
Key Points
-
An imbalance between the production and clearance of amyloid-β (Aβ) is an early, often initiating, factor in Alzheimer disease (AD)
-
Peripheral systems are suggested to be involved in Aβ production and clearance
-
The central and peripheral pathways of Aβ metabolism communicate with each other, and work synergistically to clear Aβ from the brain
-
Increasing experimental, epidemiologic and clinical evidence suggests that AD manifestations extend beyond the brain, and that AD pathogenesis is closely associated with systemic abnormalities
-
The systemic abnormalities in patients with AD might not be secondary to the cerebral degeneration; instead, they might reflect underlying disease processes
-
A systemic view of AD provides a novel perspective for understanding the role of Aβ in AD pathogenesis and offers opportunities for the development of new treatments and diagnostic biomarkers for AD
Abstract
Alzheimer disease (AD) is the most common type of dementia, and is currently incurable; existing treatments for AD produce only a modest amelioration of symptoms. Research into this disease has conventionally focused on the CNS. However, several peripheral and systemic abnormalities are now understood to be linked to AD, and our understanding of how these alterations contribute to AD is becoming more clearly defined. This Review focuses on amyloid-β (Aβ), a major hallmark of AD. We review emerging findings of associations between systemic abnormalities and Aβ metabolism, and describe how these associations might interact with or reflect on the central pathways of Aβ production and clearance. On the basis of these findings, we propose that these abnormal systemic changes might not only develop secondary to brain dysfunction but might also affect AD progression, suggesting that the interactions between the brain and the periphery have a crucial role in the development and progression of AD. Such a systemic view of the molecular pathogenesis of AD could provide a novel perspective for understanding this disease and present new opportunities for its early diagnosis and treatment.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


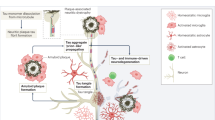
Change history
13 October 2017
In the version of this article originally published online, Figure 1 incorrectly labelled the main Aβ molecule in the peripheral pool as Aβ42 instead of Aβ40. This error has been corrected in the HTML and PDF versions of the article.
References
Mangialasche, F., Solomon, A., Winblad, B., Mecocci, P. & Kivipelto, M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 9, 702–716 (2010).
Berk, C., Paul, G. & Sabbagh, M. Investigational drugs in Alzheimer's disease: current progress. Expert Opin. Investig. Drugs 23, 837–846 (2014).
Selkoe, D. J. & Hardy, J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol. Med. 8, 595–608 (2016). This article reviews new evidence supporting the concept that an imbalance between production and clearance of Aβ is a very early, often initiating factor, in AD — a widely debated issue.
Yankner, B. A. & Mesulam, M. M. Seminars in medicine of the Beth Israel Hospital, Boston. β-Amyloid and the pathogenesis of Alzheimer's disease. N. Engl. J. Med. 325, 1849–1857 (1991).
Roher, A. E. et al. Amyloid β peptides in human plasma and tissues and their significance for Alzheimer's disease. Alzheimers Dement. 5, 18–29 (2009). This study evaluates Aβ levels in brain, peripheral organs and tissues, suggesting that brain as well as plasma Aβ levels are the consequence of intricate relationships between central and peripehral sources.
Li, Q. X., Fuller, S. J., Beyreuther, K. & Masters, C. L. The amyloid precursor protein of Alzheimer disease in human brain and blood. J. Leukoc. Biol. 66, 567–574 (1999).
Toledo, J. et al. Factors affecting Aβ plasma levels and their utility as biomarkers in ADNI. Acta Neuropathol. 122, 401–413 (2011).
Mehta, P. D., Pirttila, T., Patrick, B. A., Barshatzky, M. & Mehta, S. P. Amyloid β protein 1–40 and 1–42 levels in matched cerebrospinal fluid and plasma from patients with Alzheimer disease. Neurosci. Lett. 304, 102–106 (2001).
Delvaux, E., Bentley, K., Stubbs, V., Sabbagh, M. & Coleman, P. Differential processing of amyloid precursor protein in brain and in peripheral blood leukocytes. Neurobiol. Aging 34, 1680–1686 (2013).
Evin, G., Zhu, A., Holsinger, R. M., Masters, C. & Li, Q.-X. Proteolytic processing of the Alzheimer's disease amyloid precursor protein in brain and platelets. J. Neurosci. Res. 74, 386–392 (2003).
Biere, A. L. et al. Amyloid β-peptide is transported on lipoproteins and albumin in human plasma. J. Biol. Chem. 271, 32916–32922 (1996).
Kuo, Y. M. et al. Amyloid-β peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem. Biophys. Res. Commun. 268, 750–756 (2000).
Joachim, C. L., Mori, H. & Selkoe, D. J. Amyloid β-protein deposition in tissues other than brain in Alzheimer's disease. Nature 341, 226–230 (1989).
Koronyo, Y., Salumbides, B., Black, K. & Koronyo Hamaoui, M. Alzheimer's disease in the retina: imaging retinal Aβ plaques for early diagnosis and therapy assessment. Neurodegener. Dis. 10, 285–293 (2012).
Troncone, L. et al. Aβ amyloid pathology affects the hearts of patients with Alzheimer's disease: mind the heart. J. Am. Coll. Cardiol. 68, 2395–2407 (2016). This article was the first to describe the presence of compromised myocardial function and intramyocardial deposits of Aβ in patients with AD.
Stine, W. B., Dahlgren, K., Krafft, G. & LaDu, M. In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622 (2003).
Murray, M. et al. Amyloid β protein: Aβ40 inhibits Aβ42 oligomerization. J. Am. Chem. Soc. 131, 6316–6317 (2009).
Tarasoff-Conway, J. M. et al. Clearance systems in the brain — implications for Alzheimer disease. Nat. Rev. Neurol. 11, 457–470 (2015). This review summarizes the clearance systems of Aβ and tau in the brain.
Yao, X. Q. et al. p75NTR ectodomain is a physiological neuroprotective molecule against amyloid-β toxicity in the brain of Alzheimer's disease. Mol. Psychiatry 20, 1301–1310 (2015).
Liao, M. C. et al. N-Terminal domain of myelin basic protein inhibits amyloid β-protein fibril assembly. J. Biol. Chem. 285, 35590–35598 (2010).
Qosa, H. et al. Differences in amyloid-β clearance across mouse and human blood–brain barrier models: kinetic analysis and mechanistic modeling. Neuropharmacology 79, 668–678 (2014).
Yuede, C. M. et al. Rapid in vivo measurement of β-amyloid reveals biphasic clearance kinetics in an Alzheimer's mouse model. J. Exp. Med. 213, 677–685 (2016).
Xiang, Y. et al. Physiological amyloid-β clearance in the periphery and its therapeutic potential for Alzheimer's disease. Acta Neuropathol. 130, 487–499 (2015). This article demonstrates that peripheral clearance systems are potent in clearing brain Aβ and preventing AD.
Bradshaw, E. M. et al. CD33 Alzheimer's disease locus: altered monocyte function and amyloid biology. Nat. Neurosci. 16, 848–850 (2013).
Kanekiyo, T. & Bu, G. The low-density lipoprotein receptor-related protein 1 and amyloid-β clearance in Alzheimer's disease. Front. Aging Neurosci. 6, 93 (2014).
Ghiso, J. et al. Systemic catabolism of Alzheimer's Aβ40 and Aβ42 . J. Biol. Chem. 279, 45897–45908 (2004). This article demonstrates that the liver is the major organ responsible for uptake and degradation of circulating Aβ 42 and Aβ 40 , followed by the kidney.
Ghiso, J. et al. Alzheimer's soluble amyloid β is a normal component of human urine. FEBS Lett. 408, 105–108 (1997).
Liu, Z. et al. Characterization of insulin degrading enzyme and other amyloid-β degrading proteases in human serum: a role in Alzheimer's disease? J. Alzheimers Dis. 29, 329–340 (2012).
Mackic, J. B. et al. Human blood–brain barrier receptors for Alzheimer's amyloid-β1–40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J. Clin. Invest. 102, 734–743 (1998).
Silverberg, G. D., Mayo, M., Saul, T., Rubenstein, E. & McGuire, D. Alzheimer's disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis. Lancet Neurol. 2, 506–511 (2003).
Aspelund, A. et al. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 212, 991–999 (2015).
Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523, 337–341 (2015).
Iliff, J. J., Goldman, S. A. & Nedergaard, M. Implications of the discovery of brain lymphatic pathways. Lancet Neurol. 14, 977–979 (2015).
Eisele, Y. S. et al. Peripherally applied Aβ-containing inoculates induce cerebral β-amyloidosis. Science 330, 980–982 (2010). This study suggests that peripherally derived Aβ might enter the brain and participate in AD pathogenesis.
Eisele, Y. S. et al. Multiple factors contribute to the peripheral induction of cerebral β-amyloidosis. J. Neurosci. 34, 10264–10273 (2014).
Ritchie, D. L. et al. Amyloid-β accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol. 134, 221–240 (2017).
Jaunmuktane, Z. et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature 525, 247–250 (2015).
Deane, R. et al. RAGE mediates amyloid-β peptide transport across the blood–brain barrier and accumulation in brain. Nat. Med. 9, 907–913 (2003).
Donahue, J. E. et al. RAGE, LRP-1, and amyloid-β protein in Alzheimer's disease. Acta Neuropathol. 112, 405–415 (2006).
Yan, S. D. et al. RAGE and amyloid-β peptide neurotoxicity in Alzheimer's disease. Nature 382, 685–691 (1996).
Zenaro, E. et al. Neutrophils promote Alzheimer's disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 21, 880–886 (2015).
Frenkel, D. et al. Scara1 deficiency impairs clearance of soluble amyloid-β by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nat. Commun. 4, 2030 (2013).
Krabbe, G. et al. Functional impairment of microglia coincides with β-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 8, e60921 (2013).
Zaghi, J. et al. Alzheimer disease macrophages shuttle amyloid-β from neurons to vessels, contributing to amyloid angiopathy. Acta Neuropathol. 117, 111–124 (2009).
Gu, B. J. et al. Innate phagocytosis by peripheral blood monocytes is altered in Alzheimer's disease. Acta Neuropathol. 132, 377–389 (2016). This human study demonstrates that innate immunity is compromised in patients with AD.
Darlington, D. et al. Human umbilical cord blood-derived monocytes improve cognitive deficits and reduce amyloid-β pathology in PSAPP mice. Cell Transplant. 24, 2237–22350 (2015).
Baruch, K. et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer's disease. Nat. Med. 22, 135–137 (2016).
Prokop, S. et al. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer's disease-like mice. J. Exp. Med. 212, 1811–1818 (2015).
Hollingworth, P. et al. Common variants at ABCA7. MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat. Genet. 43, 429–435 (2011).
Jonsson, T. et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 (2013).
Jay, T. R. et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J. Exp. Med. 212, 287–295 (2015).
Bartos, A., Fialova, L., Svarcova, J. & Ripova, D. Patients with Alzheimer disease have elevated intrathecal synthesis of antibodies against tau protein and heavy neurofilament. J. Neuroimmunol. 252, 100–105 (2012).
Liu, Y. H. et al. Immunity and Alzheimer's disease: immunological perspectives on the development of novel therapies. Drug Discov. Today 18, 1212–1220 (2013).
Wang, T. et al. Naturally occurring autoantibodies against Aβ oligomers exhibited more beneficial effects in the treatment of mouse model of Alzheimer's disease than intravenous immunoglobulin. Neuropharmacology 105, 561–576 (2016).
DeMarshall, C. A. et al. Detection of Alzheimer's disease at mild cognitive impairment and disease progression using autoantibodies as blood-based biomarkers. Alzheimers Dement. (Amst.) 3, 51–62 (2016).
Monning, U. E. A. in Alzheimer's Disease: Basic Mechanisms, Diagnosis and Therapeutic Strategies (eds Iqbal, K. et al.) 557–563 (Wiley–Blackwell, 1991).
Sevigny, J. et al. The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537, 50–56 (2016).
Marsh, S. E. et al. The adaptive immune system restrains Alzheimer's disease pathogenesis by modulating microglial function. Proc. Natl Acad. Sci. USA 113, E1316–E1325 (2016).
Baruch, K. et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer's disease pathology. Nat. Commun. 6, 7967 (2015).
Rosenberg, R. N. et al. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch. Neurol. 54, 139–144 (1997).
Di Luca, M. et al. Abnormal pattern of platelet APP isoforms in Alzheimer disease and Down syndrome. Arch. Neurol. 53, 1162–1166 (1996).
Srisawat, C. et al. The platelet amyloid precursor protein ratio as a diagnostic marker for Alzheimer's disease in Thai patients. J. Clin. Neurosci. 20, 644–648 (2013).
Doecke, J. D. et al. Blood-based protein biomarkers for diagnosis of Alzheimer disease. Arch. Neurol. 69, 1318–1325 (2012).
Rogers, J. et al. Peripheral clearance of amyloid β peptide by complement C3-dependent adherence to erythrocytes. Neurobiol. Aging 27, 1733–1739 (2006).
Chen, S. H. et al. Altered peripheral profile of blood cells in Alzheimer disease: a hospital-based case–control study. Medicine (Baltimore) 96, e6843 (2017).
Loffredo, F. S. et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 153, 828–839 (2013).
Castellano, J. M. et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 544, 488–492 (2017).
Villeda, S. A. et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat. Med. 20, 659–663 (2014). This article suggests that anti-ageing molecules exist in young blood. Identification of these protective components could be important in understanding the pathogenesis of AD and in developing systemic rejuvenation therapeutics.
Villeda, S. A. et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94 (2011).
Katsimpardi, L. et al. Vascular and neurogenic rejuvenation of the aging mouse brain by young systemic factors. Science 344, 630–634 (2014).
Middeldorp, J. et al. Preclinical assessment of young blood plasma for Alzheimer disease. JAMA Neurol. 73, 1325–1333 (2016).
Xu, W. et al. Meta-analysis of modifiable risk factors for Alzheimer's disease. J. Neurol. Neurosurg. Psychiatry 86, 1299–1306 (2015).
Velayudhan, L. et al. Risk of developing dementia in people with diabetes and mild cognitive impairment. Br. J. Psychiatry 196, 36–40 (2010).
Tamaki, C., Ohtsuki, S. & Terasaki, T. Insulin facilitates the hepatic clearance of plasma amyloid β-peptide (1–40) by intracellular translocation of low-density lipoprotein receptor-related protein 1 (LRP-1) to the plasma membrane in hepatocytes. Mol. Pharmacol. 72, 850–855 (2007).
Kang, S., Lee, Y. H. & Lee, J. E. Metabolism-centric overview of the pathogenesis of Alzheimer's disease. Yonsei Med. J. 58, 479–488 (2017).
Gasparini, L. et al. Stimulation of β-amyloid precursor protein trafficking by insulin reduces intraneuronal β-amyloid and requires mitogen-activated protein kinase signaling. J. Neurosci. 21, 2561–2570 (2001).
Salameh, T. S., Shah, G. N., Price, T. O., Hayden, M. R. & Banks, W. A. Blood–brain barrier disruption and neurovascular unit dysfunction in diabetic mice: protection with the mitochondrial carbonic anhydrase inhibitor topiramate. J. Pharmacol. Exp. Ther. 359, 452–459 (2016).
Leuner, K. et al. Mitochondrion-derived reactive oxygen species lead to enhanced amyloid β formation. Antioxid. Redox Signal. 16, 1421–1433 (2012).
Moreno-Gonzalez, I. et al. Molecular interaction between type 2 diabetes and Alzheimer's disease through cross-seeding of protein misfolding. Mol. Psychiatry 22, 1327–1334 (2017). This study proposes a new molecular interaction between AD and diabetes mellitus: misfolded amylin (generated in the pancreas in type 2 diabetes mellitus) and Aβ accelerate or exacerbate the misfolding and aggregation of each other by cross-seeding.
Biessels, G. J. & Reijmer, Y. D. Brain changes underlying cognitive dysfunction in diabetes: what can we learn from MRI? Diabetes 63, 2244–2252 (2014).
Lesser, G. T. Association of Alzheimer disease pathology with abnormal lipid metabolism: the Hisayama study. Neurology 78, 1280 (2012).
Sato, N. & Morishita, R. The roles of lipid and glucose metabolism in modulation of β-amyloid, tau, and neurodegeneration in the pathogenesis of Alzheimer disease. Front. Aging Neurosci. 7, 199 (2015).
Yu, J. T., Tan, L. & Hardy, J. Apolipoprotein E in Alzheimer's disease: an update. Annu. Rev. Neurosci. 37, 79–100 (2014).
Lee, C. Y., Tse, W., Smith, J. D. & Landreth, G. E. Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 287, 2032–2044 (2012).
Zissimopoulos, J. M., Barthold, D., Brinton, R. D. & Joyce, G. Sex and race differences in the association between statin use and the incidence of Alzheimer disease. JAMA Neurol. 74, 225–232 (2016).
Reed, B. et al. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 71, 195–200 (2014).
Zubenko, G. S. et al. Platelet membrane fluidity in Alzheimer's disease and major depression. Am. J. Psychiatry 144, 860–868 (1987).
Collins, J. M., Scott, R. B., McClish, D. K., Taylor, J. R. & Grogan, W. M. Altered membrane anisotropy gradients of plasma membranes of living peripheral blood leukocytes in aging and Alzheimer's disease. Mech. Ageing Dev. 59, 153–162 (1991).
Zubenko, G. S. & Howland, R. Markedly increased platelet membrane fluidity in Down syndrome with a (14q, 21q) translocation. J. Geriatr. Psychiatry Neurol. 1, 218–219 (1988).
Scott, R. B., Collins, J. M. & Hunt, P. A. Alzheimer's disease and Down syndrome: leukocyte membrane fluidity alterations. Mech. Ageing Dev. 75, 1–10 (1994).
Yassine, H. N. et al. Association of serum docosahexaenoic acid with cerebral amyloidosis. JAMA Neurol. 73, 1208–1216 (2016).
Nishihira, J. et al. Associations between serum omega-3 fatty acid levels and cognitive functions among community-dwelling octogenarians in Okinawa, Japan: the KOCOA study. J. Alzheimers Dis. 51, 857–866 (2016).
Rusanen, M. et al. Heart diseases and long-term risk of dementia and Alzheimer's disease: a population-based CAIDE study. J. Alzheimers Dis. 42, 183–191 (2014).
Qiu, C. et al. Heart failure and risk of dementia and Alzheimer disease: a population-based cohort study. Arch. Intern. Med. 166, 1003–1008 (2006).
Jefferson, A. L. et al. Low cardiac index is associated with incident dementia and Alzheimer disease: the Framingham Heart Study. Circulation 131, 1333–1339 (2015).
Luchsinger, J. A. et al. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology 65, 545–551 (2005).
Li, J. et al. Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology 76, 1485–1491 (2011).
Jin, W. S. et al. Reduced cardiovascular functions in patients with Alzheimer's disease. J. Alzheimers Dis. 58, 919–925 (2017).
Okamoto, Y. et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol. 123, 381–394 (2012).
Zetterberg, H. et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS ONE 6, e28263 (2011).
Wang, L. et al. Chronic cerebral hypoperfusion induces memory deficits and facilitates Aβ generation in C57BL/6J mice. Exp. Neurol. 283, 353–364 (2016).
Cermakova, P. et al. Heart failure and Alzheimer's disease. J. Intern. Med. 277, 406–425 (2015).
Mattsson, N. et al. Association of brain amyloid-β with cerebral perfusion and structure in Alzheimer's disease and mild cognitive impairment. Brain 137, 1550–1561 (2014).
Leeuwis, A. E. et al. Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer's disease. Alzheimers Dement. 13, 531–540 (2017).
Marnane, M. & Hsiung, G. Y. Could better phenotyping small vessel disease provide new insights into Alzheimer disease and improve clinical trial outcomes? Curr. Alzheimer Res. 13, 750–763 (2016).
Kester, M. I. et al. Associations between cerebral small-vessel disease and Alzheimer disease pathology as measured by cerebrospinal fluid biomarkers. JAMA Neurol. 71, 855–862 (2014).
Mackic, J. B. et al. Cerebrovascular accumulation and increased blood-brain barrier permeability to circulating Alzheimer's amyloid-β peptide in aged squirrel monkey with cerebral amyloid angiopathy. J. Neurochem. 70, 210–215 (1998).
Wang, Y. R. et al. Associations between hepatic functions and plasma amyloid-β levels.— implications for the capacity of liver in peripheral amyloid-β clearance. Mol. Neurobiol. 54, 2338–2344 (2017).
Sehgal, N. et al. Withania somnifera reverses Alzheimer's disease pathology by enhancing low-density lipoprotein receptor-related protein in liver. Proc. Natl Acad. Sci. USA 109, 3510–3515 (2012).
Ghersi-Egea, J. F. et al. Fate of cerebrospinal fluid-borne amyloid β-peptide: rapid clearance into blood and appreciable accumulation by cerebral arteries. J. Neurochem. 67, 880–883 (1996).
Arvanitakis, Z., Lucas, J. A., Younkin, L. H., Younkin, S. G. & Graff-Radford, N. R. Serum creatinine levels correlate with plasma amyloid β protein. Alzheimer Dis. Assoc. Disord. 16, 187–190 (2002).
Liu, Y. H. et al. Association between serum amyloid-β and renal functions: implications for roles of kidney in amyloid-β clearance. Mol. Neurobiol. 52, 115–119 (2015).
Gronewold, J. et al. Factors responsible for plasma β-amyloid accumulation in chronic kidney disease. Mol. Neurobiol. 53, 3136–3145 (2016).
Deckers, K. et al. Dementia risk in renal dysfunction: a systematic review and meta-analysis of prospective studies. Neurology 88, 198–208 (2017).
Sakai, K. et al. Patients that have undergone hemodialysis exhibit lower amyloid deposition in the brain: evidence supporting a therapeutic strategy for Alzheimer's disease by removal of blood amyloid. J. Alzheimers Dis. 51, 997–1002 (2016).
Emamian, F. et al. The association between obstructive sleep apnea and Alzheimer's disease: a meta-analysis perspective. Front. Aging Neurosci. 8, 78 (2016).
Brunnstrom, H. R. & Englund, E. M. Cause of death in patients with dementia disorders. Eur. J. Neurol. 16, 488–492 (2009).
Pan, W. & Kastin, A. J. Can sleep apnea cause Alzheimer's disease? Neurosci. Biobehav. Rev. 47, 656–669 (2014).
Yaffe, K. et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA 306, 613–619 (2011).
Osorio, R. S. et al. Sleep-disordered breathing advances cognitive decline in the elderly. Neurology 84, 1964–1971 (2015).
Bu, X. L. et al. Serum amyloid-β levels are increased in patients with obstructive sleep apnea syndrome. Sci. Rep. 5, 13917 (2015).
Bu, X. L. et al. Serum amyloid-β levels are increased in patients with chronic obstructive pulmonary disease. Neurotox. Res. 28, 346–351 (2015).
Osorio, R. S. et al. Interaction between sleep-disordered breathing and apolipoprotein E genotype on cerebrospinal fluid biomarkers for Alzheimer's disease in cognitively normal elderly individuals. Neurobiol. Aging 35, 1318–1324 (2014).
Rosenzweig, I. et al. Sleep apnoea and the brain: a complex relationship. Lancet Respir. Med. 3, 404–414 (2015).
Musiek, E. S. & Holtzman, D. M. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354, 1004–1008 (2016).
Cedernaes, J. et al. Candidate mechanisms underlying the association between sleep–wake disruptions and Alzheimer's disease. Sleep Med. Rev. 31, 102–111 (2017).
Gareau, M. Microbiota–gut–brain axis and cognitive function. Adv. Exp. Med. Biol. 817, 357–371 (2014).
Zhan, X. et al. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 87, 2324–2332 (2016).
Akbari, E. et al. Effect of probiotic supplementation on cognitive function and metabolic status in Alzheimer's disease: a randomized, double-blind and controlled trial. Front. Aging Neurosci. 8, 256 (2016).
Bu, X. L. et al. A study on the association between infectious burden and Alzheimer's disease. Eur. J. Neurol. 22, 1519–1525 (2015). This study offers the first evidence that an increased infectious burden is associated with AD, supporting the role of systemic infection and/or inflammation in the aetiopathogenesis of AD.
Harris, S. A. & Harris, E. A. Herpes simplex virus type 1 and other pathogens are key causative factors in sporadic Alzheimer's disease. J. Alzheimers Dis. 48, 319–353 (2015).
Abbayya, K., Puthanakar, N. Y., Naduwinmani, S. & Chidambar, Y. S. Association between periodontitis and Alzheimer's disease. N. Am. J. Med. Sci. 7, 241–246 (2015).
Wallin, K. et al. Midlife rheumatoid arthritis increases the risk of cognitive impairment two decades later: a population-based study. J. Alzheimers Dis. 31, 669–676 (2012).
Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 9, 429–439 (2009).
Gao, H. M. & Hong, J. S. Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol. 29, 357–365 (2008).
Wang, J. et al. Anti-inflammatory drugs and risk of Alzheimer's disease: an updated systematic review and meta-analysis. J. Alzheimers Dis. 44, 385–396 (2015).
Lövheim, H. et al. Plasma concentrations of free amyloid-β cannot predict the development of Alzheimer's disease. Alzheimers Dement. 13, 778–782 (2017).
Mattsson, N. et al. Plasma tau in Alzheimer disease. Neurology 87, 1827–1835 (2016).
Wood, H. Alzheimer disease: biomarkers of AD risk — the end of the road for plasma amyloid-β? Nat. Rev. Neurol. 12, 613 (2016).
Herskovits, A. Z., Locascio, J. J., Peskind, E. R., Li, G. & Hyman, B. T. A. Luminex assay detects amyloid β oligomers in Alzheimer's disease cerebrospinal fluid. PLoS ONE 8, e67898 (2013).
Sengupta, U. et al. Tau oligomers in cerebrospinal fluid in Alzheimer's disease. Ann. Clin. Transl Neurol. 4, 226–235 (2017).
Shen, Y. et al. Increased plasma β-secretase 1 may predict conversion to Alzheimer's disease dementia in individuals with mild cognitive impairment. Biol. Psychiatry http://dx.doi.org/10.1016/j.biopsych.2017.02.007 (2017).
Brooks, M. One target, one treatment? Not for Alzheimer's disease. Medscape http://www.medscape.com/viewarticle/848322 (2015).
Wang, Y. J. Alzheimer disease: lessons from immunotherapy for Alzheimer disease. Nat. Rev. Neurol. 10, 188–189 (2014).
Larson, E. B., Yaffe, K. & Langa, K. M. New insights into the dementia epidemic. N. Engl. J. Med. 369, 2275–2277 (2013).
Wu, Y. T. et al. Dementia in western Europe: epidemiological evidence and implications for policy making. Lancet Neurol. 15, 116–124 (2016).
Satizabal, C. L. et al. Incidence of dementia over three decades in the Framingham heart study. N. Engl. J. Med. 374, 523–532 (2016). This study finds that the incidence of dementia has decreased over the past three decades, suggesting that control of systemic comorbidities and risk factors, as well as maintenance of body homeostasis, could bring improved results for AD prevention.
Langa, K. M. et al. A comparison of the prevalence of dementia in the United States in 2000 and 2012. JAMA Intern. Med. 177, 51–58 (2017).
Haag, M. D., Hofman, A., Koudstaal, P. J., Stricker, B. H. & Breteler, M. M. Statins are associated with a reduced risk of Alzheimer disease regardless of lipophilicity. The Rotterdam Study. J. Neurol. Neurosurg. Psychiatry 80, 13–17 (2009).
Papadopoulos, P., Tong, X. K. & Hamel, E. Selective benefits of simvastatin in bitransgenic APPSwe, Ind/TGF-β1 mice. Neurobiol. Aging 35, 203–212 (2014).
Richardson, K. et al. Statins and cognitive function: a systematic review. Ann. Intern. Med. 159, 688–697 (2013).
Ancoli-Israel, S. et al. Cognitive effects of treating obstructive sleep apnea in Alzheimer's disease: a randomized controlled study. J. Am. Geriatr. Soc. 56, 2076–2081 (2008).
Cooke, J. R. et al. Sustained use of CPAP slows deterioration of cognition, sleep, and mood in patients with Alzheimer's disease and obstructive sleep apnea: a preliminary study. J. Clin. Sleep Med. 5, 305–309 (2009).
Boada, M. et al. Amyloid-targeted therapeutics in Alzheimer's disease: use of human albumin in plasma exchange as a novel approach for Aβ mobilization. Drug News Perspect. 22, 325–339 (2009).
Jin, W. S. et al. Peritoneal dialysis reduces amyloid-β plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 134, 207–220 (2017).
Liu, Y. et al. Expression of neprilysin in skeletal muscle reduces amyloid burden in a transgenic mouse model of Alzheimer disease. Mol. Ther. 17, 1381–1386 (2009).
Liu, Y. H. et al. Clearance of amyloid-β in Alzheimer's disease: shifting the action site from center to periphery. Mol. Neurobiol. 51, 1–7 (2015).
Winston, C. N. et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement. (Amst.) 3, 63–72 (2016).
Goetzl, E. J. et al. Cargo proteins of plasma astrocyte-derived exosomes in Alzheimer's disease. FASEB J. 30, 3853–3859 (2016).
Veitinger, M. et al. A platelet protein biochip rapidly detects an Alzheimer's disease-specific phenotype. Acta Neuropathol. 128, 665–677 (2014). This article demonstrates platelet changes in AD, providing potential biomarkers for early diagnosis of AD.
Burnham, S. C. et al. A blood-based predictor for neocortical Aβ burden in Alzheimer's disease: results from the AIBL study. Mol. Psychiatry 19, 519–526 (2014).
Mattsson, N., Andreasson, U., Zetterberg, H., Blennow, K. & Alzheimer's Disease Neuroimaging Initiative. Association of plasma neurofilament light with neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 74, 557–566 (2017).
Chaves, M. L. et al. Serum levels of S100B and NSE proteins in Alzheimer's disease patients. J. Neuroinflammation 7, 6 (2010).
Teunissen, C. E. et al. Brain-specific fatty acid-binding protein is elevated in serum of patients with dementia-related diseases. Eur. J. Neurol. 18, 865–871 (2011).
Zhang, R. et al. Systemic immune system alterations in early stages of Alzheimer's disease. J. Neuroimmunol. 256, 38–42 (2013).
Kayano, M. et al. Plasma microRNA biomarker detection for mild cognitive impairment using differential correlation analysis. Biomark. Res. 4, 22 (2016).
Lu, R. et al. Reduced TRPC6 mRNA levels in the blood cells of patients with Alzheimer's disease and mild cognitive impairment. Mol. Psychiatry http://dx.doi.org/10.1038/mp.2017.136 (2017).
Roberts, B. R. et al. Biochemically-defined pools of amyloid-β in sporadic Alzheimer's disease: correlation with amyloid PET. Brain 140, 1486–1498 (2017).
Bush, A. I. et al. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J. Biol. Chem. 265, 15977–15983 (1990).
Li, Q. X. et al. Secretion of Alzheimer's disease Aβ amyloid peptide by activated human platelets. Lab. Invest. 78, 461–469 (1998).
Citron, M. et al. Excessive production of amyloid β-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc. Natl Acad. Sci. USA 91, 11993–11997 (1994).
Li, S., Liu, B., Zhang, L. & Rong, L. Amyloid β peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone 61, 164–175 (2014).
Kuo, Y. M. et al. Elevated Aβ42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AβPP metabolism. Am. J. Pathol. 156, 797–805 (2000).
Zhang, X. et al. Hypoxia-inducible factor 1α (HIF-1α)-mediated hypoxia increases BACE1 expression and β-amyloid generation. J. Biol. Chem. 282, 10873–10880 (2007).
Sun, X. et al. Hypoxia facilitates Alzheimer's disease pathogenesis by up-regulating BACE1 gene expression. Proc. Natl Acad. Sci. USA 103, 18727–18732 (2006).
Acknowledgements
The authors' research work is supported by the National Natural Science Foundation of China (grants 81471296 and 81625007 to Y.-J.W., and grant 81600949 to J.W.), and the Chinese Ministry of Science and Technology (grant 2016YFC1306401 to Y.-J.W.). The authors thank Dr J. Piña-Crespo and Dr H. Xu at Sanford Burnham Prebys Medical Discovery Institute, USA, for critical reading of the paper.
Author information
Authors and Affiliations
Contributions
All authors contributed to researching data for the article, discussion of the content, writing the article, and to review and/or editing of the manuscript before submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Wang, J., Gu, B., Masters, C. et al. A systemic view of Alzheimer disease — insights from amyloid-β metabolism beyond the brain. Nat Rev Neurol 13, 612–623 (2017). https://doi.org/10.1038/nrneurol.2017.111
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneurol.2017.111
This article is cited by
-
Therapeutics for neurodegenerative diseases by targeting the gut microbiome: from bench to bedside
Translational Neurodegeneration (2024)
-
Identification of immunogenic cell death-related genes involved in Alzheimer’s disease
Scientific Reports (2024)
-
Flavonoids and Alzheimer’s disease: reviewing the evidence for neuroprotective potential
Molecular and Cellular Biochemistry (2024)
-
Kamikihito reduces β-amyloid25–35-induced axon damage via neurotrophic factors
Journal of Natural Medicines (2024)
-
Myelin in Alzheimer’s disease: culprit or bystander?
Acta Neuropathologica Communications (2023)
