
Key Points
-
Passive antibody therapy is not a new technique. Behring and Kitasato discovered that specific antibodies could protect against bacterial toxins in the early 1890s and, by the 1930s, serum therapy was being widely used to treat a variety of infectious diseases. However, the increase in the popularity of serum therapy occurred at about the same time as the first antibiotics were developed, and as antibiotics became more widely available, so the use of serum therapy declined. By the late 1940s it had largely been abandoned.
-
In recent years there has been renewed interest in using passive antibody therapy to treat infectious diseases. However, at present, although immunoglobulin preparations are available to treat some infections, such as hepatitis B, rabies and varicella–zoster virus, only one monoclonal antibody (palivizumab) has been licensed to prevent an infectious disease.
-
The advantages of using antibody molecules to treat infectious diseases include their specificity and versatility. Antibodies are capable of mediating a variety of different biological effects including both those that are independent of other components of the host immune system, such as neutralizing toxins and viruses and activating complement, and effects that involve other components of the host immune system, such as antibody-dependent cellular cytotoxicity and opsonization. Additionally, the effects of antibodies can be synergistic with those of conventional antimicrobial therapies, and the time to develop therapeutic antibody preparations would be considerably shorter than the development time for a vaccine.
-
One of the most important advantages of using antibodies is that they can be easily modified to target host cells. One such strategy is radioimmunotherapy, in which a radionuclide is attached to an antibody molecule. As an intact immune system is not required, radioimmunotherapy could be particularly effective in immunocompromised hosts. As infected cells can be killed by a 'crossfire' effect, radioimmunotherapy might also be useful to target intracellular pathogens and chronic infections.
-
The high specificity of antibodies can also be a disadvantage when considering antibody-based therapies because accurate diagnosis of the causative microbial agent of an infection is necessary and a 'cocktail' of different antibodies might be required to treat infections with a microorganism that undergoes antigenic variation. As the efficacy of therapeutic antibody preparations decreases with time, this might mean that they are best applied to infections where early diagnosis is possible. Additionally, the costs associated with antibody treatments can be higher than treatment with conventional antimicrobial agents; however, the increased costs of the treatment should be offset against the lower rates of resistance associated with antibody therapy.
Abstract
Antibody-based therapies are currently undergoing a renaissance. After being developed and then largely abandoned in the twentieth century, many antibody preparations are now in clinical use. However, most of the reagents that are available target non-infectious diseases. Interest in using antibodies to treat infectious diseases is now being fuelled by the wide dissemination of drug-resistant microorganisms, the emergence of new microorganisms, the relative inefficacy of antimicrobial drugs in immunocompromised hosts and the fact that antibody-based therapies are the only means to provide immediate immunity against biological weapons. Given the need for new antimicrobial therapies and many recent technological advances in the field of immunoglobulin research, there is considerable optimism regarding renewed applications of antibody-based therapy for the prevention and treatment of infectious diseases.
Similar content being viewed by others


Main
Passive antibody therapy was the first consistently effective antimicrobial strategy. The ability of specific antibodies to protect against bacterial toxins was discovered by Behring and Kitasato in the early 1890s (Ref. 1), and this observation led to the rapid development of antibody therapy for the treatment of various infectious diseases2,3 (Timeline). As all antibody preparations were derived from the serum of immunized animals or immune human donors, this form of therapy was known as 'serum therapy'. Serum therapy was effective, but the administration of large amounts of animal proteins was often associated with side effects that ranged from immediate HYPERSENSITIVITY REACTIONS to serum sickness, which is a form of ANTIGEN–ANTIBODY COMPLEX DISEASE. By the 1930s, improvements in antibody purification methods allowed the production of antibody preparations with reduced toxicity, and serum therapy was an effective means of treating many infectious diseases. However, after 1935, the use of serum therapy declined rapidly due to the introduction of sulphonamides and, soon thereafter, other classes of antimicrobial chemotherapy. By the late 1940s, serum was largely abandoned as an antibacterial agent, but antibody-based therapies retained a niche as a treatment for venoms, toxins and certain viral infections.

One of the paradoxes of the history of serum therapy is that its abandonment coincided with advances in antibody purification technology that significantly reduced the toxicity of antibody preparations4. One can speculate that these technological advances might have allowed serum therapy to remain competitive with the new antimicrobial agents had the heyday of serum therapy been a decade earlier or had antimicrobial chemotherapy been developed just a few years later. In fact, there were indications at the time that the combination of serum therapy and antimicrobial therapy was effective3,5. Unfortunately, the toxicity and complexity of serum therapy was such that the benefits of combined therapy were not sufficient to justify its continued use to treat diseases for which antimicrobial therapy was available. However, in the second half of the twentieth century, the inability to treat certain viral diseases drove efforts to develop antibody preparations derived from immunized human donors for the prophylaxis and treatment of rabies, hepatitis A and B, varicella–zoster virus and pneumonia caused by respiratory syncytial virus (RSV).
A technological revolution
In 1975, the discovery of a method to produce MONOCLONAL ANTIBODIES (mAbs) by immortalizing B cells, which developed into HYBRIDOMA TECHNOLOGY, revolutionized antibody therapeutics6. For the first time, it was possible to produce large quantities of an immunoglobulin of a defined specificity and a single ISOTYPE in vitro. This innovation allowed the generation of homogeneous antibodies in almost unlimited quantities, eliminating the need for large animal or human donors. Together with the development of new methods for cloning, and recombinant DNA technology, the development of hybridoma technology was supplemented by techniques to genetically modify antibody molecules, including the synthesis of mouse–human chimeric and HUMANIZED ANTIBODIES. In the last decades of the twentieth century, there were several new technological advances, including the immortalization of human peripheral B cells, direct cloning of variable genes into phage expression libraries7 and the creation of transgenic mice that produce only human antibodies8. Although each of these technologies has inherent limitations, together they provide the means to produce mAbs against almost any antigen.
Hybridoma technology was rapidly exploited for clinical use, and a mAb to CD3 was introduced into clinical practice in the mid-1980s to prevent organ rejection. After the introduction of mAbs, there was hope for the rapid development of many therapeutic applications, especially in the field of oncology. However, the pace of discovery and development was slowed by the complexity of successfully targeting tumours and the well-publicized failures of two anti-endotoxin antibodies9,10; this failure might have been due to an insufficient understanding of microbial pathogenesis and the mechanisms of antibody action, areas in which studies using mAbs have recently provided a wealth of new information. Nonetheless, by the late 1990s many mAbs were in advanced clinical development. Today, more than twelve mAbs are licensed for therapeutic use, including two that are labelled with radionuclides to deliver tumoricidal radiation (Table 1). However, it is striking that only one mAb, palivizumab, has been licensed for an infectious disease (RSV infection), despite the fact that antibodies have proven to be good antimicrobial agents.
Infectious diseases provide a wealth of targets
Passive antibody therapy has been used against many microorganisms that are responsible for human disease, including representatives of the viral, bacterial, fungal and parasitic microbial groups (Table 2). In contrast to the use of mAb therapy to treat malignancies, which depends on discriminating between self-antigens that are expressed by normal and tumour cells, passive antibody therapy for infectious diseases is aided by the large antigenic differences between the microorganism and the host. Historically, antibodies have been effective when directed against either microbial antigens or their products, such as toxins. In some microbial diseases, antibodies provide a component of the humoral immune response to natural infection, whereas host defence against other microorganisms relies primarily on cell-mediated immune mechanisms. Nonetheless, there is now considerable evidence to indicate that it is possible to generate mAbs that are protective against microorganisms — such as Mycobacterium tuberculosis 11,12, Listeria monocytogenes 13, Leishmania mexicana14 and Histoplasma capsulatum15 — for which the activation of humoral immunity is not important for the development of resistance to natural infection. Even intracellular microorganisms can be susceptible to antibodies16.
In the pre-antibiotic era, antibody therapies were developed against a wide variety of infectious diseases because there were no alternative therapies3. Today, although antimicrobial drugs are available, microbial resistance, the emergence of microorganisms that are not susceptible to existing drugs and the fact that antimicrobial drugs are often ineffective in immunocompromised hosts can compromise the efficacy of these drugs. The latter is exemplified by the lack of success in treating the infectious diseases that arise in the setting of severe immunosuppression, such as bone marrow and organ transplantation, and AIDS. Interestingly, on the basis of evidence that mechanisms of antibody efficacy can include the regulation or induction of cellular immune responses17, antibody therapy with or without additional immunomodulators might have promise for treating infectious diseases in immunocompromised hosts. So, antibodies represent a new, although historically validated, approach to the development of therapies against microorganisms that cause disease in individuals with impaired immunity and/or for which there are no available drugs.
Antibody-based therapies: pros and cons
The advantages and disadvantages of antibody-based therapies are often compared with those of conventional antimicrobial drugs. However, immunoglobulins are sufficiently different in their physical characteristics and modes of action to be regarded as a distinct therapeutic class.
Advantages. Antibody-based therapies that use human or humanized antibodies have low toxicities and high specificities. The high specificity of antibodies is both an advantage and a disadvantage. The advantage of high specificity is that antibody-based therapies target only the microorganism that causes disease and, therefore, should not alter the host flora or select for resistance among non-targeted microorganisms. However, high specificity also means that more than one antibody preparation might be required to target microorganisms with high antigenic variation. In fact, in the case of serum therapy, numerous type-specific sera were developed for the treatment of pneumococcal pneumonia because only type-specific sera were effective against pneumococci2,5. In theory, a disadvantage of high specificity is the emergence of variants that lack the determinant that the antibody recognizes, such as viral escape mutants. The use of cocktails of antibodies that are specific for several antigens could obviate this concern. However, this approach would also have the drawback of increasing the cost of production and the complexity of regulatory issues involving efficacy and safety.
The high specificity of antibody molecules is complemented by their versatility, which allows an antibody that binds a single determinant to mediate various different biological effects (Fig. 1). As natural products of the immune system, antibodies can interact with other immune components. Some mechanisms of antibody action, such as toxin and virus neutralization and complement activation, and direct antimicrobial functions, such as the generation of oxidants, are independent of other host immune components. By contrast, antibody-dependent cellular cytotoxicity and opsonization are dependent on cellular and other host mediators. In recent years, the recognition that antibodies can be immunomodulators, bridging the innate, acquired, cellular and humoral immune responses, has revealed new mechanisms of antibody-mediated immunity and has provided a better understanding of how and why antibodies are effective against microorganisms for which they do not mediate a direct biological effect17. In fact, there is evidence that B cells and antibodies can protect against certain infectious diseases by reducing host damage resulting from the inflammatory response17. This could partly explain the efficacy of intravenous immunoglobulin, which is used to treat certain inflammatory conditions. Evidence that specific immunoglobulin G (IgG)–Fc receptor interactions can inhibit the inflammatory response18 indicates that antibody therapy could be effective against certain infectious diseases by reducing the damage that results from the host inflammatory response.

Toxin and virus neutralization, complement activation and direct antimicrobial functions such as the generation of oxidants are independent of other components of the host immune system, whereas antibody-dependent cellular cytotoxicity and opsonization depend on other host cells and mediators.
In addition, the use of antibodies as therapeutic reagents has the advantage that there are several isotypes, which can function therapeutically in either an intact form or as fragments. In the intact molecule, the variable region (Fab) binds antigen, whereas the constant region (Fc) determines the biological properties of the immunoglobulin molecule, such as serum half-life, interaction with cellular Fc receptors and the ability to activate complement. When the binding of antibody to a target antigen is sufficient to mediate an effect, which can occur when an antibody is functioning as an antitoxin or antiviral agent, an antibody fragment can be sufficient for efficacy. However, when antibody efficacy is dependent on immunomodulation or interaction with effector cells to mediate phagocytosis, complement activation or antibody-dependent cellular cytotoxicity, an intact immunoglobulin molecule is required for efficacy. Whether a Fab fragment or intact antibody is suitable as a therapeutic agent also depends on the microorganism that is being targeted and the immunological status of the host. As biological effects that depend on the Fc receptor could require intact host immunological function, antibodies that have direct antimicrobial effects or that mediate beneficial effects by the binding of Fab alone might be more useful in immunocompromised hosts. Antibodies with direct antimicrobial properties have recently been described against several microorganisms, including Borrelia spp.19, Candida albicans20,21 and Cryptococcus neoformans22.
An important potential advantage of antibody therapies is that they can be synergistic or additive when combined with conventional antimicrobial chemotherapy against bacterial and viral diseases (reviewed in Refs 3,5). In addition, recent studies suggest that combinations of antibodies and drugs are more effective against fungal infections than when either therapy is used alone15,20,23. Consequently, antibody-based therapies could easily be incorporated into existing treatment protocols; however, demonstrating the advantages of combination therapy in rigorous clinical trials can be logistically and practically difficult, and the use of combination therapy would be more expensive.
In addition to their advantages as therapeutic agents, antibodies have had a central role in vaccine development. Historically, vaccine development for numerous infectious diseases was fuelled by antibody-based therapies and research into antibody-mediated immunity. Antibody therapy can be protective against an infectious disease, which suggests that a vaccine that elicits similar antibodies could be protective against the relevant pathogen. For example, successful passive antibody therapy against pneumococcal pneumonia and diphtheria preceded the development of vaccines against these diseases. More recently, the generation of protective mAbs against C. neoformans and C. albicans identified polysaccharide antigens that were then used to design effective conjugate vaccines24,25. Protective antibodies to microbial polysaccharides can be used to identify PEPTIDE MIMOTOPES that elicit protective antibody responses26, and antibodies that elicit protective ANTI-IDIOTYPIC RESPONSES can be used directly as immunogens27. Efforts to develop antibody-based therapies can, therefore, promote vaccine development.
Disadvantages. As antibodies are natural products they must be produced in cell lines or other live expression systems. This raises the theoretical concern that there could be contamination of antibody preparations by infectious agents such as prions or viruses. Although tight regulation and regulatory vigilance and surveillance can reduce this concern, the need for ongoing monitoring and testing for contamination contributes to the high cost of developing and administering antibody therapies. In addition, antibody-based therapies require considerable logistical support. As antibodies are proteins, they cannot be given orally, except for those used to treat certain types of mucosal infectious diseases, such as Cryptosporidium parvum-associated diarrhoea, and therefore, systemic administration is required.
Owing to their high specificity, antibodies have activity against the microorganism to which they bind. Antibody therapy therefore requires knowledge of the causative microbial agent, which in turn requires rapid microbiological diagnosis. Additionally, because antibody efficacy is highest when given early in the course of infection, rapid diagnosis is essential for the success of antibody therapy. For example, the efficacy of serum therapy for pneumococcal pneumonia is markedly reduced after the first three days of symptoms2,5. In the first decades of the antibiotic era, the lack of innovation in microbiological diagnosis was tolerated owing to the availability of broad-spectrum antimicrobial agents. However, the need for rapid diagnostic techniques has assumed greater urgency with the emergence of fungi in immunocompromised hosts and nosocomial infections, resistant bacteria and previously unknown viral diseases for which the available antimicrobial armamentarium is inadequate. At the same time, the development of PCR and other rapid diagnostic techniques has provided new options that could support antibody-based therapies. Importantly, the efficacy of anti-infective antibody-based therapies can be assessed relatively easily, as there are well-defined clinical end points that can be used to determine whether therapy has been successful.
A peculiar characteristic of antibody-based therapies is that their efficacy diminishes rapidly as the duration of infection increases. Antibody reagents with therapeutic potential are often evaluated by administration to naive hosts before infection. Although this approach to select antibody preparations was well established during the development of serum therapy, it is noteworthy that serum was effective in humans even when administered several days after the onset of symptoms, despite having little or no therapeutic efficacy in mouse models2. However, even in humans, the efficacy of therapeutic antibodies diminished rapidly after the onset of symptoms2,5. The mechanism responsible for this is not well understood, but might reflect a rapid increase in the microbial burden in the animal models used, which are usually selected on the basis of their marked susceptibility to the agent in question28. A loss of efficacy with increased duration of infection or disease could limit the application of antibody-based strategies to prophylaxis and/or conditions where an early diagnosis is possible.
One of the greatest advantages of antibody therapy, namely high specificity, means that the potential market for a reagent is likely to be small — as the size of the market is proportional to the number of affected individuals. Given the large expenses that are associated with drug discovery and development, it is likely that development of antibody-based therapies will focus mainly on infectious diseases that are sufficiently common to provide financial rewards. In practice, this means that, although antibody therapy could be effective, such therapy is unlikely to be developed for relatively rare infectious diseases because the costs are considered prohibitive.
The high costs of production, storage and administration of antibodies are disadvantages of antibody-based therapies. For example, in the United Kingdom, the cost of palivizumab therapy for the prevention of RSV disease is estimated at UK £2,500, which affects the cost/benefit ratio29. In the field of infectious diseases, discussion of the costs of antibody therapy is often affected by the fact that they are compared with relatively cheap antimicrobial drugs. However, a true comparison of the costs must include the fact that nonspecific drug therapy selects for resistant organisms and predisposes individuals to super-infection, which in turn incurs additional costs for prolonged hospitalization, therapy and patient follow-up. As the high specificity of antibody therapies makes it unlikely that they will select for resistance in non-targeted microorganisms, they should not markedly impact on the resident microflora. So, high costs could be offset by lower levels of resistance and fewer nosocomial infections. The cost of antibody development notwithstanding, it is notable that the time to development of a potential antibody therapy, provided an appropriate antigen is available, is considerably shorter than that needed to develop a vaccine30.
New directions in anti-infective antibody therapy
A great advantage of antibody-based anti-infective therapies is their inherent flexibility — in addition to the availability of nine natural isotypes with different half-lives, the ability to activate complement and the ability to interact with different Fc receptors, it is also possible to modify immunoglobulins to have new antimicrobial capabilities. One strategy is to target infected host cells by linking cellular toxins to antibodies against microbial antigens that are expressed on the surfaces of host cells. Along these lines, antibodies to murine cytomegalovirus that are linked to a deglycosylated ricin A chain have been shown to target cytomegalovirus-infected cells31. Similarly, viral envelope proteins that are expressed on the surface of HIV-infected, virus-synthesizing cells can be targeted with antibodies linked to the ricin A chain32 or Pseudomonas spp. exotoxin A33. Immunotoxins are particularly attractive for the therapy of infectious diseases in which the pathogen is intracellular and uses this environment to reproduce. However, they are not necessarily active against extracellular microorganisms as the antibodies must be internalized relatively rapidly and the covalent attachment of a toxin to the immunoglobulin molecule has the potential to elicit an antibody response in treated hosts, which would limit repeated use.
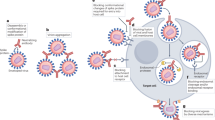
Another strategy for antibody targeting is to link radionuclides to specific antibodies such that the immunoglobulin molecule targets and delivers microbicidal radiation to the microorganism (Fig. 2). This approach is known as radioimmunotherapy and has been successfully used in cancer treatment34. A proof-of-principle for the use of this strategy to treat an infection was established by demonstrating that mAbs to the C. neoformans capsular glucuronoxylomannan labelled with 213Bi or 188Re could be used to treat murine cryptococcosis35. The administration of radiolabelled mAbs prolonged survival and reduced the organ fungal burden in this model, whereas an irrelevant radiolabelled mAb or unlabelled specific mAb had no effect35. Apart from a transient drop in serum platelet counts, no measurable toxicity was detected in mice that were treated with radiolabelled mAbs36. The efficacy of radioimmunotherapy against murine pneumococcal infections has also been established, showing the applicability of this approach to a bacterium with a fast doubling time37. Analysis of the susceptibility of fungal cells to radiolabelled mAbs that bind to surface antigens in vitro, using both C. neoformans and H. capsulatum, showed markedly greater susceptibility to killing by antibody-delivered particulate (β- and α-particles) radiation than to external γ-radiation38. Although this is not well understood, it is possible that particulate radiation that is delivered in close proximity to microbial cells has greater killing power than γ-photons. Alternatively, antibody effects, such as the recently described ability to generate oxygen-related oxidants39, might synergistically increase the killing power of locally emitted radiation.

Biological distribution of 111Indium-labelled whole 18B7 monoclonal antibody (mAb) (a), F(ab)2 (b) and Fab (c) 24 h post-injection. AJ/Cr mice were infected intravenously with 105 C. neoformans cells 24 h before injection with radiolabelled mAbs. The radiation localizes to the lungs, which are heavily infected with C. neoformans. Activity in the lungs is seen for all three carriers. For methodology, see Ref. 35.
Attaching a radionuclide to an immunoglobulin converts the antibody into a microbicidal molecule, even if the antibody is not protective independently. Methods have been developed for the stable attachment of radionuclides to immunoglobulins such that in vivo hydrolysis is not a major problem40. Consequently, radiolabelling has the potential to enhance the power of passive antibody therapy by conferring the power to kill the targeted microorganism on any specific antibody. Therefore, radioimmunotherapy for infectious diseases should theoretically be effective in immunocompromised hosts and might also be effective against chronically infected cells that express microbial antigens on their surface (Fig. 3), which could be a powerful means to eliminate latent microorganisms that might be harboured by cells and that avoid host defence mechanisms. As particulate radiation also kills infected cells through a 'crossfire' effect (radiation emanating from a cell hits an adjacent or a distant cell), not every microbial cell in the infected area needs to be bound by a labelled antibody molecule to be killed. In contrast to immunotoxins, radiolabelled human antibodies do not need to be internalized, are unlikely to elicit significant immune responses that would limit subsequent use and the unlikely separation of the chelator–radiometal label would not produce a toxic product. However, the application of this technology to infectious diseases is in its infancy, and the extent of its usefulness and potential toxicity remain to be defined.

a | For Cryptococcus neoformans infection, a radiolabelled antibody binds to both capsular and shed polysaccharide, and results in localized fungicidal radiation. Macrophages infected with replicating intracellular fungi can also be killed by a crossfire effect. b | The proposed mechanism by which radiolabelled antibody is effective against an intracellular pathogen such as a virus that expresses microbial antigens on the cell surface after cellular infection.
Another approach to confer additional biological properties to an immunoglobulin is to create a bispecific antibody in which one arm of the Fab fragment recognizes a microbial epitope, and the other recognizes a host immune component, which is often a relevant receptor. Numerous reports of bispecific antibodies with effective antimicrobial action have been published. For example, bispecific antibodies consisting of a pathogen-binding Fab and a complement-receptor-binding Fab have been shown to be effective in promoting the clearance of bacteriophage41 and Pseudomonas aeruginosa42.
Unsolved problems in antibody therapy
After more than 110 years of use in humans, there are still many unsolved problems that limit the widespread application of antibody-based therapies. The development of therapeutic antibodies remains, for the most part, an empirical science. For many infectious diseases, the current understanding of microbial pathogenesis is insufficient to predict the microbial antigens against which therapeutic antibodies should be raised. In addition, current immunological knowledge is insufficient to predict which antibodies are effective against specific microorganisms, particularly in immunocompromised hosts. The relationship between antibody isotype and efficacy is unclear for many microorganisms, and generalizations are difficult. For example, murine IgG3 mAbs have been shown to be protective against Streptococcus pneumoniae 43 and M. tuberculosis11, but are relatively ineffective against C. neoformans44. Human IgM has been found to be highly protective against experimental C. neoformans and S. pneumoniae infections45,46, although in vaccine development the presence of specific serum IgG is used as a surrogate for immunity. However, we do not know if insights into isotype function that have been gained from animal studies can be applied to humans. It is clear that antibody binding to certain epitopes on a given antigen can result in protection, whereas binding to other epitopes is ineffective, but we cannot currently predict which epitopes elicit protective antibodies. Another difficult problem is estimating the amount of antibody to use for therapy. Administration of too little antibody can produce no therapeutic effect, whereas administration of too much antibody can produce disconcerting PROZONE-like effects, whereby antibody efficacy is lost and antibody administration can be detrimental to the host47,48,49. This seems to be a result of an excess of antibody interfering with the host microbicidal mechanisms and changes in cytokine expression47,48.
The near and far horizons
Passive antibody administration is currently used to treat and prevent diseases caused by hepatitis B virus, rabies virus, RSV, Clostridium tetani , Clostridium botulinum , vaccinia virus, echovirus and enterovirus. Antibody therapies against HIV, rotavirus, bacterial sepsis, cytomegalovirus, C. neoformans and C. albicans are in clinical development. Furthermore, there are many monoclonal antibodies against infectious diseases in advanced preclinical development, and one can confidently expect that many more antibodies will be developed for clinical use. In this regard, the realization that passive antibody therapy can provide immediate immunity against biological weapons has spurred the search for, and development of, protective antibodies against many selected agents including Bacillus anthracis toxins, Ebola virus and the C. botulinum toxins. Consequently, current efforts to develop countermeasures to biological weapons could be an important stimulus for the development of antibody therapies for infectious diseases. However, the combination of manufacturing and economic hurdles, the need for a cold chain, intravenous administration, rapid diagnosis and pathogen specificity, and the continuing availability of antimicrobial drugs indicates that the development of anti-infective antibody therapies will progress slowly and will almost certainly lag behind the application of antibody therapies to non-infectious diseases for which no therapy is available and where the potential market is larger. Nevertheless, we predict that, in the future, the use of this proven antimicrobial strategy will increase, and anticipate a time when antibody therapy, antimicrobial chemotherapy and possibly other forms of immunotherapy are used in combination to treat a wide variety of infectious diseases.
References
Behring, E. A. & Kitasato, S. Ueber das zustandekommen der diptherie-immunität und der tetanus-immunität bei thieren. Deutch. Med. Woch. 49, 1113–1114 (1890).
Casadevall, A. & Scharff, M. D. Serum therapy revisited: animal models of infection and the development of passive antibody therapy. Antimicrob. Agents Chemother. 38, 1695–1702 (1994).
Casadevall, A. & Scharff, M. D. Return to the past: the case for antibody-based therapies in infectious diseases. Clin. Infect. Dis. 21, 150–161 (1995).
Good, R. A. & Lorenz, E. Historic aspects of intravenous immunoglobulin therapy. Cancer 68, 1415–1421 (1991).
Buchwald, U. K. & Pirofski, L. Immune therapy for infectious diseases at the dawn of the twenty-first century: the past, present and future role of antibody therapy, therapeutic vaccination and biological response modifiers. Curr. Pharm. Des. 9, 945–968 (2003).
Kohler, G. & Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495–497 (1975). The first description of hybridoma technology.
Kang, A. S., Burton, D. R. & Lerner, R. A. Combinatorial immunoglobulin libraries in phage. Methods Companion Methods Enzymol. 2, 111–118 (1991).
Green, L. L. et al. Antigen-specific human monoclonal antibodies from mice engineered with human Ig heavy and light chain YACs. Nature Genet. 7, 13–21 (1994). Demonstrated that transgenic mice express B cells producing human antibodies that can be used to generate hybridomas and monoclonal antibodies.
Angus, D. C. et al. E5 murine monoclonal antiendotoxin antibody in Gram-negative sepsis: a randomized controlled trial. E5 Study Investigators. JAMA 283, 1723–1730 (2000).
Derkx, B., Wittes, J. & McCloskey, R. Randomized, placebo-controlled trial of HA-1A, a human monoclonal antibody to endotoxin, in children with meningococcal septic shock. European Pediatric Meningococcal Septic Shock Trial Study Group. Clin. Infect. Dis 28, 770–777 (1999).
Teitelbaum, R. et al. A monoclonal antibody recognizing a surface antigen of Mycobacterium tuberculosis enhances host survival. Proc. Natl Acad. Sci. USA 95, 15688–15693 (1998).
Pethe, K. et al. The heparin-binding haemagglutinin of M. tuberculosis is required for extrapulmonary dissemination. Nature 412, 190–194 (2001).
Edelson, B. T., Cossart, P. & Unanue, E. R. Cutting edge: paradigm revisited: antibody provides resistance to Listeria infection. J. Immunol. 163, 4087–4090 (1999).
Anderson, S., David, J. R. & McMahon-Pratt, D. In vivo protection against Leishmania mexicana mediated by monoclonal antibodies. J. Immunol. 131, 1616–1618 (1983).
Nosanchuk, J. D., Steenbergen, J. N., Shi, L., Deepe, G. S. Jr & Casadevall, A. Antibodies to a cell surface histone-like protein protect against Histoplasma capsulatum. J. Clin. Invest. 112, 1164–1175 (2003).
Casadevall, A. Antibody-mediated immunity against intracellular pathogens: two-dimensional thinking comes full circle. Infect. Immun. 71, 4225–4228 (2003).
Casadevall, A. & Pirofski, L. A. Antibody-mediated regulation of cellular immunity and the inflammatory response. Trends Immunol. 24, 474–478 (2003). Proposes that an important function of antibody responses is to regulate cellular immunity and the inflammatory response.
Samulsson, A., Towers, T. L. & Ravetch, J. V. Anti-inflammatory activity of IVIG mediated through the inhibitory Fc receptor. Science 291, 484–486 (2001).
Connolly, S. E. & Benach, J. L. Cutting edge: the spirochetemia of murine relapsing fever is cleared by complement-independent bactericidal antibodies. J. Immunol. 167, 3029–3032 (2001). Reports that certain antibodies can kill bacteria directly by altering their surface structures.
Matthews, R. C. et al. Preclinical assessment of the efficacy of mycograb, a human recombinant antibody against fungal HSP90. Antimicrob. Agents Chemother. 47, 2208–2216 (2003).
Moragues, M. D. et al. A monoclonal antibody directed against a Candida albicans cell wall mannoprotein exerts three anti-C. albicans activities. Infect. Immun. 71, 5273–5279 (2003).
Rodrigues, M. L. et al. Human antibodies against a purified glucosylceramide from Cryptococcus neoformans inhibit cell budding and fungal growth. Infect. Immun. 68, 7049–7060 (2000).
Mukherjee, J., Zuckier, L., Scharff, M. D. & Casadevall, A. Therapeutic efficacy of monoclonal antibodies to Cryptococcus neoformans glucuronoxylomannan alone and in combination with amphotericin B. Antimicrob. Agents Chemother. 38, 580–587 (1994).
Devi, S. J. N. Preclinical efficacy of a glucuronoxylomannan–tetanus toxoid conjugate vaccine of Cryptococcus neoformans in a murine model. Vaccine 14, 841–842 (1996).
Han, Y., Ulrich, M. A. & Cutler, J. E. Candida albicans mannan extract–protein conjugates induce a protective immune response against experimental candidiasis. J. Infect. Dis. 179, 1477–1484 (1999).
Pirofski, L. Polysaccharides, mimotopes and vaccines for fungal and encapsulated pathogens. Trends Microbiol. 9, 445–451 (2001).
Polonelli, L. et al. Idiotypic intravaginal vaccination to protect against candidal vaginitis by secretory, yeast killer toxin-like anti-idiotypic antibodies. J. Immunol. 152, 3175–3181 (1994).
Robbins, J. B., Schneerson, R. & Szu, S. C. Perspective: hypothesis: serum IgG antibody is sufficient to confer protection against infectious diseases by inactivating the inoculum. J. Infect. Dis. 171, 1387–1398 (1995). Proposes the hypothesis that the generation of specific antibody responses is sufficient for vaccine efficacy.
Handforth, J., Sharland, M. & Friedland, J. S. Prevention of respiratory syncytial virus infection in infants. BMJ 328, 1026–1027 (2004).
Hassani, M., Patel, M. C. & Pirofski, L. A. Vaccines for the prevention of diseases caused by potential bioweapons. Clin. Immunol. 111, 1–15 (2004).
Barnett, B. B., Smee, D. F., Malek, S. M. & Sidwell, R. W. Selective cytotoxicity of ricin A chain immunotoxins towards murine cytomegalovirus-infected cells. Antimicrob. Agents Chemother. 40, 470–472 (1996).
Till, M. A. et al. Human immunodeficiency virus-infected T cells and monocytes are killed by monoclonal human anti-gp41 antibodies coupled to ricin A chain. Proc. Natl Acad. Sci. USA 86, 1987–1991 (1989).
McHugh, L. et al. Increased affinity and stability of an anti-HIV-1 envelope immunotoxin by structure-based mutagenesis. J. Biol. Chem. 277, 34383–34390 (2002).
Waldmann, T. A. Immunotherapy: past, present and future. Nature Med. 9, 269–277 (2003). Excellent review of the development of this promising technology.
Dadachova, E., Nakouzi, A., Bryan, R. A. & Casadevall, A. Ionizing radiation delivered by specific antibody is therapeutic against a fungal infection. Proc. Natl Acad. Sci. USA 100, 10942–10947 (2003). Showed that labelling antibodies with radionuclides converts them into microbicidal molecules that can be used in therapy of fungal infections.
Dadachova, E. et al. Evaluation of acute hematologic and long-term pulmonary toxicities of radioimmunotherapy of Cryptococcus neoformans infection in murine models. Antimicrob. Agents Chemother. 48, 1004–1006 (2004).
Dadachova, E. et al. Feasibility of radioimmunotherapy of experimental pneumococcal infection. Antimicrob. Agents Chemother. 48, 1624–1629 (2004).
Dadachova, E. et al. Susceptibility of the human pathogenic fungi Cryptococcus neoformans and Histoplasma capsulatum to γ-radiation versus radioimmunotherapy with α- and β-emitting radioisotopes. J. Nucl. Med. 45, 313–320 (2004).
Wentworth, P. Jr et al. Evidence for antibody-catalyzed ozone formation in bacterial killing and inflammation. Science 298, 2195–2199 (2002). Showed that antibodies can catalyse the formation of oxygen-derived oxidants, which might contribute to their antimicrobial properties.
Gansow, O. A. Newer approaches to the radiolabeling of monoclonal antibodies by use of metal chelates. Int. J. Rad. Appl. Instrum. B 18, 369–381 (1991).
Taylor, R. P. et al. Bispecific monoclonal antibody complexes facilitate erythrocyte binding and liver clearance of a prototype particulate pathogen in a monkey model. J. Immunol. 159, 4035–4044 (1997).
Lindorfer, M. A. et al. Targeting of Pseudomonas aeruginosa in the bloodstream with bispecific monoclonal antibodies. J. Immunol. 167, 2240–2249 (2001).
Briles, D. E., Claffin, J. L., Schroer, K. & Forman, C. Mouse IgG3 antibodies are highly protective against infection with Streptococcus pneumoniae. Nature 294, 88–89 (1981).
Yuan, R., Casadevall, A., Spira, G. & Scharff, M. D. Isotype switching from IgG3 to IgG1 converts a non-protective murine antibody to C. neoformans into a protective antibody. J. Immunol. 154, 1810–1816 (1995).
Chang, Q., Zhong, Z., Lees, A., Pekna, M. & Pirofski, L. Structure–function relationships for human antibodies to pneumococcal capsular polysaccharide from transgenic mice with human immunoglobulin loci. Infect. Immun. 70, 4977–4986 (2002).
Fleuridor, R., Zhong, Z. & Pirofski, L. A human IgM monoclonal antibody prolongs survival of mice with lethal cryptococcosis. J. Infect. Dis. 178, 1213–1216 (1998).
Taborda, C. P. & Casadevall, A. Immunoglobulin M efficacy against Cryptococcus neoformans: mechanism, dose dependence and prozone-like effects in passive protection experiments. J. Immunol. 66, 2100–2107 (2001).
Taborda, C. P., Rivera, J., Zaragoza, O. & Casadevall, A. More is not necessarily better: 'prozone-like' effects in passive immunization with immunoglobulin G. J. Immunol. 140, 3621–3630 (2003).
Maitta, R. et al. Protective and non-protective human IgM monoclonal antibodies to Cryptococcus neoformans glucuronoxylomannan manifest different specificity and gene usage. Infect. Immun. (in the press).
Lucchesi, P. F. & Gildersleeve, N. The treatment of anthrax. JAMA 14, 1506–1508 (1941).
McGuinness, A. C., Armstrong, J. G. & Felton, H. M. Hyperimmune whooping cough serum. J. Pediatr. 24, 249–258 (1944).
Blake, P. A., Feldman, R. A., Buchanan, T. M., Brooks, G. F. & Bennett, J. V. Serologic therapy of tetanus in the United States. JAMA 235, 42–44 (1976).
Tacket, C. O., Shandera, W. X., Mann, J. M., Hargrett, N. T. & Blake, P. A. Equine antitoxin use and other factors that predict outcome in type A foodborne botulism. Am. J. Med. 76, 794–798 (1984).
Gordon, M. A. & Casadevall, A. Serum therapy of cryptococcal meningitis. Clin. Infect. Dis. 21, 1477–1479 (1995).
Greenberg, P. D. & Cello, J. P. Treatment of severe diarrhea caused by Cryptosporidium parvum with oral bovine immunoglobulin concentrate in patients with AIDS. J. AIDS 13, 348–354 (1996).
Bodensteiner, J. B., Morris, H. H., Howell, J. T. & Schochet, S. S. Chronic ECHO type 5 virus meningoencephalitis in X-linked hypo-γ-globulinemia: treatment with immune plasma. Neurology 29, 815–819 (1979).
Blake, F. G. & Trask, J. D. Observations on therapeutic value of scarlatinal antitoxin. NY State J. Med. 25, 1093–1099 (1925).
McGory, R. W. et al. Improved outcome of orthotopic liver transplantation for chronic hepatitis B cirrhosis with aggressive passive immunization. Transplantation 61, 1358–1364 (1996).
Gallagher, J. R. Use of convalescent measles serum to control measles in a preparatory school. Am. J. Public. Health 25, 595–598 (1935).
Glatman-Freedman, A. & Casadevall, A. Serum therapy for tuberculosis revisited: a reappraisal of the role of antibody-mediated immunity against Mycobacterium tuberculosis. Clin. Microbiol. Rev. 11, 514–532 (1998).
Flexner, S. The results of the serum treatment in thirteen hundred cases of epidemic meningitis. J. Exp. Med. 17, 553 (1913).
Frickhofen, N. et al. Persistent B19 parvovirus infection in patients infected with human immunodeficiency virus type 1 (HIV-1): a treatable cause of anemia in AIDS. Ann. Intern. Med. 113, 926–933 (1990).
Hattwick, M. A., Corey, L. & Creech, W. B. Clinical use of human globulin immune to rabies virus. J. Infect. Dis. 133, A266–A272 (1976).
roothuis, J. R. et al. Prophylactic administration of respiratory syncytial virus immune globulin to high-risk infants and young children. N. Engl. J. Med. 329, 1524–1530 (1993).
Weech, A. A. The prophylaxis of varicella with convalescent's serum. JAMA 82, 1245–1246 (1924).
Kempe, C. H. et al. The use of vaccinia hyperimmune γ-globulin in the prophylaxis of smallpox. Bull. World Health Organ. 25, 41–48 (1961).
Acknowledgements
The authors would like to thank R. Bryan and T. Moadel for obtaining images of C. neoformans-infected animals.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Related links
DATABASES
Entrez
Infectious Disease Information
FURTHER INFORMATION
Arturo Casadevall's laboratory
Liise-anne Pirofski's laboratory
Ekaterina Dadachova's laboratory
Encyclopedia of Life Sciences
Glossary
- HYPERSENSITIVITY REACTION
-
An inappropriate reaction to an allergen that can be immediate (types I, II and III) or delayed (type IV). Different hypersensitivity reactions involve different antibody classes and effector cells.
- ANTIGEN–ANTIBODY COMPLEX DISEASE
-
An immune complex disease that is caused by the administration of foreign serum or serum proteins, and is characterized by fever, lymphadenopathy and skin welts.
- MONOCLONAL ANTIBODY
-
A highly specific, purified antibody that is derived from only one clone of cells and which recognizes a single antigen.
- HYBRIDOMA TECHNOLOGY
-
The technology that led to the production of monoclonal antibodies and which involved the generation of an antibody-secreting B-cell line by fusing splenic-derived B cells with an immortal myeloid cell line.
- ISOTYPE
-
The class — or type — of antibody as determined by structural features of the heavy chain constant regions. In humans there are five main isotypes: IgA (2 subclasses), IgD, IgE, IgG (4 subclasses) and IgM.
- HUMANIZED ANTIBODIES
-
Murine monoclonal antibodies are recognized as foreign by the human immune system. To avoid this, humanized antibodies can be constructed in which rodent hypervariable regions (antigen-binding site) are grafted into a human antibody framework.
- CD3
-
A polypeptide complex that is associated with the T-cell receptor and is involved in signal transduction. A CD3-specific monoclonal antibody blocks T-cell activation.
- PEPTIDE MIMOTOPES
-
Peptides that mimic natural epitopes.
- ANTI-IDIOTYPIC RESPONSES
-
The antigen-binding site of an antibody is also known as the idiotype. An antibody response to this region can generate antibodies that bear the image of the original immunogen or antigen.
- PROZONE EFFECT
-
A decrease in an antigen–antibody reaction that occurs as the concentration of antibody or antigen increases.
Rights and permissions
About this article
Cite this article
Casadevall, A., Dadachova, E. & Pirofski, La. Passive antibody therapy for infectious diseases. Nat Rev Microbiol 2, 695–703 (2004). https://doi.org/10.1038/nrmicro974
Issue Date:
DOI: https://doi.org/10.1038/nrmicro974
This article is cited by
-
Nanotherapeutics with immunoregulatory functions for the treatment of bacterial infection
Biomaterials Research (2023)
-
Efficacy of COVID-HIGIV in animal models of SARS-CoV-2 infection
Scientific Reports (2022)
-
COVID-19 and Solid Organ Transplantation: Role of Anti-SARS-CoV-2 Monoclonal Antibodies
Current Transplantation Reports (2022)
-
COVID-19 one year into the pandemic: from genetics and genomics to therapy, vaccination, and policy
Human Genomics (2021)
-
A new emerging pandemic of severe fever with thrombocytopenia syndrome (SFTS)
VirusDisease (2021)
