
Key Points
-
Base excision repair (BER) is a highly conserved process that primarily repairs oxidative DNA damage, and human-adapted bacterial pathogens have evolved specialized mechanisms for BER.
-
Mycobacterium tuberculosis displays striking redundancy in the enzymes that prevent incorporation of oxidized guanines into the DNA backbone and excise nucleotides mispaired with this damaged base.
-
By contrast, Helicobacter pylori, which inhabits the gastric mucosa, has a minimal complement of BER enzymes. A network of enzymes recognize and repair DNA damage by BER in Neisseria meningitidis.
-
Expression of the BER enzymes is constitutive in Neisseria meningitidis, which might reflect the high selective pressure of oxidative stress in its habitat in the aerobic upper airways.
-
Further work is required to understand mechanisms of BER in other pathogens and related human commensal species, which should provide insights into whether specialization in BER contributes to colonization and/or human disease.
Abstract
During colonization and disease, bacterial pathogens must survive the onslaught of the host immune system. A key component of the innate immune response is the generation of reactive oxygen and nitrogen species by phagocytic cells, which target and disrupt pathogen molecules, particularly DNA, and the base excision repair (BER) pathway is the most important mechanism for the repair of such oxidative DNA damage. In this Review, we discuss how the human-specific pathogens Mycobacterium tuberculosis, Helicobacter pylori and Neisseria meningitidis have evolved specialized mechanisms of DNA repair, particularly their BER pathways, compared with model organisms such as Escherichia coli. This specialization in DNA repair is likely to reflect the distinct niches occupied by these important human pathogens in the host.
Similar content being viewed by others


Main
All living organisms have evolved mechanisms to repair DNA damage to prevent genome instability and limit the accumulation of deleterious mutations. Genomes are under continual threat from endogenous and exogenous factors that result in aberrant base pairing, DNA crosslinks, modified bases, apurinic or apyrimidinic (AP) sites (also known as abasic sites), single-strand breaks (SSBs), double-strand breaks (DSBs) and the insertion of bulky adducts (Box 1). These lesions reduce viability by impairing the transcription of essential genes and the stalling of DNA replication; furthermore, the incorporation of incorrect bases can itself be harmful and affect the viability of progeny. Given its fundamental importance, the basis of DNA repair has been extensively studied in both prokaryotes and eukaryotes, particularly in model organisms such as Escherichia coli and Saccharomyces cerevisiae1,2,3. Owing to the diverse nature of DNA damage, several repair pathways have evolved to rectify this damage and to restore lost genetic information (Box 2).
Throughout their association with a host, pathogenic microorganisms must survive the immune responses that they trigger during the colonization of mucosal surfaces and following the penetration of the epithelial barrier4. Innate immune responses against invading pathogens lead to the production of genotoxic molecules, including reactive oxygen species (ROS) and reactive nitrogen species (RNS), which are generated by epithelial cells and following phagocytosis of microorganisms by neutrophils and macrophages in a 'respiratory burst' (also known as an 'oxidative burst')5,6,7,8. Activated neutrophils and macrophages show a rapid increase in oxygen consumption, which is subsequently reduced into superoxide anions (O2−) by a membrane-associated NADPH-dependent oxidase and secreted into the phagosome. The importance of this aspect of innate immunity is highlighted by individuals with chronic granulomatous disease (CGD), who are deficient in this NADPH-dependent oxidase. CGD is associated with increased rates of chronic infections caused by bacteria and fungi, including Staphylococcus aureus and Candida albicans9. In the phagosome, superoxide anions are subsequently converted into other more destructive oxygen, nitrogen and chlorine species that cause damage to macromolecules such as DNA, proteins and lipids. For example, superoxide dismutates into hydrogen peroxide and subsequently into hydroxyl radicals (OH•) by the Fenton reaction, or into hypochlorous acid through the action of myeloperoxidase (which is expressed by neutrophils and, to a lesser extent, macrophages10); genetic deficiency of myeloperoxidase is associated with susceptibility to certain fungal, but not bacterial, infections11. Furthermore, superoxide reacts with the antimicrobial molecule nitric oxide to form the highly reactive oxidant peroxynitrite (ONOO−)12. Nitric oxide synthase deficiency has not yet been described in humans, but knockout mice are more prone to bacterial and fungal infections11, which is also observed following the administration of nitric oxide synthase inhibitors. Interestingly, knockout mice that lack both NADPH oxidase and nitric oxide synthase show extreme sensitivity to infections (including those caused by Listeria monocytogenes and Salmonella enterica) and are typically killed by overgrowth of the indigenous microbiota13, indicating synergistic antimicrobial activity between superoxide and nitric oxide.
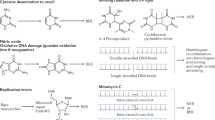
The type of DNA lesion caused by these insults varies according to the particular ROS or RNS produced by the host cell and includes SSBs, DSBs and crosslinking of DNA strands14. However, the most frequent types of damage are chemical modifications of single DNA bases and the generation of AP sites. Superoxide anions and hydrogen peroxide are relatively unreactive with DNA, but hypochlorous acid is able to chlorinate nucleobases and generally forms 5-chlorocytosine, 8-chloroadenine and 8-chloroguanine14. Furthermore, hydroxyl radicals are highly reactive and produce a wide range of oxidized nucleobases, including 7,8-dihydro-8-oxoguanine (8-oxoG), 5-hydroxycytosine, 4,6-diamino-5-formamidopyrimidine (FapyA) and 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) (reviewed in Ref. 14). Nitric oxide can cause DNA damage through nitrosation reactions with primary amines on the bases of DNA, which in downstream reactions lead to deamination15. In addition to deamination reactions, peroxynitrite oxidizes DNA and preferentially reacts with guanine to form 8-oxoG16. The majority of these chemical modifications and nucleobase lesions are typically resolved by the base excision repair (BER) pathway17 (Fig. 1).

a | Damaged bases in DNA are recognized by DNA glycosylases (such as formamidopyrimidine DNA glycosylase (MutM), Nth, Nei, MutY, Ung, AlkA, Mug, and Tag) and are removed. During excision, the damaged base is removed via the action of DNA glycosylases. Bifunctional glycosylases also cleave the DNA backbone (termed incision) after base excision; this occurs through β-elimination (by Nth) or δ-elimination (by MutM or Nei). Apurinic/apyrimidinic (AP) endonucleases (XthA or members of the End family) also cleave the DNA backbone during base excision repair (BER). In short-patch BER, the termini generated by cleavage are processed by an AP endonuclease (XthA or an End enzyme), a single-stranded DNA-specific exonuclease (RecJ) or a δ-eliminating DNA glycosylase (MutM or Nei), resulting in 5′-phosphate and 3′-hydroxyl groups. The gap is filled by DNA polymerase I (PolA), and the ends are ligated by LigA. In long-patch BER, 2–10 nucleotides are added by PolA (in a process known as displacement synthesis), and the resulting flap is removed by Fen1; the ends are then ligated by LigA. b | The GO system specifically recognizes and repairs the oxidized form of guanine, 7,8-dihydro-8-oxoguanine (8-oxoG). This base is recognized and removed by the DNA glycosylase MutM, and the resulting AP site is repaired by BER. When adenine is misincorporated (instead of cytosine) opposite an 8-oxoG, the DNA glycosylase MutY excises the adenine, and the AP site is subsequently repaired by BER, leading to the correct incorporation of a cytosine or another misincorporation of an adenine. To prevent the misincorporation of 8-oxoG, the 8-oxoG triphosphatase MutT hydrolyses 8-oxoGTP into 8-oxoGMP.
In general, fundamental processes, such as DNA replication and repair, are highly conserved in bacteria. However, there is increasing evidence that human-adapted bacterial pathogens have evolved specialized pathways of DNA repair, including the BER pathway. It has also become evident that BER facilitates microbial survival in the hostile environment of the host and contributes to virulence. In this Review, we illustrate this emerging concept by comparing the different strategies used by the three bacterial pathogens, Mycobacterium tuberculosis, Helicobacter pylori and Neisseria meningitidis (commonly known as meningococcus), to repair oxidative DNA damage, with an emphasis on the BER pathway. These three pathogens can all successfully colonize a given individual for several months (in the case of meningococcus) or even decades (for M. tuberculosis and H. pylori), so they exist in an intimate, long-term association with their human host. However, they occupy distinct sites in the body; N. meningitidis and M. tuberculosis are found in the upper and lower respiratory tract, respectively, whereas H. pylori colonizes the gastric mucosa. Furthermore, of these three microorganisms, only M. tuberculosis is thought to be predominantly an intracellular pathogen. We outline below how these different lifestyles are reflected in the organization of the BER pathway and the ways in which this pathway interacts with other mechanisms of DNA repair.
The BER pathway
The BER pathway repairs non-distorting DNA lesions following oxidation, deamination or alkylation of bases in the DNA backbone18. Initially, a damaged base is recognized and removed by a DNA glycosylase that cleaves the N-glycosidic bond between the base and the DNA backbone, generating an AP site (Fig. 1a). Although certain DNA glycosylases are lesion specific, others recognize a wide range of substrates and some share common substrates with other enzymes17. Glycosylases can be classified as monofunctional or bifunctional: monofunctional enzymes possess glycosylase activity only, whereas bifunctional enzymes also have lyase activity for cleavage of the DNA backbone at the AP site. Lyase activity can either generate a 3′-α,β-unsaturated aldehyde (through β-elimination) and a 5′-phosphate, or result in a 3′-phosphate (through γ-elimination)19. Alternatively, AP sites are cleaved by AP endonucleases, which incise the DNA backbone and leave a 3′-hydroxyl and a 5′-deoxyribose phosphate (dRP)20. Subsequently, these products must be processed into 3′-hydroxyl and 5′-phosphate groups before the repair process is complete19,20. In short-patch repair, single-nucleotide gaps are filled by the activity of DNA polymerase I (PolA) and are sealed by DNA ligase (LigA). Alternatively, in long-patch repair, PolA replaces up to 10 nucleotides using the 5′-dRP-containing strand as a template21,22; the flap is then processed by an endonuclease such as Fen1 before the nick is sealed23.
The GO system forms part of the BER pathway and specifically prevents oxidation of guanine or repairs DNA containing 8-oxoG and associated mismatches (Fig. 1b). As guanine is one of the most frequent oxidized nucleobases24, the GO system is particularly important for host-adapted bacteria, as they are continuously exposed to ROS and RNS, both of which generate 8-oxoG. The presence of 8-oxoG can result in the misincorporation of adenine, instead of cytosine, into the DNA backbone. Typically, the GO system consists of two DNA glycosylases, Fapy DNA glycosylase (MutM; also known as Fpg) and MutY, as well as an 8-oxoG triphosphatase (MutT). The DNA glycosylase MutM removes 8-oxoG25, whereas MutY removes adenines that are base-paired with 8-oxoG to enable the correct insertion of cytosine26. MutT hydrolyses intracellular pools of 8-oxoGTP to produce 8-oxoGMP, which reduces the likelihood that 8-oxoGTP is introduced into newly synthesized DNA27.
The intracellular lifestyle of M. tuberculosis
M. tuberculosis, the causative agent of tuberculosis (TB), is one of the most widespread bacterial pathogens and infects one-third of the global population, causing 1.3–1.4 million deaths each year28. This bacterium initially colonizes the mucosal surface of the lower respiratory epithelium and is subsequently phagocytized by alveolar macrophages. As one member of a select group of pathogens that survives and replicates in an intracellular environment29,30, M. tuberculosis is primarily found within phagosomes and is able to prevent the maturation of phagosomes into phagolysosomes, which promotes bacterial proliferation31. Within these activated macrophages, the bacterium is continuously exposed to ROS, RNS and other antimicrobial factors32, which control the spread of M. tuberculosis. Consistent with this, mice lacking components of enzyme complexes that are responsible for the generation of ROS in phagocytes or missing nitric oxide synthase (which is required for the production of RNS), as well as individuals with CGD who have a defective respiratory burst33,34,35, show increased susceptibility to TB. These findings indicate that throughout its life cycle, M. tuberculosis is exposed to a variety of DNA damaging agents, and as such, the bacterium has the complete repertoire of DNA repair systems found in other bacteria (with the exception of the mismatch repair (MMR) system) (Table 1). The enzymes involved in M. tuberculosis DNA repair are regulated by the SOS response through both RecA-dependent and RecA-independent pathways36,37. In the RecA-dependent pathway, RecA is activated by DSBs or SSBs and cleaves the repressor LexA, which facilitates the transcription of genes required for DNA repair. The mechanisms underlying the RecA-independent pathway have not been identified, although the binding sites of LexA have been mapped across the genome38. However, clear specialization of BER is evident in M. tuberculosis, reflecting its intracellular niche and its high GC content, which renders its genome susceptible to oxidation of guanine and deamination of cytosine.
Redundancy in mycobacterial BER. M. tuberculosis has evolved remarkable redundancy in its GO system39,40 (Fig. 2), which repairs 8-oxoG. For example, the bacterium harbours four MutT orthologues (MutT1–MutT4); although MutT1 and MutT2 show high levels of hydrolase activity for 8-oxoGTP, MutT4 exhibits only a low level of hydrolysis41,42,43. Notably, a mutT1 deletion mutant shows an increase in mutation rate41, even though MutT1 shares overlapping biochemical activity with MutT2. The multiple MutT orthologues that are encoded by this bacterium emphasize the specialization of BER for dealing with ROS by reducing the cytoplasmic pool of 8-oxoGTP.

M. tuberculosis colonizes the respiratory epithelium and is subsequently phagocytized by infiltrating alveolar macrophages and other phagocytic cells. Inside the phagosome of activated macrophages, the bacterium is exposed to reactive oxygen species (ROS) and reactive nitrogen species (RNS), resulting in many modifications of nucleobases through oxidation (via hydroxyl radicals and peroxynitrite), deamination (via nitric oxide and peroxynitrite) and alkylation (via nitrous anhydride and nitric oxide) reactions. The most frequently encountered oxidized nucleobases include 7,8-dihydro-8-oxoguanine (8-oxoG), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG), 4,6-diamino-5-formamidopyrimidine (FapyA), thymine glycol (Tg), 5,6-dihydrothymine (DHT), 5,6-dihydrouracil (DHU), 5-hydroxyuracil (5-OHU) and 5-hydroxycytosine (5-OHC). The most common deaminated nucleobases include xanthine (Xa), hypoxanthine (Hx) and uracil, and the most frequent alkylated nucleobases are 7-methylguanine (7-mG), 3-methyladenine (3-mA) and ethenocytosine (ɛC). These single-nucleotide lesions are generally repaired by base excision repair (BER), and M. tuberculosis has evolved remarkable functional redundancy in this pathway. It expresses a large number of DNA glycosylases that recognize and excise a wide variety of damaged nucleobases. For example, M. tuberculosis expresses MutY, which excises adenine, guanine and thymine following their mispairing with 8-oxoG. M. tuberculosis also harbours four genes encoding potential 8-oxoG triphosphatase (MutT) orthologues, and it expresses the two apurinic or apyrimidinic (AP) endonucleases XthA and End for the repair of AP sites (not shown).
Biochemical analyses of the mycobacterial glycosylase MutY have shown that this enzyme has an extended range of substrates compared with the E. coli enzyme and can remove adenine, guanine and thymine bases that are mispaired with 8-oxoG44,45. Interestingly, M. tuberculosis also has two genes that potentially encode MutM glycosylases for removing 8-oxoG from the DNA backbone. The substrate specificity of one of these enzymes, MutM1 formamidopyrimidine-DNA glycosylase 1 (Fpg1), is similar to that of its E. coli counterpart44, whereas the second MutM orthologue, Fpg2, does not bind to DNA, lacks glycosylase activity and seems to be encoded by a pseudogene46. In addition, M. tuberculosis has two endonuclease VIII (Nei) orthologues that usually excise oxidized pyrimidines, although E. coli Nei also excises 8-oxoG mispaired with an adenine or a guanine47,48. The M. tuberculosis enzymes, Nei1 and Nei2, can both excise 8-oxoG when paired with guanine46,49, albeit at low levels. Given this inefficiency, the Nei enzymes probably function in conjunction with the endonuclease Nth, which is thought to remove 8-oxoG from the DNA backbone during replication50. Nei1 also excises uracil from the DNA backbone46, which arises through the deamination of cytosine.
In addition, the high GC content of the genome may make the bacterium particularly susceptible to deamination of cytosine, which results in its conversion into uracil; alternatively, efficient repair of deamination might be responsible for the high GC content of the genome. Incorporation of uracil into the genome is minimized by the presence of the mycobacterial dUTPase, which hydrolyses dUTP and thereby reduces the intracellular pool51; interestingly, dUTPase also deaminates dCTP52. In E. coli, uracil is excised from the genome by the glycosylase Ung (also known as Udg), which is a component of BER53,54. Similarly, mycobacterial Ung efficiently excises uracil from both single- and double-stranded DNA55. Notably, Ung is required for full virulence of M. tuberculosis in a murine model of infection56, and for the growth of non-pathogenic Mycobacterium smegmatis in murine macrophages57. In addition to Ung, M. tuberculosis contains a second uracil DNA glycosylase, UdgB58, which recognizes a broad range of substrates, including hypoxanthine and ethenocytosine59. Furthermore, UdgB has the unusual property of being able to bind to AP sites, which are cytotoxic and mutagenic; thus, it has been suggested that UdgB might shield AP sites until the downstream BER enzymes process these lesions40,58.
Inside phagosomes, bacteria are also exposed to DNA alkylating agents that arise from nitrosation reactions between nitrous anhydride and amines or amides60. Both E. coli and M. tuberculosis contain an O6-alkylguanine DNA alkyltransferase (known as Ada and Ogt, respectively), which reverses the O6-alkylation of guanine61. In E. coli, alkylated bases can also be repaired by the 3-methyladenine DNA glycosylases, TagA and AlkA, which are part of the BER pathway. Although TagA is a highly specific enzyme that only excises 3-methyladenine and 3-methylguanine, AlkA recognizes a wide range of alkylated bases62. Interestingly, in M. tuberculosis the genes ada and alkA are found in the same operon as ogt, and ada and alkA encode a fusion protein60,63. Mutants lacking this operon are sensitive to alkylating agents but do not show virulence attenuation in mice60. Consistent with this, M. tuberculosis has two other putative 3-methyladenine DNA glycosylases, TagA and Mpg63, which could provide functional redundancy for repairing damage following nucleobase alkylation39.
AP sites can result in mutation or stalling of the replication fork64. In E. coli, AP sites are repaired by endonucleases belonging to the exonuclease III (XthA) and endonuclease IV (End; also known as Nfo) families. Similarly, homologues of XthA and End have been identified in M. tuberculosis63. However, in E. coli, XthA contributes 90% of the AP endonuclease activity, whereas in M. tuberculosis, End is the major AP endonuclease and XthA functions predominantly as a 3′-to-5′ exonuclease65. Interestingly, both of the M. tuberculosis AP endonucleases preferentially process AP sites that form opposite cytosine residues, whereas the E. coli AP endonucleases do not exhibit a substrate preference65,66. The additional bias of the M. tuberculosis BER pathway towards the repair of 8-oxoG and associated mismatches (those that involve mispairing with adenine primarily) further emphasizes how the bacterium has adapted to resist oxidative stress and to counteract the risks associated with a high GC content. Consistent with this, M. tuberculosis mutants lacking End and XthA display increased sensitivity to oxidative stress65, and an xthA mutant also has attenuated virulence in mice56.
In conclusion, as an intracellular bacterium, M. tuberculosis is continuously exposed to ROS and RNS and, as the majority of the damage caused by these oxidants is single-nucleobase lesions, M. tuberculosis has evolved extraordinary redundancy in its BER pathway. This functional redundancy is demonstrated by the presence of no fewer than four mutT orthologues, two nei orthologues, two mutM orthologues and two ung orthologues, which highlights the specialization of M. tuberculosis in its ability to repair oxidative DNA damage.
Surviving in the gastric mucosa: H. pylori
H. pylori colonizes the gastric mucosa in approximately 50% of the global population and causes chronic gastroduodenal disorders such as gastric cancer, peptic ulcers and gastritis67. The precise mode of transmission of H. pylori remains unknown, but it has been suggested that direct oral–oral or faecal–oral transmission are the most probable modes68. H. pylori has been detected in saliva, vomit and faeces; the bacterium is highly sensitive to conditions outside the body, such as atmospheric oxygen pressure and temperatures below 34 °C67. After penetration of the gastric mucus, H. pylori resides in close proximity to the gastric epithelial layer and elicits innate immune responses, resulting in subsequent exposure to ROS and RNS produced by epithelial cells and recruited phagocytic cells, such as neutrophils69,70,71 (Fig. 3; Table 1). To survive these insults, H. pylori must detect and repair DNA damage; however, little is known about the regulation of enzymes required for DNA repair. There is surprisingly limited redundancy in DNA repair pathways, including BER, and the bacterium also lacks functional MMR and non-homologous end-joining (NHEJ) pathways72,73.

H. pylori colonizes the gastric mucosa and can also be phagocytized by infiltrating neutrophils. Both intracellular and extracellular H. pylori are exposed to reactive oxygen species (ROS) and reactive nitrogen species (RNS) that are generated by a strong 'respiratory burst' from neutrophils. ROS and RNS damage DNA by oxidation reactions (mediated by hydroxyl radicals and peroxynitrite), which generate 7,8-dihydro-8-oxoguanine (8-oxoG), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG), 4,6-diamino-5-formamidopyrimidine (FapyA), thymine glycol (Tg), 5,6-dihydrothymine (DHT), 5,6-dihydrouracil (DHU), 5-hydroxyuracil (5-OHU) and 5-hydroxycytosine (5-OHC); deamination (mediated by nitric oxide and peroxynitrite), which generates xanthine (Xa), hypoxanthine (Hx) and uracil; and alkylation (mediated by nitrous anhydride and nitric oxide), which generates 7-methylguanine (7-mG), 3-methyladenine (3-mA) and ethenocytosine (ɛC). These single-nucleobase lesions are repaired by the base excision repair (BER) pathway. H. pylori has three DNA glycosylases (Nth, Ung and MagIII) that recognize and excise several of the most cytotoxic nucleobases. Interestingly, these DNA glycosylases do not recognize and excise nucleobase lesions that are mutagenic. However, H. pylori MutY excises adenine mispaired with 8-oxoG and thereby reduces the level of spontaneous mutations. Furthermore, H. pylori has the apurinic or apyrimidinic (AP) endonuclease XthA for the repair of AP sites (not shown).
Functional specialization of BER. H. pylori contains the minimal complement of enzymes needed to carry out each step of the BER pathway, all of which are necessary to repair single-base lesions caused by ROS and RNS (Fig. 3). Interestingly, the BER components of H. pylori seem to have higher specificity for cytotoxic base lesions than mutagenic base lesions. This specialization might contribute to the high genetic variability that is observed among H. pylori strains and between individual bacteria during long-term colonization. It has been suggested that this high genetic variability, combined with a high mutation rate, provides H. pylori with an adaptive advantage in the gastric environment of the human host67. A high mutation rate, owing to the reduced proofreading capacity of its DNA polymerase74, may increase the chance of H. pylori acquiring mutations that confer a fitness benefit under stressful conditions, despite the inherent cost of deleterious mutations. As there is little competition with other microorganisms in the gastric environment, loss of a fraction of the population due to deleterious mutations might not be a major drawback for H. pylori, and the high mutation rate might therefore be more beneficial for H. pylori than for pathogens that inhabit crowded sites of the body. Consistent with this, a study of human volunteers and primates challenged with H. pylori indicated that the bacterium has an increased mutation rate on exposure to the host inflammatory response75. Thus, this might increase the emergence of genetic variants of H. pylori that could survive the selective pressure imposed by the innate immune response of the host.
H. pylori contains a single DNA glycosylase, Nth, for the removal of oxidized nucleobases. Nth is a bifunctional glycosylase that initially excises oxidized pyrimidines and subsequently excises the AP site. Although oxidized purines (such as 8-oxoG) are far more abundant than oxidized pyrimidines, the former tend to be mostly mutagenic. By contrast, oxidized pyrimidines have the propensity to block DNA replication and transcription; therefore, they tend to be cytotoxic76. Consistent with this, H. pylori strains lacking Nth are sensitive to oxidizing agents, are preferentially killed by activated macrophages and are impaired in their ability to colonize or persist in a mouse model of infection76. At the level of recognition and excision of damaged bases, BER is different in E. coli, as this organism contains the DNA glycosylases MutM and Nei, which provide some redundancy for removing oxidized nucleobases. A similar lack of redundancy is found in the ability of H. pylori to remove methylated nucleobases from the DNA backbone. E. coli contains two 3-methyladenine glycosylases, TagA and AlkA, whereas H. pylori contains a single inducible, monofunctional 3-methyladenine DNA glycosylase (MagIII) that belongs to a different enzyme class77,78. MagIII contains structural motifs found in AlkA DNA glycosylases, but its substrates are similar to those recognized by E. coli TagA. MagIII is highly specific for 3-methyladenines owing to the structure of its binding pocket, which excludes 7-methylguanine and other methylated bases78. 3-Methyladenines are particularly harmful because they block DNA synthesis, whereas 7-methylguanine is mostly mutagenic78. Furthermore, MagIII is required for survival during alkylation stress and for gastric colonization77,79. In addition, H. pylori has a potential uracil DNA glycosylase Ung; although this enzyme has not been characterized in detail, it is required for gastric colonization79. Thus, at the level of DNA glycosylases, there seems to be little redundancy in H. pylori BER.
Other features of the minimal BER pathway found in H. pylori include its single AP endonuclease73,80,81 and the absence of the GO system, although the organism does encode the glycosylase MutY, which excises bases mismatched with 8-oxoG. Specifically, MutY excises adenines that are base-paired with 8-oxoG but, unlike E.coli MutY, it does not excise adenines that are paired with guanine80,82. Despite this limited range of activity, MutY reduces the level of spontaneous mutations in H. pylori, which is evident from the increased mutation rate of a mutY deletion mutant80,81. Furthermore, MutY contributes to gastric colonization of H. pylori82. Interestingly, mutY contains a homopolymeric tract of eight adenines81, and slipped-strand mispairing during DNA replication can lead to frameshift mutations, which leads to the production of non-functional MutY and a mutator phenotype. This highlights how H. pylori maintains a delicate balance between DNA mutagenesis and repair: MutY phase variation results in a subpopulation of H. pylori with an increased mutation rate and potentially a higher propensity of acquiring adaptive mutations. Furthermore, H. pylori does not contain an intact MMR system, even though it does express a MutS2 homologue83. However, this enzyme inhibits homologous recombination83, has high affinity for double-stranded DNA that contains 8-oxoG and protects cells against oxidative stress through unknown mechanisms84.
In summary, H. pylori is found in the gastric mucosa in close association with epithelial cells and infiltrating neutrophils, and is subsequently exposed to a strong respiratory burst. H. pylori has evolved a highly specialized BER pathway to repair the resulting oxidative DNA damage. The BER components of this pathogen show high specificity for nucleobase lesions that are cytotoxic, whereas mutagenic lesions are generally untargeted. This specialization might be related to the high genetic variability and mutation rate that are typically observed in H. pylori strains67.
Networks of DNA repair in N. meningitidis
The natural habitat of N. meningitidis is the upper respiratory tract85,86 where the bacterium causes asymptomatic infection in 10–40% of healthy individuals85,87, and carriage of a single strain can last for several months. During infection, the bacterium crosses the epithelial barrier in the upper respiratory tract and enters the bloodstream, resulting in septicaemia and spread to the meningeal space. In contrast to M. tuberculosis and H. pylori, meningococcus is exposed to atmospheric oxygen concentrations during colonization of the mucosal surface of the nasopharynx during its life cycle. Therefore, this bacterium is exposed to substantial oxidative stress owing to the high extracellular oxygen concentration and its presence inside immune cells (including neutrophils) that generate ROS and RNS88. Consistent with this constant exposure to oxidative stress, enzymes involved in BER are constitutively expressed89,90, rather than being under the control of the SOS response (as in E. coli)91. Furthermore, similarly to its close relative the gonococcus, N. meningitidis can survive within the hostile environment of neutrophils88, which is thought to promote a rapidly progressive and severe disease, leading to extremely high bacterial densities in the bloodstream. The adaptation of the bacterium to the upper airways and its interactions with immune cells have probably provided selective pressure for the evolution of a distinct BER pathway in N. meningitidis compared with model organisms such as E. coli92 (Fig. 4).

N. meningitidis colonizes the human nasopharynx, where it is exposed to reactive oxygen species (ROS) and reactive nitrogen species (RNS) produced by atmospheric oxygen and by the 'respiratory burst' of phagocytes. Oxidized nucleobases include 7,8-dihydro-8-oxoguanine (8-oxoG), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG), 4,6-diamino-5-formamidopyrimidine (FapyA), thymine glycol (Tg), 5,6-dihydrothymine (DHT), 5,6-dihydrouracil (DHU), 5-hydroxyuracil (5-OHU) and 5-hydroxycytosine (5-OHC). Deaminated bases include xanthine (Xa), hypoxanthine (Hx) and uracil; alkylated nucleobases include 7-methylguanine (7-mG), 3-methyladenine (3-mA) and ethenocytosine (ɛC). N. meningitidis has several DNA glycosylases with overlapping functions (such as MutM and Nth) that excise oxidized nucleobases. N. meningitidis harbours a putative uracil DNA glycosylase (Ung) for the removal of uracil (it is unknown whether this enzyme also removes the other deaminated bases Xa and Hx) and a putative 3-methyladenine DNA glycosylase (Tag) that removes 3-methyladenine (it is unknown whether this enzyme also removes the other alkylated bases 7-mG and ɛC). In addition, N. meningitidis MutY excises adenine, guanine and thymine mispaired with 8-oxoG; the bacterium contains a single 8-oxoG triphosphatase (MutT) homologue, which hydrolyses cellular 8-oxoGTP to 8-oxoGMP. Finally, meningococcal NApe is a true apurinic/apyrimidinic (AP) endonuclease, whereas NExo lacks AP endonuclease activity but instead is a specialized 3′-phosphatase (not shown).
Specialization and integration of meningococcal BER. The initial recognition and processing of DNA lesions in meningococcus involve the overlapping activity of DNA glycosylases90, as shown by the redundancy of the bifunctional glycosylases, MutM and Nth90,93. These enzymes recognize and process a variety of DNA lesions: both enzymes eliminate 8-oxoG, 5′-OH substituted uracil (5′-OHU) and thymine glycol (Tg) from the DNA backbone and have similar levels of activity. However, unlike Nth, MutM activity is inhibited by the presence of its products, including the incised DNA backbone with a 3′-phosphate, which reduces the processivity of this enzyme90. Therefore, Nth is more effective than MutM in conditions of higher oxidative stress and DNA damage, such as during exposure to a respiratory burst inside neutrophils or in the aerobic conditions of the upper respiratory tract.
By contrast, there is exquisite specialization of the enzymes involved in subsequent steps of the repair process. Meningococcus has two AP endonuclease homologues, NApe and NExo, which show a high level of structural homology89; both of these enzymes contribute to bacterial survival during systemic disease and inside neutrophils89. Despite their structural similarity, NApe is the sole active AP endonuclease in meningococcus, whereas NExo is unable to cleave AP sites. Instead, NExo plays a part in BER through its 3′-phosphatase activity94, which contributes to the repair of residual lesions, the 3′-phosphate and 3′-unsaturated aldehyde, following the lyase activity of MutM or spontaneous base loss through direct DNA damage. Remarkably, this specialization of activity is altered by a single amino acid substitution in each enzyme; substitution of His167 in NExo with either Gly or Ser (which are found in other known AP endonucleases, such as those in E. coli) confers AP endonuclease activity on NExo, and introducing His167 into NApe is sufficient for it to function as a 5′-phosphatase94. By contrast, both of these activities are carried out by a single enzyme, ExoIII, in E. coli95. Therefore, the meningococcal processing enzymes NApe and NExo have a crucial role in bacterial survival in the presence of oxidative stress because the groups that they recognize (3′-phosphate, which is recognized by NExo, and 5′-dRP, which is recognized by both) block DNA replication and are therefore cytotoxic.
It has been suggested that potentially cytotoxic intermediates of BER are passed from one enzyme to the next in a defined series of steps during DNA repair. However, there seems to be little integration between the initial and subsequent stages of BER in meningococcus. For example, functional studies of the glycosylases Nth and MutM have shown that the presence of a processing enzyme (such as NApe or NExo) does not promote their activity and processing of the lesion90. Thus, BER in meningococcus does not seem to be arranged as an ordered pathway, as proposed for other bacteria, in which the products of the repair process are passed in a sequential manner from one enzyme to the next (Fig. 1). Instead, a collection of constitutively expressed glycosylases, including MutM and Nth, with overlapping activities identify and excise the damaged bases, and the subsequent steps of repair are not determined by complexes of cooperating enzymes but by the activities and abundance of the processing enzymes. Indeed, consistent with this, there is evidence that both MMR and NER contribute to meningococcal survival following oxidative DNA damage90,96. Thus, meningococcus integrates multiple enzymes and mechanisms of DNA repair, which are organized as a network rather than a pathway, to restore genomic integrity following oxidative DNA damage.
GO or stop during DNA repair? N. meningitidis contains a rudimentary GO system that consists of the glycosylases MutM and MutY, as well as a single MutT homologue90,97. A meningococcal strain lacking all three enzymes is viable and is not hypersensitive to oxidizing agents, so the GO system does not seem to be a major contributor to the repair of oxidative DNA damage. By contrast, in other bacteria, such as Pseudomonas aeruginosa, loss of MutY, MutT or MutM impairs bacterial survival in vivo and confers hypersensitivity to ROS98. By removing 8-oxoG and reducing the misincorporation of bases, the GO system in meningococcus limits the emergence of spontaneous point mutations, which are detected by the appearance of spontaneous streptomycin-resistant colonies (owing to amino acid substitutions in the ribosomal protein RpsL). As these changes do not affect DNA replication, loss of the GO system does not impair survival under conditions of oxidative stress, but it does increase mutation rate. By contrast, enzymes that process intermediate products in BER (such as NApe and NExo) and that can block replication when they are absent, have major effects on bacterial viability but have only a minor effect on mutation rate90. The meningococcal genome also contains genes encoding putative enzymes, Ung and Tag, which excise deaminated and alkylated bases, respectively.
Finally, NER also seems to contribute to the repair of oxidative DNA damage in meningococcus, which is consistent with observations in gonococcus on exposure to low levels of oxidizing agents99. However, the loss of NER seems to have a more detrimental effect on the survival of meningococcus than gonococcus in the presence of oxidizing agents90. Given the role of NER in repairing damage caused by ultraviolet radiation, this difference might reflect the different body sites at which these bacteria are found in the human host, with meningococcus (in the respiratory tract) being more likely to be directly exposed to ultraviolet radiation than gonococcus, which is found in the genital tract.
Conclusions and future prospects
In the past, the majority of research on pathogenic bacteria has focused on their ability to invade the host and promote virulence. However, as we learn more about how microorganisms are adapted to particular microenvironments during the different stages of the disease process, it is becoming clear that activities that were once considered 'housekeeping' are also specialized and fit for purpose. This is evident even when considering a highly conserved process such as BER, which is found in humans and prokaryotes. Even between the three bacterial pathogens considered here, there is substantial variation in the complement and function of BER enzymes, which reflects the diversity of habitats that they inhabit in the human body. Furthermore, some accepted paradigms of DNA repair do not hold for these organisms, which can survive in close association with a hostile host. For example, a RecA-dependent SOS response, which has a crucial role in the induction of DNA repair in E. coli, is not found in N. meningitidis, and a RecA-independent pathway of induction is found in M. tuberculosis. Little is known about the regulation of DNA repair in H. pylori, except that RecA is subject to post-translational modification100 and that it is constitutively expressed101. Further studies on the regulation of enzymes in BER in these bacteria might reveal the environmental cues that trigger gene expression. Furthermore, DNA repair pathways are thought to operate independently of each other in E. coli. The extent of interaction between BER and NER in N. meningitidis suggests that future work will uncover other examples of integration of repair pathways in pathogens.
Distinctive mechanisms of DNA repair may well have evolved in other human-adapted microorganisms, including commensal species and other pathogens. For example, bioinformatic analyses have shown that the commensal species Neisseria lactamica and Neisseria polysaccharea have the same repertoire of BER enzymes as meningococcus (S. v. d. V. and C. M. T., unpublished observations), so their evolution is likely to have been shaped by survival at the mucosal surface. However, these enzymes have several polymorphisms and their activities are poorly understood, so future work in the field should determine whether these polymorphisms affect enzyme function and whether their regulation is altered compared to pathogens. This should provide insights into whether DNA repair is specific for the pathogenic lifestyle or reflects general adaptations that both commensals and pathogens have evolved to enable colonization and transmission. Furthermore, Streptococcus pneumoniae can secrete hydrogen peroxide, which modulates host inflammatory responses102; the bacterium withstands this genotoxic agent without having a catalase enzyme. Although S. pneumoniae has other mechanisms to eliminate ROS, little is known about its ability to correct DNA damage through BER. Haemophilus influenzae is another pathogen found in the nasopharynx, and comparative genome analysis indicates that it has a similar complement of BER enzymes as meningococcus103. Therefore, further studies on H. influenzae have the potential to demonstrate whether the network model of DNA repair is a more general phenomenom or whether it is restricted to N. meningitidis.
Several technical challenges remain. Appropriate animal models to study the behaviour of human-adapted pathogens in physiologically relevant settings are lacking, and the construction of bacterial strains with multiple mutations is demanding but necessary to examine functional redundancy in repair systems. Furthermore, live cell imaging methods104, to assess the behaviour of single proteins in a bacterium during DNA repair, have not yet been adapted for use in microorganisms within the context of their hosts. Despite these limitations, it is clear that the BER pathways differ between the three bacterial pathogens discussed here. Compared to the minimal system in H. pylori (which lacks the MMR and NHEJ pathways), DNA repair in M. tuberculosis is characterized by remarkable redundancy, especially in the GO system. In addition, DNA repair enzymes in meningococcus display functional specialization for the repair of AP sites, but these enzymes function within a network of interacting repair pathways. An increase in our understanding of the mechanisms involved in BER in these and other human-adapted pathogens should provide unparalleled insights into the fundamental process of DNA repair and offer potential opportunities for the development of novel therapeutics.
References
Eisen, J. A. & Hanawalt, P. C. A phylogenomic study of DNA repair genes, proteins, and processes. Mutat. Res. 435, 171–213 (1999).
Boiteux, S. & Guillet, M. Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair (Amst.) 3, 1–12 (2004).
Daley, J. M., Zakaria, C. & Ramotar, D. The endonuclease IV family of apurinic/apyrimidinic endonucleases. Mutat. Res. 705, 217–227 (2010).
Eisele, N. A. & Anderson, D. M. Host defense and the airway epithelium: frontline responses that protect against bacterial invasion and pneumonia. J. Pathog. 2011, 249802 (2011).
Swanson, P. A. et al. Enteric commensal bacteria potentiate epithelial restitution via reactive oxygen species-mediated inactivation of focal adhesion kinase phosphatases. Proc. Natl Acad. Sci. USA 108, 8803–8808 (2011).
Kinnula, V. L., Adler, K. B., Ackley, N. J. & Crapo, J. D. Release of reactive oxygen species by guinea pig tracheal epithelial cells in vitro. Am. J. Physiol. 262, L708–L712 (1992).
Sibille, Y. & Reynolds, H. Y. Macrophages and polymorphonuclear neutrophils in lung defense and injury. Am. Rev. Respir. Dis. 141, 471–501 (1990).
Fialkow, L., Wang, Y. & Downey, G. P. Reactive oxygen and nitrogen species as signaling molecules regulating neutrophil function. Free Radic. Biol. Med. 42, 153–164 (2007).
Goldblatt, D. & Thrasher, A. J. Chronic granulomatous disease. Clin. Exp. Immunol. 122, 1–9 (2000).
Gaut, J. P. et al. Neutrophils employ the myeloperoxidase system to generate antimicrobial brominating and chlorinating oxidants during sepsis. Proc. Natl Acad. Sci. USA 98, 11961–11966 (2001).
Nathan, C. & Shiloh, M. U. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl Acad. Sci. USA 97, 8841–8848 (2000). This excellent review summarizes evidence for the role of ROS and RNS in protection against microbial infections.
Missall, T. A., Lodge, J. K. & McEwen, J. E. Mechanisms of resistance to oxidative and nitrosative stress: implications for fungal survival in mammalian hosts. Eukaryot. Cell 3, 835–846 (2004).
Shiloh, M. U. et al. Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity 10, 29–38 (1999).
Cadet, J. & Wagner, J. R. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 5, a012559 (2013).
Burney, S., Caulfield, J. L., Niles, J. C., Wishnok, J. S. & Tannenbaum, S. R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. 424, 37–49 (1999).
Szabo, C. & Ohshima, H. DNA damage induced by peroxynitrite: subsequent biological effects. Nitr. Oxide 1, 373–385 (1997).
Huffman, J. L., Sundheim, O. & Tainer, J. A. DNA base damage recognition and removal: new twists and grooves. Mutat. Res. 577, 55–76 (2005).
Krokan, H. E. & Bjoras, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 5, a012583 (2013).
Svilar, D., Goellner, E. M., Almeida, K. H. & Sobol, R. W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal 14, 2491–2507 (2011).
Dianov, G. & Lindahl, T. Reconstitution of the DNA base excision-repair pathway. Curr. Biol. 4, 1069–1076 (1994).
Mosbaugh, D. W. & Linn, S. Characterization of the action of Escherichia coli DNA polymerase I at incisions produced by repair endodeoxyribonucleases. J. Biol. Chem. 257, 575–583 (1982).
Sung, J. S. & Mosbaugh, D. W. Escherichia coli uracil- and ethenocytosine-initiated base excision DNA repair: rate-limiting step and patch size distribution. Biochemistry 42, 4613–4625 (2003).
Yao, M. & Kow, Y. W. Cleavage of insertion/deletion mismatches, flap and pseudo-Y DNA structures by deoxyinosine 3′-endonuclease from Escherichia coli. J. Biol. Chem. 271, 30672–30676 (1996).
Singh, P. et al. Carbon nanotube-nucleobase hybrids: nanorings from uracil-modified single-walled carbon nanotubes. Chemistry 17, 6772–6780 (2011).
Tchou, J. et al. 8-oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity. Proc. Natl Acad. Sci. USA 88, 4690–4694 (1991).
Tsai-Wu, J. J., Liu, H. F. & Lu, A. L. Escherichia coli MutY protein has both N-glycosylase and apurinic/apyrimidinic endonuclease activities on A.C and A.G mispairs. Proc. Natl Acad. Sci. USA 89, 8779–8783 (1992).
Nakamura, T. et al. Structural and dynamic features of the MutT protein in the recognition of nucleotides with the mutagenic 8-oxoguanine base. J. Biol. Chem. 285, 444–452 (2010).
Dye, C., Scheele, S., Dolin, P., Pathania, V. & Raviglione, M. C. Consensus statement. Global burden of tuberculosis: estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA 282, 677–686 (1999).
Subbian, S., Mehta, P. K., Cirillo, S. L., Bermudez, L. E. & Cirillo, J. D. A. Mycobacterium marinum mel2 mutant is defective for growth in macrophages that produce reactive oxygen and reactive nitrogen species. Infect. Immun. 75, 127–134 (2007).
Li, Q. et al. Differences in rate and variability of intracellular growth of a panel of Mycobacterium tuberculosis clinical isolates within a human monocyte model. Infect. Immun. 70, 6489–6493 (2002).
Sullivan, J. T., Young, E. F., McCann, J. R. & Braunstein, M. The Mycobacterium tuberculosis SecA2 system subverts phagosome maturation to promote growth in macrophages. Infect. Immun. 80, 996–1006 (2012).
Nicholson, S. et al. Inducible nitric oxide synthase in pulmonary alveolar macrophages from patients with tuberculosis. J. Exp. Med. 183, 2293–2302 (1996).
Adams, L. B., Dinauer, M. C., Morgenstern, D. E. & Krahenbuhl, J. L. Comparison of the roles of reactive oxygen and nitrogen intermediates in the host response to Mycobacterium tuberculosis using transgenic mice. Tuber Lung Dis. 78, 237–246 (1997).
MacMicking, J. D. et al. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl Acad. Sci. USA 94, 5243–5248 (1997). This study provides direct evidence for the protective role of the host nitric oxide synthase against TB.
Lee, P. P. et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr. Infect. Dis. J. 27, 224–230 (2008).
Rand, L. et al. The majority of inducible DNA repair genes in Mycobacterium tuberculosis are induced independently of RecA. Mol. Microbiol. 50, 1031–1042 (2003).
Davis, E. O. et al. DNA damage induction of recA in Mycobacterium tuberculosis independently of RecA and LexA. Mol. Microbiol. 46, 791–800 (2002).
Smollett, K. L. et al. Global analysis of the regulon of the transcriptional repressor LexA, a key component of SOS response in Mycobacterium tuberculosis. J. Biol. Chem. 287, 22004–22014 (2012).
Dos Vultos, T., Mestre, O., Tonjum, T. & Gicquel, B. DNA repair in Mycobacterium tuberculosis revisited. FEMS Microbiol. Rev. 33, 471–487 (2009).
Kurthkoti, K. & Varshney, U. Distinct mechanisms of DNA repair in mycobacteria and their implications in attenuation of the pathogen growth. Mech. Ageing Dev. 133, 138–146 (2012).
Dos Vultos, T., Blazquez, J., Rauzier, J., Matic, I. & Gicquel, B. Identification of Nudix hydrolase family members with an antimutator role in Mycobacterium tuberculosis and Mycobacterium smegmatis. J. Bacteriol. 188, 3159–3161 (2006).
Patil, A. G., Sang, P. B., Govindan, A. & Varshney, U. Mycobacterium tuberculosis MutT1 (Rv2985) and ADPRase (Rv1700) proteins constitute a two-stage mechanism of 8-oxo-dGTP and 8-oxo-GTP detoxification and adenosine to cytidine mutation avoidance. J. Biol. Chem. 288, 11252–11262 (2013).
Sang, P. B. & Varshney, U. Biochemical properties of MutT2 proteins from Mycobacterium tuberculosis and M. smegmatis and their contrasting antimutator roles in Escherichia coli. J. Bacteriol. 195, 1552–1560 (2013).
Jain, R., Kumar, P. & Varshney, U. A distinct role of formamidopyrimidine DNA glycosylase (MutM) in down-regulation of accumulation of G, C mutations and protection against oxidative stress in mycobacteria. DNA Repair (Amst.) 6, 1774–1785 (2007).
Olsen, I. et al. Characterization of the major formamidopyrimidine-DNA glycosylase homolog in Mycobacterium tuberculosis and its linkage to variable tandem repeats. FEMS Immunol. Med. Microbiol. 56, 151–161 (2009).
Guo, Y. et al. The oxidative DNA glycosylases of Mycobacterium tuberculosis exhibit different substrate preferences from their Escherichia coli counterparts. DNA Repair (Amst.) 9, 177–190 (2010). This study provides a thorough comparative biochemical analysis of DNA glycosylases and highlights differences between M. tuberculosis and E. coli.
Hazra, T. K. et al. Characterization of a novel 8-oxoguanine-DNA glycosylase activity in Escherichia coli and identification of the enzyme as endonuclease VIII. J. Biol. Chem. 275, 27762–27767 (2000).
Zharkov, D. O. et al. Structural analysis of an Escherichia coli endonuclease VIII covalent reaction intermediate. EMBO J. 21, 789–800 (2002).
Sidorenko, V. S., Rot, M. A., Filipenko, M. L., Nevinsky, G. A. & Zharkov, D. O. Novel DNA glycosylases from Mycobacterium tuberculosis. Biochem. (Mosc) 73, 442–450 (2008).
Matsumoto, Y., Zhang, Q. M., Takao, M., Yasui, A. & Yonei, S. Escherichia coli Nth and human hNTH1 DNA glycosylases are involved in removal of 8-oxoguanine from 8-oxoguanine/guanine mispairs in DNA. Nucleic Acids Res. 29, 1975–1981 (2001).
Vertessy, B. G. & Toth, J. Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 42, 97–106 (2009).
Helt, S. S. et al. Mechanism of dTTP inhibition of the bifunctional dCTP deaminase:dUTPase encoded by Mycobacterium tuberculosis. J. Mol. Biol. 376, 554–569 (2008).
Lindahl, T. An N-glycosidase from Escherichia coli that releases free uracil from DNA containing deaminated cytosine residues. Proc. Natl Acad. Sci. USA 71, 3649–3653 (1974).
Lindahl, T., Ljungquist, S., Siegert, W., Nyberg, B. & Sperens, B. D.N. A. N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli. J. Biol. Chem. 252, 3286–3294 (1977).
Purnapatre, K. & Varshney, U. Uracil DNA glycosylase from Mycobacterium smegmatis and its distinct biochemical properties. Eur. J. Biochem. 256, 580–588 (1998).
Sassetti, C. M. & Rubin, E. J. Genetic requirements for mycobacterial survival during infection. Proc. Natl Acad. Sci. USA 100, 12989–12994 (2003). By screening a mutant library, this study identified 194 genes that are important for mycobacterial growth in vivo.
Venkatesh, J., Kumar, P., Krishna, P. S., Manjunath, R. & Varshney, U. Importance of uracil DNA glycosylase in Pseudomonas aeruginosa and Mycobacterium smegmatis, G+C-rich bacteria, in mutation prevention, tolerance to acidified nitrite, and endurance in mouse macrophages. J. Biol. Chem. 278, 24350–24358 (2003).
Srinath, T., Bharti, S. K. & Varshney, U. Substrate specificities and functional characterization of a thermo-tolerant uracil DNA glycosylase (UdgB) from Mycobacterium tuberculosis. DNA Repair (Amst.) 6, 1517–1528 (2007).
Kumar, P., Bharti, S. K. & Varshney, U. Uracil excision repair in Mycobacterium tuberculosis cell-free extracts. Tuberculosis (Edinb.) 91, 212–218 (2011).
Durbach, S. I. et al. DNA alkylation damage as a sensor of nitrosative stress in Mycobacterium tuberculosis. Infect. Immun. 71, 997–1000 (2003).
Miggiano, R. et al. Biochemical and structural studies of the Mycobacterium tuberculosis O6-methylguanine methyltransferase and mutated variants. J. Bacteriol. 195, 2728–2736 (2013).
O'Brien, P. J. & Ellenberger, T. The Escherichia coli 3-methyladenine DNA glycosylase AlkA has a remarkably versatile active site. J. Biol. Chem. 279, 26876–26884 (2004).
Cole, S. T. et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 (1998).
Loeb, L. A. Apurinic sites as mutagenic intermediates. Cell 40, 483–484 (1985).
Puri, R. V., Singh, N., Gupta, R. K. & Tyagi, A. K. Endonuclease IV is the major apurinic/apyrimidinic endonuclease in Mycobacterium tuberculosis and is important for protection against oxidative damage. PLoS ONE 8, e71535 (2013).
Takeuchi, M., Lillis, R., Demple, B. & Takeshita, M. Interactions of Escherichia coli endonuclease IV and exonuclease III with abasic sites in DNA. J. Biol. Chem. 269, 21907–21914 (1994).
Kusters, J. G., van Vliet, A. H. & Kuipers, E. J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 19, 449–490 (2006).
Salama, N. R., Hartung, M. L. & Muller, A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori. Nature Rev. Microbio. 11, 385–399 (2013).
Allen, L. A., Beecher, B. R., Lynch, J. T., Rohner, O. V. & Wittine, L. M. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. J. Immunol. 174, 3658–3667 (2005). This study shows that H. pylori disrupts targeting by NADPH oxidase in the phagosome and redirects superoxide for release to the extracellular environment.
Chaturvedi, R. et al. Induction of polyamine oxidase 1 by Helicobacter pylori causes macrophage apoptosis by hydrogen peroxide release and mitochondrial membrane depolarization. J. Biol. Chem. 279, 40161–40173 (2004).
Xu, H. et al. Spermine oxidation induced by Helicobacter pylori results in apoptosis and DNA damage: implications for gastric carcinogenesis. Cancer Res. 64, 8521–8525 (2004).
Alm, R. A. et al. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397, 176–180 (1999).
Tomb, J. F. et al. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547 (1997).
García-Ortíz, M. V. et al. Unexpected role for Helicobacter pylori DNA polymerase I as a source of genetic variability. PLoS Genet. 7, e1002152 (2011).
Linz, B. et al. A mutation burst during the acute phase of Helicobacter pylori infection in humans and rhesus macaques. Nature Commun. 5, 4165 (2014).
O'Rourke, E. J. et al. Pathogen DNA as target for host-generated oxidative stress: role for repair of bacterial DNA damage in Helicobacter pylori colonization. Proc. Natl Acad. Sci. USA 100, 2789–2794 (2003). This study provides evidence for host-induced oxidative DNA damage of the H. pylori genome during colonization.
O'Rourke, E. J. et al. A novel 3-methyladenine DNA glycosylase from Helicobacter pylori defines a new class within the endonuclease III family of base excision repair glycosylases. J. Biol. Chem. 275, 20077–20083 (2000).
Eichman, B. F., O'Rourke, E. J., Radicella, J. P. & Ellenberger, T. Crystal structures of 3-methyladenine DNA glycosylase MagIII and the recognition of alkylated bases. EMBO J. 22, 4898–4909 (2003).
Baldwin, D. N. et al. Identification of Helicobacter pylori genes that contribute to stomach colonization. Infect. Immun. 75, 1005–1016 (2007). This study screened a library of H. pylori mutants in a mouse model of infection to identify genes that are important for gastric colonization.
Mathieu, A., O'Rourke, E. J. & Radicella, J. P. Helicobacter pylori genes involved in avoidance of mutations induced by 8-oxoguanine. J. Bacteriol. 188, 7464–7469 (2006).
Huang, S., Kang, J. & Blaser, M. J. Antimutator role of the DNA glycosylase mutY gene in Helicobacter pylori. J. Bacteriol. 188, 6224–6234 (2006).
Eutsey, R., Wang, G. & Maier, R. J. Role of a MutY DNA glycosylase in combating oxidative DNA damage in Helicobacter pylori. DNA Repair (Amst.) 6, 19–26 (2007).
Pinto, A. V. et al. Suppression of homologous and homeologous recombination by the bacterial MutS2 protein. Mol. Cell 17, 113–120 (2005).
Wang, G., Alamuri, P., Humayun, M. Z., Taylor, D. E. & Maier, R. J. The Helicobacter pylori MutS protein confers protection from oxidative DNA damage. Mol. Microbiol. 58, 166–176 (2005).
Stephens, D. S. Uncloaking the meningococcus: dynamics of carriage and disease. Lancet 353, 941–942 (1999).
Cartwright, K. A., Stuart, J. M., Jones, D. M. & Noah, N. D. The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica. Epidemiol. Infect. 99, 591–601 (1987).
Christensen, H., May, M., Bowen, L., Hickman, M. & Trotter, C. L. Meningococcal carriage by age: a systematic review and meta-analysis. Lancet Infect. Dis. 10, 853–861 (2010).
Criss, A. K. & Seifert, H. S. A bacterial siren song: intimate interactions between Neisseria and neutrophils. Nature Rev. Microbiol. 10, 178–190 (2012).
Carpenter, E. P. et al. AP endonuclease paralogues with distinct activities in DNA repair and bacterial pathogenesis. EMBO J. 26, 1363–1372 (2007). This study demonstrates that the two distinct N. meningitidis AP endonuclease paralogues differ in functional activity owing to a single polymorphism.
Nagorska, K. et al. A network of enzymes involved in repair of oxidative DNA damage in Neisseria meningitidis. Mol. Microbiol. 83, 1064–1079 (2012). This study highlights that BER in N. meningitidis consists of a flexible network of enzymes with both overlapping and distinct activities.
Morelle, S., Carbonnelle, E., Matic, I. & Nassif, X. Contact with host cells induces a DNA repair system in pathogenic Neisseriae. Mol. Microbiol. 55, 853–861 (2005).
Davidsen, T. & Tønjum, T. Meningococcal genome dynamics. Nature Rev. Microbiol. 4, 11–22 (2006).
Tibballs, K. L. et al. Characterization of the meningococcal DNA glycosylase Fpg involved in base excision repair. BMC Microbiol. 9, 7 (2009).
Silhan, J. et al. Specialization of an Exonuclease III family enzyme in the repair of 3′ DNA lesions during base excision repair in the human pathogen Neisseria meningitidis. Nucleic Acids Res. 40, 2065–2075 (2012).
Demple, B. & Harrison, L. Repair of oxidative damage to DNA: enzymology and biology. Annu. Rev. Biochem. 63, 915–948 (1994).
Davidsen, T., Tuven, H. K., Bjoras, M., Rodland, E. A. & Tonjum, T. Genetic interactions of DNA repair pathways in the pathogen Neisseria meningitidis. J. Bacteriol. 189, 5728–5737 (2007). This study provides evidence for interactions between DNA repair pathways in N. meningitidis.
Davidsen, T., Bjoras, M., Seeberg, E. C. & Tonjum, T. Antimutator role of DNA glycosylase MutY in pathogenic Neisseria species. J. Bacteriol. 187, 2801–2809 (2005).
Sanders, L. H., Sudhakaran, J. & Sutton, M. D. The GO system prevents ROS-induced mutagenesis and killing in Pseudomonas aeruginosa. FEMS Microbiol. Lett. 294, 89–96 (2009).
LeCuyer, B. E., Criss, A. K. & Seifert, H. S. Genetic characterization of the nucleotide excision repair system of Neisseria gonorrhoeae. J. Bacteriol. 192, 665–673 (2010).
Fischer, W. & Haas, R. The RecA protein of Helicobacter pylori requires a posttranslational modification for full activity. J. Bacteriol. 186, 777–784 (2004).
Orillard, E., Radicella, J. P. & Marsin, S. Biochemical and cellular characterization of Helicobacter pylori RecA, a protein with high-level constitutive expression. J. Bacteriol. 193, 6490–6497 (2011).
Yesilkaya, H., Andisi, V. F., Andrew, P. W. & Bijlsma, J. J. Streptococcus pneumoniae and reactive oxygen species: an unusual approach to living with radicals. Trends Microbiol. 21, 187–195 (2013).
Ambur, O. H. et al. Genome dynamics in major bacterial pathogens. FEMS Microbiol. Rev. 33, 453–470 (2009).
Lesterlin, C., Ball, G., Schermelleh, L. & Sherratt, D. J. RecA bundles mediate homology pairing between distant sisters during DNA break repair. Nature 506, 249–253 (2014).
Pitcher, R. S. et al. NHEJ protects mycobacteria in stationary phase against the harmful effects of desiccation. DNA Repair (Amst.) 6, 1271–1276 (2007).
Prieto, A. I., Ramos-Morales, F. & Casadesus, J. Repair of DNA damage induced by bile salts in Salmonella enterica. Genetics 174, 575–584 (2006).
Thompson, S. A. & Blaser, M. J. Isolation of the Helicobacter pylori recA gene and involvement of the recA region in resistance to low pH. Infect. Immun. 63, 2185–2193 (1995).
Thompson, S. A., Latch, R. L. & Blaser, J. M. Molecular characterization of the Helicobacter pylori uvr B gene. Gene 209, 113–122 (1998).
Yi, C. & He, C. DNA repair by reversal of DNA damage. Cold Spring Harb. Perspect. Biol. 5, a012575 (2013).
Krokan, H. E., Standal, R. & Slupphaug, G. DNA glycosylases in the base excision repair of DNA. Biochem. J. 325, 1–16 (1997).
Friedman, J. I. & Stivers, J. T. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry 49, 4957–4967 (2010).
Berti, P. J. & McCann, J. A. Toward a detailed understanding of base excision repair enzymes: transition state and mechanistic analyses of N-glycoside hydrolysis and N-glycoside transfer. Chem. Rev. 106, 506–555 (2006).
Mol, C. D., Hosfield, D. J. & Tainer, J. A. Abasic site recognition by two apurinic/apyrimidinic endonuclease families in DNA base excision repair: the 3′ ends justify the means. Mutat. Res. 460, 211–229 (2000).
Kisker, C., Kuper, J. & Van Houten, B. Prokaryotic nucleotide excision repair. Cold Spring Harb. Perspect. Biol. 5, a012591 (2013).
Deaconescu, A. M. et al. Structural basis for bacterial transcription-coupled DNA repair. Cell 124, 507–520 (2006).
Jiricny, J. Postreplicative mismatch repair. Cold Spring Harb. Perspect. Biol. 5, a012633 (2013).
Ayora, S. et al. Double-strand break repair in bacteria: a view from Bacillus subtilis. FEMS Microbiol. Rev. 35, 1055–1081 (2011).
Jasin, M. & Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 5, a012740 (2013).
Pitcher, R. S., Brissett, N. C. & Doherty, A. J. Nonhomologous end-joining in bacteria: a microbial perspective. Annu. Rev. Microbiol. 61, 259–282 (2007).
Nohmi, T. Environmental stress and lesion-bypass DNA polymerases. Annu. Rev. Microbiol. 60, 231–253 (2006).
Acknowledgements
Work in C.M.T.'s laboratory is supported by a Wellcome Trust Senior Investigator Award, the Medical Research Council and Action Medical Research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Apurinic or apyrimidinic (AP) sites
-
Regions of the DNA that lack nucleobases, generally as a result of DNA damage, spontaneous loss or inefficient DNA repair.
- Reactive oxygen species
-
(ROS). Reactive molecules that contain oxygen (such as superoxide, hydrogen peroxide and hydroxyl radicals), which damage macromolecules such as DNA, proteins and lipids.
- Reactive nitrogen species
-
(RNS). Nitrogen-containing oxides, such as peroxynitrite and nitric oxide radicals, which are highly reactive molecules that damage DNA and proteins through oxidation and nitrosation reactions.
- Phagocytosis
-
The mechanism by which phagocytes, such as macrophages and neutrophils, engulf and destroy microorganisms, foreign material and cellular debris.
- Respiratory burst
-
Rapid release of reactive oxygen species and reactive nitrogen species from immune cells, such as macrophages and neutrophils, to attack invading microorganisms. It is initiated by the NADPH-dependent oxidase that converts oxygen into superoxide, which subsequently reacts with other molecules such as nitric oxide to form the highly reactive oxidants peroxynitrite and hydroxyl radials.
- Activated neutrophils and macrophages
-
Neutrophils and macrophages that undergo morphological changes following triggers (such as cytokines) and that are able to extend pseudopods that assist phagocytosis. In addition, they can rapidly release reactive oxygen species and reactive nitrogen species in a 'respiratory burst', thereby killing engulfed microorganisms.
- Superoxide anions
-
(O2−). A common reactive form of oxygen that is generated when molecular oxygen gains an electron. It is a common intermediate in biological processes and is generated by phagocytes to kill microorganisms in a 'respiratory burst'.
- Phagosome
-
Intracellular compartment of a phagocyte that contains phagocytised microorganisms, foreign material or cellular debris.
- Chronic granulomatous disease
-
(CGD). A genetic deficiency of phagocyte oxidase components that is characterized by recurrent infections.
- Hydroxyl radicals
-
(OH•). Highly reactive oxygen-containing molecules that cause severe damage to macromolecules. They are produced by phagocytes through the conversion of water into superoxide and hydrogen peroxide to kill microorganisms in a 'respiratory burst'.
- Fenton reaction
-
Reaction between hydrogen peroxide and iron salts, mostly via iron–sulfur protein clusters, which generates hydroxyl radicals.
- Peroxynitrite
-
(ONOO−). A highly reactive molecule that damages macromolecules, such as DNA and proteins, through oxidation and nitrosation reactions. In phagocytes, it is produced during the 'respiratory burst' through the reaction of superoxide with nitric oxide.
- Deamination
-
Removal of an amine group from nucleobases, amino acids or other molecules.
- Alkylation
-
Transfer of alkyl groups, such as a methyl group or chains with more carbons, between molecules.
- β-elimination
-
Cleavage of the DNA backbone at the 3′ end of an apurinic or apyrimidinic site by a bifunctional DNA glycosylase after removal of a damaged nucleobase. Cleavage produces a 3′-unsaturated aldehyde and a 5′-phosphate group.
- γ-elimination
-
Cleavage of the DNA backbone at the 5′ end of an apurinic or apyrimidinic (AP) site by a bifunctional DNA glycosylase. Cleavage removes the AP site and leaves a phosphate group at both termini.
- SOS response
-
An inducible pathway in bacteria that is activated on the accumulation of single-stranded DNA as a result of DNA damage or the collapse of replication forks. This response typically involves DNA repair proteins and translesion DNA polymerases.
- UvrABC complex
-
A multicomponent enzyme complex that has endonucleolytic activity and is involved in the nucleotide excision repair pathway.
- Holliday junction
-
An assembly of four DNA strands that forms during certain types of genetic recombination.
- Hypoxanthine
-
A deamination product of adenine that is highly mutagenic owing to its ability to base-pair with cytosine and adenine during replication.
- Ethenocytosine
-
A highly mutagenic adduct that is formed by the alkylation of cytosine.
- Phase variation
-
A heritable change in the level of expression of a protein. It often occurs through an alteration in repetitive DNA sequences.
Rights and permissions
About this article

Cite this article
van der Veen, S., Tang, C. The BER necessities: the repair of DNA damage in human-adapted bacterial pathogens. Nat Rev Microbiol 13, 83–94 (2015). https://doi.org/10.1038/nrmicro3391
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro3391
This article is cited by
-
Bacterial DNA excision repair pathways
Nature Reviews Microbiology (2022)
-
ROS induces NETosis by oxidizing DNA and initiating DNA repair
Cell Death Discovery (2021)
-
Genome instability in pathogenesis of tuberculosis
Genome Instability & Disease (2021)
-
“Gene accordions” cause genotypic and phenotypic heterogeneity in clonal populations of Staphylococcus aureus
Nature Communications (2020)
-
Suicide inactivation of the uracil DNA glycosylase UdgX by covalent complex formation
Nature Chemical Biology (2019)
