
Key Points
-
Monoamine neurotransmitter disorders are under-recognized and often misdiagnosed, as many mimic cerebral palsy and other neurological disorders
-
'Red flag' symptoms of monoamine neurotransmitter disorders include diurnal variation of symptoms, a mixed movement disorder, autonomic disturbance, involvement of the eyes (ptosis, oculogyric crisis) and levodopa responsiveness
-
Many monoamine neurotransmitter disorders are amenable to treatment; appropriate therapy is curative in some disorders
-
Analysis of cerebrospinal fluid neurotransmitter levels aids identification of the specific monoamine pathway defect and is vital for accurate diagnosis of most primary neurotransmitter disorders and selection of appropriate disease-specific pharmacotherapy
-
Research in the past few years has identified novel monoamine neurotransmitter disorders that involve defects in dopamine transport and monoamine vesicle packaging
-
Discoveries of novel genetic defects and biomarkers in monoamine neurotransmitter disorders, together with novel disease models, will improve our understanding of pathophysiological mechanisms and facilitate the development of new treatments
Abstract
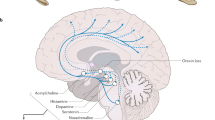
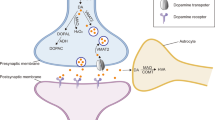
The monoamine neurotransmitter disorders are important genetic syndromes that cause disturbances in catecholamine (dopamine, noradrenaline and adrenaline) and serotonin homeostasis. These disorders result in aberrant monoamine synthesis, metabolism and transport. The clinical phenotypes are predominantly neurological, and symptoms resemble other childhood neurological disorders, such as dystonic or dyskinetic cerebral palsy, hypoxic ischaemic encephalopathy and movement disorders. As a consequence, monoamine neurotransmitter disorders are under-recognized and often misdiagnosed. The diagnosis of monoamine neurotransmitter disorders requires detailed clinical assessment, cerebrospinal fluid neurotransmitter analysis and further supportive diagnostic investigations. Prompt and accurate diagnosis of neurotransmitter disorders is paramount, as many are responsive to treatment. The treatment is usually mechanism-based, with the aim to reverse disturbances of monoamine synthesis and/or metabolism. Therapeutic intervention can lead to complete resolution of motor symptoms in some conditions, and considerably improve quality of life in others. In this Review, we discuss the clinical features, diagnosis and management of monoamine neurotransmitter disorders, and consider novel concepts, the latest advances in research and future prospects for therapy.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout




Similar content being viewed by others

References
Goldstein, D. S. Catecholamines in the periphery. Overview. Adv. Pharmacol. 42, 529–539 (1998).
Yildiz, O., Smith, J. R. & Purdy, R. E. Serotonin and vasoconstrictor synergism. Life Sci. 62, 1723–1732 (1998).
Hoffmann, G. F. & Blau, N. (Eds) Congenital Neurotransmitter Disorders: A Clinical Approach (Nova Science Publishers, 2014).
Blau, N. & Thöny, B. PND database [online] (2015).
Pearl, P. L., Capp, P. K., Novotny, E. J. & Gibson, K. M. Inherited disorders of neurotransmitters in children and adults. Clin. Biochem. 38, 1051–1058 (2005).
Assmann, B., Surtees, R. & Hoffmann, G. F. Approach to diagnosis of neurotransmitter diseases exemplified by the differential diagnosis of childhood-onset dystonia. Ann. Neurol. 54 (Suppl. 6), S18–S24 (2003).
Kurian, M. A., Gissen, P, Smith, M., Heales, S. Jr & Clayton, P. T. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol. 10, 721–733 (2011).
Fernández-Alvarez, E. Movement disorders in children: recent advances in management. Indian J. Pediatr. 76, 531–536 (2009).
Marín-Valencia, I. et al. Biochemical diagnosis of dopaminergic disturbance in paediatric patients: analysis of cerebrospinal fluid homovanillic acid and other biogenic amines. Clin. Biochem. 41, 1306–1315 (2008).
Garelis, E., Young, S. N., Lal, S. & Sourkes, T. L. Monoamine metabolites in lumbar CSF: the question of their origin in relation to clinical studies. Brain Res. 79, 1–8 (1974).
McConnell, H. & Bianchine, J. Cerebrospinal fluid. in Neurology and Psychiatry. Ch. 3 35–42 (Chapman & Hall Medical, 1994).
Bräutigam, C., Weykamp, C., Hoffmann, G. F. & Wevers, R. A. Neurotransmitter metabolites in CSF: an external quality control scheme. J. Inherit. Metab. Dis. 25, 287–298 (2002).
Hyland, K. et al. Cerebrospinal fluid concentrations of pterins and metabolites of serotonin and dopamine in a pediatric reference population. Pediatr. Res. 34, 10–14 (1993).
Segawa, M. Hereditary progressive dystonia with marked diurnal fluctuation. Brain Dev. 33, 195–201 (2011).
Opladen, T., Hoffmann, G. F. & Blau, N. An international survey of patients with tetrahydrobiopterin deficiencies presenting with hyperphenylalaninaemia. J. Inherit. Metab. Dis. 35, 963–973 (2012).
Willemsen, M. A. et al. Tyrosine hydroxylase deficiency: a treatable disorder of brain catecholamine biosynthesis. Brain 133, 1810–1822 (2010).
Yeung, W. L., Lam, C. W., Hui, J., Tong, S. F. & Wu, S. P. Galactorrhea—a strong clinical clue towards the diagnosis of neurotransmitter disease. Brain Dev. 28, 389–391 (2006).
Aitkenhead, H. & Heales, S. J. Establishment of paediatric age-related reference intervals for serum prolactin to aid in the diagnosis of neurometabolic conditions affecting dopamine metabolism. Ann. Clin. Biochem. 50, 156–158 (2013).
Brun, L. et al. Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology 75, 64–71 (2010).
Kurian, M. A. et al. Homozygous loss-of-function mutations in the gene encoding the dopamine transporter are associated with infantile parkinsonism–dystonia. J. Clin. Invest. 119, 1595–1603 (2009).
Kurian, M. A. et al. Clinical and molecular characterisation of hereditary dopamine transporter deficiency syndrome: an observational cohort and experimental study. Lancet Neurol. 10, 54–62 (2011).
Rilstone, J. J., Alkhater, R. A. & Minassian, B. A. Brain dopamine–serotonin vesicular transport disease and its treatment. N. Engl. J. Med. 368, 543–550 (2013).
Saunders-Pullman, R. et al. Phenylalanine loading as a diagnostic test for DRD: interpreting the utility of the test. Mol. Genet. Metab. 83, 207–212 (2004).
Guldberg, P., Henriksen, K. F., Lou, H. C. & Güttler, F. Aberrant phenylalanine metabolism in phenylketonuria heterozygotes. J. Inherit. Metab. Dis. 21, 365–372 (1998).
Opladen, T., Hoffmann, G. F, Kühn, A. A. & Blau, N. Pitfalls in phenylalanine loading test in the diagnosis of dopa-responsive dystonia. Mol. Genet. Metab. 108, 195–197 (2013).
Segawa, M. Autosomal dominant GTP cyclohydrolase 1 (AD GCH 1) deficiency (Segawa disease, dystonia 5; DYT 5). Chang Gung Med. J. 32, 1–11 (2009).
Friedman, J. et al. Sepiapterin reductase deficiency: a treatable mimic of cerebral palsy. Ann. Neurol. 71, 520–530 (2012).
Clot, F. et al. Exhaustive analysis of BH4 and dopamine biosynthesis genes in patients with Dopa-responsive dystonia. Brain 132, 1753–1763 (2009).
Pons, R. et al. Levodopa-induced dyskinesias in tyrosine hydroxylase deficiency. Mov. Disord. 28, 1058–1063 (2013).
López-Laso, E., Beyer, K., Opladen, T., Artuch, R. & Saunders-Pullman, R. Dyskinesias as a limiting factor in the treatment of Segawa disease. Pediatr. Neurol. 46, 404–406 (2012).
Leuzzi, V. et al. Phenotypic variability, neurological outcome and genetics background of 6-pyruvoyl-tetrahydrobiopterin synthase deficiency. Clin. Genet. 77, 249–257 (2010).
Ng, J. et al. Dopamine transporter deficiency syndrome: phenotypic spectrum from infancy to adulthood. Brain 137, 1107–1119 (2014).
Timmers, H. J., Deinum, J., Wevers, R. A. & Lenders, J. W. Congenital dopamine-β-hydroxylase deficiency in humans. Ann. N. Y. Acad. Sci. 1018, 520–523 (2004).
Blau, N., van Spronsen, F. J. & Levy, H. L. Phenylketonuria. Lancet 376, 1417–1427 (2010).
Furukawa, Y. GTP cyclohydrolase 1-deficient dopa-responsive dystonia. GeneReviews [online] (updated 2015).
Segawa, M., Hosaka, A., Miyagawa, F., Nomura, Y. & Imai, H. Hereditary progressive dystonia with marked diurnal fluctuation. Adv. Neurol. 14, 215–233 (1976).
Trender-Gerhard, I. et al. Autosomal-dominant GTPCH1-deficient DRD: clinical characteristics and long-term outcome of 34 patients. J. Neurol. Neurosurg. Psychiatry 80, 839–845 (2009).
Lee, J. H., Ki, C. S., Kim, D. S., Cho, J. W., Park, K. P. & Kim, S. Dopa-responsive dystonia with a novel initiation codon mutation in the GCH1 gene misdiagnosed as cerebral palsy. J. Korean Med. Sci. 26, 1244–1246 (2011).
Jan, M. M. Misdiagnoses in children with dopa-responsive dystonia. Pediatr. Neurol. 31, 298–303 (2004).
Dale, R. C. et al. Familial paroxysmal exercise-induced dystonia: atypical presentation of autosomal dominant GTP-cyclohydrolase 1 deficiency. Dev. Med. Child Neurol. 52, 583–586 (2010).
Mencacci, N. E. et al. Parkinson's disease in GTP cyclohydrolase 1 mutation carriers. Brain 137, 2480–2492 (2014).
Lewthwaite, A. J. et al. Novel GCH1 variant in Dopa-responsive dystonia and Parkinson's disease. Parkinsonism Relat. Disord. 21, 394–397 (2015).
Thöny, B & Blau, N. Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum. Mutat. 27, 870–878 (2006).
Steinberger, D. et al., Utility of MLPA in deletion analysis of GCH1 in dopa-responsive dystonia. Neurogenetics 8, 51–55 (2007).
Hwang, W. J., Calne, D. B., Tsui, J. K. & de la Fuente-Fernández, R. The long-term response to levodopa in dopa-responsive dystonia. Parkinsonism Relat. Disord. 8, 1–5 (2001).
Sumi-Ichinose, C. et al. Catecholamines and serotonin are differently regulated by tetrahydrobiopterin. A study from 6-pyruvolytetrahydrobiopterin synthase knockout mice. J. Biol. Chem. 276, 41150–41160 (2001).
Opladen, T. et al. Clinical and biochemical characterization of patients with early infantile onset of autosomal recessive GTP cyclohydrolase I deficiency without hyperphenylalaninemia. Mov. Disord. 26, 157–161 (2011).
Bonafé, L., Thöny, B., Leimbacher, W., Kierat, L. & Blau, N. Diagnosis of dopa-responsive dystonia and other tetrahydrobiopterin disorders by the study of biopterin metabolism in fibroblasts. Clin. Chem. 47, 477–485 (2001).
Horvath, G. A. et al. Autosomal recessive GTP cyclohydrolase I deficiency without hyperphenylalaninemia: evidence of a phenotypic continuum between dominant and recessive forms. Mol. Genet. Metab. 94, 127–131 (2008).
Longo, N. Disorders of biopterin metabolism. J. Inherit. Metab. Dis. 32, 333–342 (2009).
Smith, I. & Dhondt, J. L. Birthweight in patients with defective biopterin metabolism. Lancet 325, 818 (1985).
Jäggi, L. et al. Outcome and long-term follow up of 36 patients with tetrahydrobiopterin deficiency. Mol. Genet. Metab. 93, 295–305 (2008).
Porta, F., Mussa, A., Concolino, D., Spada, M. & Ponzone, A. Dopamine agonists in 6-pyruvoyl tetrahydrobiopterin synthase deficiency. Neurology 73, 633–637 (2009).
Dill, P. et al. Child neurology: paroxysmal stiffening, upward gaze, and hypotonia: hallmarks of sepiapterin reductase deficiency. Neurology 78, e29–e32 (2012).
Leuzzi, V. et al. Very early pattern of movement disorders in sepiapterin reductase deficiency. Neurology 81, 2141–2142 (2013).
Friedman, J., Hyland, K., Blau, N. & MacCollin, M. Dopa-responsive hypersomnia and mixed movement disorder due to sepiapterin reductase deficiency. Neurology 67, 2032–2035 (2006).
Neville, B. G., Parascandalo, R., Farrugia, R. & Felice, A. Sepiapterin reductase deficiency: a congenital dopa-responsive motor and cognitive disorder. Brain 128, 2291–2296 (2005).
Bonafé, L., Thöny, B., Penzien, J. M., Czarnecki, B. & Blau, N. Mutations in the sepiapterin reductase gene cause a novel tetrahydrobiopterin-dependent monamine-neurotransmitter deficiency without hyperphenylalaninemia. Am. J. Hum. Genet. 69, 269–277 (2001).
Crabtree, M. J. et al. Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin–eNOS stoichiometry and biopterin redox status: insights from cells with tet-regulated GTP cyclohydroxylase I expression. J. Biol. Chem. 284, 1136–1144 (2009).
Thöny, B. et al., Mutations in the pterin-4α-carbinolamine dehydratase (PCBD) gene cause a benign form of hyperphenylalaninemia. Hum. Genet. 103, 162–167 (1998).
Mendel, D. B. et al. Characterization of a cofactor that regulates dimerization of a mammalian homeodomain protein. Science 254, 1762–1767 (1991).
Ferrè, S. et al. Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J. Am. Soc. Nephrol. 25, 574–586 (2014).
Simaite, D. et al. Recessive mutations in PCBD1 cause a new type of early-onset diabetes. Diabetes 63, 3557–3564 (2014).
Mak, C. M. et al. Biochemical and molecular characterization of tyrosine hydroxylase deficiency in Hong Kong Chinese. Mol. Genet. Metab. 99, 431–433 (2010).
Giovanniello, T. et al. Tyrosine hydroxylase deficiency presenting with a biphasic clinical course. Neuropediatrics 38, 213–215 (2007).
Stamelou, M. et al. Myoclonus-dystonia syndrome due to tyrosine hydroxylase deficiency. Neurology 79, 435–441 (2012).
Espay, A. J. & Chen, R. Myoclonus. Continuum (Minneap. Minn.) 19, 1264–1286 (2013).
Ortez, C. et al. Cerebrospinal fluid synaptic proteins as useful biomarkers in tyrosine hydroxylase deficiency. Mol. Genet. Metab. 114, 34–40 (2015).
Blanchet, P. J., Konitsiotis, S. & Chase, T. N. Amantadine reduces levodopa-induced dyskinesias in parkinsonian monkeys. Mov. Disord. 13, 798–802 (1998).
Lee, H. F., Tsai, C. R., Chi, C. S., Chang, T. M. & Lee, H. J. Aromatic L-amino acid decarboxylase deficiency in Taiwan. Eur. J. Paediatr. Neurol. 13, 135–140 (2009).
Mastrangelo, M., Caputi, C., Galosi, S., Giannini, M T. & Leuzzi, V. Transdermal rotigotine in the treatment of aromatic L-amino acid decarboxylase deficiency. Mov. Disord. 28, 556–557 (2013).
Arnoux, J. B. et al. Aromatic L-amino acid decarboxylase deficiency is a cause of long-fasting hypoglycaemia. J. Clin. Endocrinol. Metab. 98, 4279–4284 (2013).
Leuzzi, V. et al. Report of two never treated adult sisters with aromatic L-amino acid decarboxylase deficiency: a portrait of the natural history of the disease or an expanding phenotype? JIMD Rep. 15, 39–45 (2015).
Chen, P. W. et al. Diagnosis of aromatic L-amino acid decarboxylase deficiency by measuring 3-O-methyldopa concentrations in dried blood spots. Clin. Chim. Acta 431, 19–22 (2014).
Atwal, P. S. et al. Aromatic L-amino acid decarboxylase deficiency diagnosed by clinical metabolomic profiling of plasma. Mol. Genet. Metab. 115, 91–94 (2015).
Surtees, R. & Hyland, K. L-3,4-dihydroxyphenylalanine (levodopa) lowers central nervous system S-adenosylmethionine concentrations in humans. J. Neurol. Neurosurg. Psychiatry 53, 569–572 (1990).
Chang, Y. T. et al. Levodopa-responsive aromatic L-amino acid decarboxylase deficiency. Ann. Neurol. 55, 435–438 (2004).
Allen, G. F., Land, J. M. & Heales, S. J. A new perspective on the treatment of aromatic L-amino acid decarboxylase deficiency. Mol. Genet. Metab. 97, 6–14 (2009).
Manegold, C. et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and follow-up. J. Inherit. Metab. Dis. 32, 371–380 (2009).
Allen, G. F. et al. Pyridoxal 5′-phosphate deficiency causes a loss of aromatic L-amino acid decarboxylase in patients and human neuroblastoma cells, implications for aromatic L-amino acid decarboxylase and vitamin B6 deficiency states. J. Neurochem. 114, 87–96 (2010).
Pons, R. et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, treatment and prognosis. Neurology 62, 1058–1065 (2004).
Hyland, K., Surtees, R A., Rodeck, C. & Clayton, P. T. Aromatic L-amino acid decarboxylase deficiency: clinical features, diagnosis, and treatment of a new inborn error of neurotransmitter amine synthesis. Neurology 42, 1980–1988 (1992).
Maller, A., Hyland, K., Milstein, S., Biaggioni, I. & Butler, I. J. Aromatic L-amino acid decarboxylase deficiency: clinical features, diagnosis, and treatment of a second family. J. Child Neurol. 12, 349–354 (1997).
Swoboda, K. J., Saul, J. P., McKenna, C. E., Speller, N. B. & Hyland, K. Aromatic L-amino acid decarboxylase deficiency: overview of clinical features and outcomes. Ann. Neurol. 54 (Suppl. 6), S49–S55 (2003).
Ng, J., Heales, S. J. & Kurian, M. A. Clinical features and pharmacotherapy of childhood monoamine neurotransmitter disorders. Paediatr. Drugs 16, 275–291 (2014).
Cawello, W. et al. Pharmacokinetics, safety and tolerability of rotigotine transdermal patch in health Japanese and Caucasian subjects. Clin. Drug Investig. 34, 95–105 (2014).
Mills, P. B. et al. Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain 133, 2148–2159 (2010).
Mills, P. B. et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5′-phosphate oxidase. Hum. Mol. Genet. 14, 1077–1086 (2005).
Derwinska, K. et al. Clinical improvement of the aggressive neurobehavioral phenotype in a patient with a deletion of PITX3 and the absence of L-DOPA in the cerebrospinal fluid. Am. J. Med. Genet. B Neuropsychiatr. Genet. 159B, 236–242 (2012).
Stockler, S. et al. Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up. Mol. Genet. Metab. 104, 48–60 (2011).
Reddy, S. D. et al. Multiple coregulatory control of tyrosine hydroxylase gene transcription. Proc. Natl Acad. Sci. USA 108, 4200–4205 (2011).
Hwang, D. Y. et al. 3,4-dihydroxyphenylalanine reverses the motor deficits in Pitx3-deficient aphakia mice: behavioral characterization of a novel genetic model of Parkinson's disease. J. Neurosci. 25, 2132–2137 (2005).
Hwang, D. Y., Ardayfio, P., Kang, U. J., Semina, E. V. & Kim, K. S. Selective loss of dopaminergic neurons in the substantia nigra of Pitx3-deficient aphakia mice. Brain Res. Mol. Brain Res. 114, 123–131 (2003).
Lenders, J. W. et al. Specific genetic deficiencies of the A and B isoenzymes of monoamine oxidase are characterized by distinct neurochemical and clinical phenotypes. J. Clin. Invest. 97, 1010–1019 (1996).
O'Leary, R. E. et al. De novo microdeletion of Xp11.3 exclusively encompassing the monoamine oxidase A and B genes in a male infant with episodic hypotonia: a genomics approach to personalized medicine. Eur. J. Med. Genet. 55, 349–353 (2012).
Gillman. P. K. Advances pertaining to the pharmacology and interactions of irreversible nonselective monoamine oxidase inhibitors. J. Clin. Psychopharmacol. 31, 66–74 (2011).
Whibley, A. et al. Deletion of MAOA and MAOB in a male patient causes severe developmental delay, intermittent hypotonia and stereotypical hand movements. Eur. J. Hum. Genet. 18, 1095–1099 (2010).
Saito, M. et al. MAOA/B deletion syndrome in male siblings with severe developmental delay and sudden loss of muscle tonus. Brain Dev. 36, 64–69 (2014).
Collins, F. A. et al. Clinical, biochemical, and neuropsychiatric evaluation of a patient with a contiguous gene syndrome due to a microdeletion Xp11.3 including the Norrie disease locus and monoamine oxidase (MAOA and MAOB) genes. Am. J. Med. Genet. 42, 127–134 (1992).
Chen, K., Holschneider, D. P., Wu, W., Rebrin, I. & Shih, J. C. A spontaneous point mutation produces monoamine oxidase A/B knock-out mice with greatly elevated monoamines and anxiety-like behaviour. J. Biol. Chem. 279, 39645–39652 (2004).
Robertson, D. & Garland, E. M. Dopamine beta-hydroxylase deficiency. GeneReviews [online] (updated 2013).
Wassenberg, T. et al. Clinical and biochemical characteristics of dopamine beta hydroxylase deficiency: expanding the clinical spectrum caused by norepinephrine and epinephrine deficiency [poster]. 4th International Symposium on Paediatric Movement Disorders P36 (2015).
Man in 't Veld, A. J. et al. dl-Threo-3,4-dihydroxyphenylserine restores sympathetic control and cures orthostatic hypotension in dopamine beta-hydroxylase deficiency. J. Hypertens. Suppl. 6, S547–S549 (1988).
Fon, E. A. et al. Vesicular transport regulates monoamine storage and release but is not essential for amphetamine action. Neuron 19, 1271–1283 (1997).
Carlsson, A. The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol. Rev. 11, 490–493 (1959).
Molero-Luis, M. et al. Homovanillic acid in cerebrospinal fluid of 1388 children with neurological disorders. Dev. Med. Child Neurol. 55, 559–566 (2013).
Hansen, F. H. et al. Missense dopamine transporter mutations associated with adult parkinsonism and ADHD. J. Clin. Invest. 124, 3107–3120 (2014).
Bhatia, K. P. 'That DAT'gene that causes dystonia-parkinsonism: broadening the phenotype. Brain 137, 976–977 (2014).
García-Cazorla, A. et al. Secondary abnormalities of neurotransmitters in infants with neurological disorders. Dev. Med. Child Neurol. 49, 740–744 (2007).
Marín-Valencia, I. et al. Biochemical diagnosis of dopaminergic disturbances in paediatric patients: analysis of cerebrospinal fluid homovanillic acid and other biogenic amines. Clin. Biochem. 41, 1306–1315 (2008).
Duarte, S. et al. Cerebrospinal fluid pterins and neurotransmitters in early severe epileptic encephalopathies. Brain Dev. 30, 106–111 (2008).
Mericmek-Mahmutoglu, S. et al. Prevalence of inherited neurotransmitter disorders in patients with movement disorders and epilepsy: a retrospective cohort study. Orphanet J. Rare Dis. 10, 12 (2015).
De Grandis, E. et al. Cerebrospinal fluid alterations of the serotonin product, 5-hydroxyindolacetic acid, in neurological disorders. J. Inherit. Metab. Dis. 33, 803–809 (2010).
Naumann, M., Götz, M., Reiners, K., Lange, K. W. & Riederer, P. Neurotransmitters in CSF of idiopathic adult-onset dystonia: reduced 5-HIAA levels as evidence of impaired serotonergic metabolism. J. Neural Transm. 103, 1083–1091 (1996).
Assmann, B., Köhler, M., Hoffmann, G. F., Heales, S. & Surtees, R. Selective decrease in central nervous system serotonin turnover in children with dopa-nonresponsive dystonia. Pediatr. Res. 52, 91–94 (2002).
Marecos, C., Ng, J., Kurian, M. A. What is new for monoamine neurotransmitter disorders? J. Inherit. Metab. Dis. 37, 619–626 (2014).
Ng, J. et al. TH gene-negative infantile onset severe dopamine deficiency syndrome: a novel neurotransmitter disorder? [abstract]. Dev. Med. Child. Neurol. 55 (Suppl. S1), 16 (2013).
Moran, M. M., Allen, N. M, Treacy, E. P. & King, M. D. “Stiff neonate” with mitochondrial DNA depletion and secondary neurotransmitter defects. Pediatr. Neurol. 45, 403–405 (2011).
Werner, E. R. et al. Tetrahydrobiopterin biosynthesic activities in human macrophages, fibroblasts, THP-1 and T-24 cells. J. Biol. Chem. 265, 3189–3192 (1990).
Millner, M. M. et al. Neopterin concentrations in cerebrospinal fluid and serum as an aid in differentiating central nervous system and peripheral infections in children. Clin. Chem. 44, 161–167 (1998).
Hagberg, L. et al. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res. Ther. 7, 15 (2010).
Rice, G. I. et al. Assessment of interferon-related biomarkers in Aicardi-Goutières syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 12, 1159–1169 (2013).
du Moulian, M., Nürnberg, P., Crow, Y. J. & Rutsch, F. Cerebral vasculopathy is a common feature in Aicardi-Goutières syndrome associated with SAMHD1 mutations. Proc. Natl Acad. Sci. USA 108, E232 (2011).
Livingston, J. H. et al. A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1. J. Med. Genet. 51, 76–82 (2014).
Crow, Y. J. Aicardi-Goutières syndrome. GeneReviews [online] (updated 2014).
Blau, N. et al. Cerebrospinal fluid pterins and folates in Aicardi-Goutières syndrome: a new phenotype. Neurology 61, 642–647 (2003).
Kurian, M. A. What is the role of dopamine in childhood neurological disorders? Dev. Med. Child Neurol. 55, 493–494 (2013).
Graziano, C. et al. Syndromic intellectual disability: a new phenotype caused by an aromatic amino acid decarboxylase gene (DDC) variant. Gene 559, 144–148 (2015).
Montioli, R. et al. A comprehensive picture of the mutations associated with aromatic amino acid decarboxylase deficiency, from molecular mechanisms to therapy implications. Hum. Mol. Genet. 23, 5429–5440 (2014).
Nguyen, H. N. et al., LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 8, 267–280 (2011).
Reinhardt, P. et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 7, 354–367 (2013).
Lancaster, M. A. et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379 (2013).
Hwang, W. Y. et al. Efficient genome editing in zebrafish using a CRISPR–Cas system. Nat. Biotechnol. 31, 227–229 (2013).
Li, D. et al. Heritable gene targeting in the mouse and rat using a CRISPR–Cas system. Nat. Biotechnol. 31, 681–683 (2013).
Brasil, S. et al. Pseudoexon exclusion by antisense therapy in 6-pyruvoyl-tetrahydropterin synthase deficiency. Hum. Mutat. 32, 1019–1027 (2011).
Eberling, J. L. et al. Results from phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology 70, 1980–1983 (2008).
Palfi, S. et al. Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson's disease: a dose escalation, open-label, phase 1/2 trial. Lancet 383, 1138–1146 (2014).
Hwu, W. L. et al. Gene therapy for aromatic L-amino acid decarboxylase deficiency. Sci. Transl. Med. 4, 134ra61 (2012).
Lee, N. C. et al. Benefits of neuronal preferential systemic gene therapy for neurotransmitter deficiency. Mol. Ther. http://dx.doi.org/10.1038/mt.2015.122 (2015).
Acknowledgements
M.A.K. is funded by a Wellcome Intermediate Clinical Fellowship (WT098524MA) and receives funding from the Rosetrees Trust and the Gracious Heart Charity Foundation. J.N. is funded by a Medical Research Council Clinical Research Training Fellowship (MR/K02342X/1). M.A.K. and J.N. are both funded by Great Ormond Street Hospital Children's Charities. A.P. receives funding from Actelion to study undiagnosed neurodegenerative disorders, the NBIA Disorders Association and Child Brain Research. None of the authors received funding for the preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
J.N. wrote the first draft and provided substantial input into revision of the manuscript. A.P. prepared tables and figures and contributed to revision of the manuscript. S.J.H. contributed to the critique of the manuscript and figures. M.A.K. provided substantial input into the manuscript concept, design, content and revision of each draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Ng, J., Papandreou, A., Heales, S. et al. Monoamine neurotransmitter disorders—clinical advances and future perspectives. Nat Rev Neurol 11, 567–584 (2015). https://doi.org/10.1038/nrneurol.2015.172
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrneurol.2015.172
This article is cited by
-
Concentration gradients of monoamines, their precursors and metabolites in serial lumbar cerebrospinal fluid of neurologically healthy patients determined with a novel LC–MS/MS technique
Fluids and Barriers of the CNS (2023)
-
Early-onset hereditary isolated non-neurogenic orthostatic hypotension in a Swedish family
Clinical Autonomic Research (2023)
-
A conserved transcriptional fingerprint of multi-neurotransmitter neurons necessary for social behavior
BMC Genomics (2022)
-
Conformational and functional changes of the native neuropeptide somatostatin occur in the presence of copper and amyloid-β
Nature Chemistry (2022)
-
Early Life Social Stress Causes Sex- and Region-Dependent Dopaminergic Changes that Are Prevented by Minocycline
Molecular Neurobiology (2022)
