J., Leddy, H. A., McNulty, A. L., O'Conor, C. J. & Guilak, F. The mechanobiology of articular cartilage: bearing the burden of osteoarthritis. Curr. Rheumatol. Rep. 16, 451 (2014).
Verzijl, N. et al. Crosslinking by advanced glycation end products increases the stiffness of the collagen network in human articular cartilage: a possible mechanism through which age is a risk factor for osteoarthritis. Arthritis Rheum. 46, 114–123 (2002).
Vos, P. A. et al. Elevation of cartilage AGEs does not accelerate initiation of canine experimental osteoarthritis upon mild surgical damage. J. Orthop. Res. 30, 1398–1404 (2012).
Li, Y. et al. Establishment of a rabbit model to study the influence of advanced glycation end products accumulation on osteoarthritis and the protective effect of pioglitazone. Osteoarthritis Cartilage 24, 307–314 (2016).
Kim, J. H. et al. Matrix cross-linking-mediated mechanotransduction promotes posttraumatic osteoarthritis. Proc. Natl Acad. Sci. USA 112, 9424–9429 (2015).
Madej, W. et al. Ageing is associated with reduction of mechanically-induced activation of Smad2/3P signaling in articular cartilage. Osteoarthritis Cartilage 24, 146–157 (2016).
Blaney Davidson, E. N. et al. Increase in ALK1/ALK5 ratio as a cause for elevated MMP-13 expression in osteoarthritis in humans and mice. J. Immunol. 182, 7937–7945 (2009).
Allen, J. L., Cooke, M. E. & Alliston, T. ECM stiffness primes the TGFβ pathway to promote chondrocyte differentiation. Mol. Biol. Cell 23, 3731–3742 (2012).
Conboy, M. J., Conboy, I. M. & Rando, T. A. Heterochronic parabiosis: historical perspective and methodological considerations for studies of aging and longevity. Aging Cell 12, 525–530 (2013).
Greene, M. A. & Loeser, R. F. Aging-related inflammation in osteoarthritis. Osteoarthritis Cartilage 23, 1966–1971 (2015).
Loeser, R. F. Aging processes and the development of osteoarthritis. Curr. Opin. Rheumatol. 25, 108–113 (2013).
Kaeberlein, M., Rabinovitch, P. S. & Martin, G. M. Healthy aging: the ultimate preventative medicine. Science 350, 1191–1193 (2015).
Longo, V. D. et al. Interventions to slow aging in humans: are we ready? Aging Cell 14, 497–510 (2015).
Dai, D. F., Chiao, Y. A., Marcinek, D. J., Szeto, H. H. & Rabinovitch, P. S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 3, 6 (2014).
Acknowledgements
The authors' research work is funded by grants from the National Institute on Aging (RO1 AG044034 to R.F.L. and F32 AG050399 to B.O.D.).
Author information
Authors and Affiliations
Contributions
All authors researched data for the article and contributed to discussion of content, writing the article, and reviewing and editing the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
PowerPoint slides
Glossary
- Telomere attrition
-
The shortening and deterioration of the protective caps on the ends of chromosomes that is associated with ageing.
- Proteostasis
-
A state of protein homeostasis in which properly folded proteins are stabilized and damaged proteins are degraded.
- Cellular senescence
-
A phenotypic state that can emerge in response to in vitro or in vivo cellular stress. Senescent cells do not divide upon stimulation and often display an altered secretory profile along with increased lysosomal activity.
- Stem cell exhaustion
-
The inability of stem cells to provide a sufficient number of differentiated cells to maintain normal tissue function. Premature stem cell exhaustion can occur under conditions of high demand for progenitors or when aberrant signalling interrupts the maintenance of quiescence.
- Senescence-associated secretory phenotype
-
(SASP). A cellular phenotype that is characterized by the production of high levels of certain inflammatory cytokines and matrix metalloproteinases. The SASP represents a mechanism by which senescent cells may function in a non-autonomous fashion.
- Advanced glycation end-products
-
(AGEs). A diverse set of molecules formed by non-enzymatic reaction of proteins with reducing sugars such as glucose and ribose.
- Anabolic dynamic compression
-
A process by which physical compression of cells within a tissue promotes an anabolic response by the cells.
Rights and permissions
About this article

Cite this article
Loeser, R., Collins, J. & Diekman, B. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol 12, 412–420 (2016). https://doi.org/10.1038/nrrheum.2016.65
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrrheum.2016.65
This article is cited by
-
Oxymatrine protects articular chondrocytes from IL-1β-induced damage through autophagy activation via AKT/mTOR signaling pathway inhibition
Journal of Orthopaedic Surgery and Research (2024)
-
Baicalin inhibits IL-1β-induced ferroptosis in human osteoarthritis chondrocytes by activating Nrf-2 signaling pathway
Journal of Orthopaedic Surgery and Research (2024)
-
Regional disparities, age-related changes and sex-related differences in knee osteoarthritis
BMC Musculoskeletal Disorders (2024)
-
Association between niacin intake and knee osteoarthritis pain and function: a longitudinal cohort study
Clinical Rheumatology (2024)
-
Why is polymyalgia rheumatica a disease of older adults? Explanations through etiology and pathogenesis: a narrative review
Clinical Rheumatology (2024)
