
Key Points
-
Antibiotics have substantial direct and indirect effects on the intestinal microbiota; although some of the changes disappear soon after cessation of the antibiotic treatment, other changes remain indefinitely. Owing to their substantial effects on the microbiota, antibiotics have been used experimentally to identify groups of bacteria that correlate with changes in immunity and disease susceptibility.
-
Throughout the course of evolution, the host has become dependent on its microbiota for many functions, including the maintenance of intestinal immune homeostasis. Changes in the microbiota that are induced by antibiotic treatment and are important to immunity include the loss of bacterial ligands that are recognized by the host, changes in the metabolites that are produced by the microbiota and the loss of specific bacterial signals.
-
Studies using antibiotics have revealed that the microbiota not only contributes to development, but also is required for the maintenance of immune function. Many changes in immunity have been observed in response to antibiotics, including reduced secretion of antimicrobial peptides and changes in T helper cell populations. Changes in immunity that result from antibiotic treatment result in increased susceptibility to infection.
-
Antibiotics have been used to implicate the gastrointestinal microbiota in a number of systemic biological processes, including elements of systemic immunity and extra-intestinal disease susceptibility. Antibiotics have linked the intestinal microbiota to models of allergies, autism, metabolic syndrome and multiple sclerosis.
-
There are several challenges that need to be considered when using antibiotics to study host–microbial interactions. Direct effects on the immune system, reproducibility with respect to the duration and number of antibiotic exposures, antibiotic resistance and individualized responses to the same treatment all influence the outcome of antibiotic studies. Currently, most antibiotic studies remain correlative owing to the lack of targeted approaches available.
-
There is a need to develop strategies to mitigate the effects of antibiotics on the immune system. Promising approaches include bacteriotherapy and exogenous immune activation with bacterial ligands.
Abstract
Antibiotics have been used effectively as a means to treat bacterial infections in humans and animals for over half a century. However, through their use, lasting alterations are being made to a mutualistic relationship that has taken millennia to evolve: the relationship between the host and its microbiota. Host–microbiota interactions are dynamic; therefore, changes in the microbiota as a consequence of antibiotic treatment can result in the dysregulation of host immune homeostasis and an increased susceptibility to disease. A better understanding of both the changes in the microbiota as a result of antibiotic treatment and the consequential changes in host immune homeostasis is imperative, so that these effects can be mitigated.
Similar content being viewed by others
Main
The term microbiota refers to an abundant and diverse population of bacteria, archaea and fungi that reside in body sites that are exposed to the environment, with the highest concentrations of these organisms being found in the gastrointestinal tract. These mutualists contribute substantially to host phenotype, so extensively, in fact, that mammals have been described as 'superorganisms', and terms such as 'ecological development' have been coined to signify that development is a product of both host genetics and interactions with the microbiota1. With recent advances in our understanding of the intestinal microbiota and its role in regulating immunity, we are becoming increasingly aware of the effects that antibiotics have on the resident microbial ecosystem and how they can adversely affect patient health. One of the most striking examples is antibiotic-associated diarrhoea (AAD) caused by the expansion of Clostridium difficile, and subsequent pseudomembranous colitis, which can result in death2.
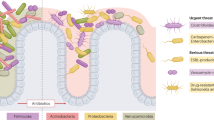
Interactions between microorganisms and the host direct the immune system locally and systemically3. Animal models demonstrate that antibiotic treatments affect both the innate and adaptive immune systems dynamically, affecting more than just immune development. Therefore, disturbances in a healthy adult microbiota can create opportunities for pathogens to infect and increase disease severity4,5,6. Antibiotics also provide excellent tools to study the role of the microbiota in protecting from and contributing to intestinal and extra-intestinal diseases, including gastroenteritis induced by Salmonella enterica subsp. enterica serovar Typhimurium, multiple sclerosis, diabetes, autism and atopic diseases7,8,9,10,11 (Fig. 1; Table 1).

Several associations between oral antibiotics and intra- or extra-intestinal diseases (indicated in the figure) have been made in clinical studies, whereas other such associations have only been made in experimental models to date. CDAD, Clostridium difficile-associated diarrhoea; EAE, experimental autoimmune encephalopathy; T1D, type 1 diabetes; T2D, type 2 diabetes; VRE, vancomycin-resistant Enterococcus.
The microbiota protects the host from incoming pathogens through various mechanisms, including competition for space or binding sites, competition for nutrients by bacteria with similar nutrient requirements, and direct inhibition through the release of inhibitory molecules (reviewed in Ref. 12). However, the focus of this Review is on how antibiotics affect the microbiota, how these changes affect immune homeostasis and how loss of immune homeostasis creates susceptibility to disease. Although the focus here is on negative impacts on the microbiota and the host, antibiotics will continue to be important tools in the battle against infectious disease. Therefore, strategies are required to allow mucosal and immunological integrity to be maintained when antibiotics are administered. New therapeutic approaches towards this aim include faecal bacteriotherapy from healthy donors following antibiotic treatment, and exogenous immune activation during the course of antibiotic therapy13,14.
Impact of antibiotics on the microbiota
To understand the consequence of antibiotics on immune homeostasis, we must first understand how they affect the intestinal microbial ecosystem. Antibiotics are selectively administered to target a specific pathogenic population. However, most antibiotics produced have broad-spectrum activity so that they can be used to treat many diseases15. Therefore, although antibiotics are designed to target pathogenic organisms, related members of the microbiota are also affected, often leaving an imprint on the gut community long after the antibiotics have been removed16,17. Antibiotic use also promotes the expansion of antibiotic-resistant strains, which can act as a lasting reservoir for resistance genes in the gut microenvironment18. Antibiotic treatment is typically followed by a decrease in the diversity of the microbiota17, but the community is resilient and resembles the pretreatment state in a matter of days or weeks19,20. Although most of the microbiota return to pretreatment levels, some members do not and are lost from the community indefinitely16,17.
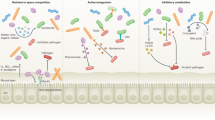
The microorganisms that are affected by antibiotic therapy are not limited to those that are directly targeted by the antibiotic. A complex network of co-dependence exists among members of the microbiota, driven by differential metabolite production and utilization pathways. These ecological interactions have been demonstrated in colonization models involving prominent members of the intestinal microbiota21. Microorganisms can be dependent on other colonizers for the provision of nutrients or secondary metabolites22, or for the removal of waste products that become toxic if they build up. For example, because of their co-dependence, the archaeon Methanobrevibacter smithii and the bacterium Bacteroides thetaiotaomicron were able to colonize germ-free mice more effectively when they were introduced together than when they were introduced alone21. Therefore, antibiotics may target specific bacteria, but other species that exchange secondary metabolites or waste products with the targeted species are indirectly affected (Fig. 2). This phenomenon was seen for treatment with vancomycin, after which some Gram-negative populations were dramatically depleted, despite the antimicrobial activity of the antibiotic being restricted to Gram-positive organisms23.

a | Mutualism exists between gut symbionts and the host in the absence of antibiotics. Co-dependence among symbionts is exemplified by the relationships shown in the intestinal lumen in the figure. A polysaccharide utilizer acquires a primary food source from the environment, obtains energy through the metabolism of that food source and produces secondary metabolites as by-products into the surrounding environment. These secondary metabolites are used as fuel by neighbouring symbionts (secondary-metabolite utilizers) that are unable to break down complex primary food sources. The secondary-metabolite utilizers produce waste products, such as H2, that are taken up by waste removers, which metabolize these waste products to make their own energy (for example, through methanogenesis). b | When antibiotics are introduced, mutualistic traits of the symbionts that are directly targeted (solid crosses) by the antibiotic are eliminated, and the microorganisms that are involved in those mutualistic associations are indirectly affected (dashed crosses). As an example, the loss of a targeted secondary-metabolite utilizer could promote the build-up of toxic secondary metabolites and/or eliminate a nutrient source for a waste remover. IEC, intestinal epithelial cell.
Antibiotics shift the composition of the microbiota in different ways depending on their spectrum of activity, a factor that can be useful in identifying which bacterial groups correlate to disease susceptibility or changes in host immunity. These antimicrobials also have different effects on diversity, bacterial biomass and the ability of the microbiota to recover post-treatment24. Although the spectrum of the antibiotic is paramount, the administrated dose is also extremely important for determining the ecological impact of a drug on the microbiota. Sub-therapeutic doses of antibiotics are used in the agriculture industry both prophylactically and to promote animal growth. Antibiotic growth promoters have been criticized because, although they do not reduce the total bacterial biomass, they shift the composition of the microbiota and promote dangerous levels of antibiotic resistance25. In piglets that are given amoxicillin 1 day after birth, the microbiota remains substantially altered 5 weeks after treatment, further emphasizing the long-term effects of antibiotics on gut homeostasis26. The therapeutic doses that are used in clinical practice are designed to minimize the effects on the host while maximizing pathogen clearance. These doses do not typically reduce the total bacterial biomass in the gut, but the elimination of subsets of the microbiota can shift the community in ways that promote colonization by opportunistic pathogens such as C. difficile27 and Candida albicans28. It should be noted that effects on the intestinal microbiota and mucosal immunity are not limited to orally administered antibiotics29. Systemically delivered antibiotics can have impacts on the intestinal microbiota, as they can reach the gut through the biliary system30. Even antibiotics that are secreted by the biliary system in only low amounts have been shown to have effects on the gut microbiota31. Antibiotic doses above therapeutic levels are used primarily for research purposes in which the depletion of the total bacterial biomass is required to test for a role of the microbiota.
Why antibiotics affect the host
Through the course of mammalian evolution, the microbiota has been selected to provide a vast number of services to the host, ranging from the provision of nutrients to the regulation of immune function. Host–microorganism interactions are surprisingly specific, as demonstrated by the inability of microorganisms from the human gut to appropriately regulate immune development of the mouse32,33. These specific interactions, which are highly co-evolved processes, are altered or lost as a consequence of antibiotic treatment, leading to effects on host health. Changes that are induced by antibiotic treatment and that are important to microbial regulation of host immunity include the loss of bacterial ligands that are recognized by the host, alterations in the metabolites produced by the microbiota and the loss of specific bacterial signals.
Loss of bacterial ligands that are important for immune induction. An important mechanism through which the microbiota affects host immunity is the activation of microorganism-associated molecular pattern (MAMP) recognition receptors, including Toll-like receptors (TLRs) and NOD-like receptors (NLRs). These pattern recognition receptors (PRRs) are activated by ligands from intestinal microorganisms, including lipopolysaccharide (LPS), lipoteichoic acid (LTA), flagellin, CpG DNA and peptidoglycan34. PRR signalling has been shown to be important in a range of homeostatic mechanisms that involve epithelial integrity and repair, as well as mucosal immune functions (reviewed in Ref. 35). Depletion of the microbiota using antibiotics certainly reduces the amount of MAMPs coming into contact with the epithelium; antibiotics targeting Gram-negative bacteria would primarily affect TLR4 and nucleotide-binding oligomerization domain-containing protein 1 (NOD1) activation, whereas antibiotics targeting Gram-positive bacteria would primarily affect TLR2 and NOD2 activation. Of note, not all LPS or LTA is equal in its ability to stimulate host responses36. A number of studies have shown that depletion of the microbiota as a consequence of antibiotic treatment results in reduced TLR signalling and downstream regulation of innate defences5,37.
Changes in bacterial metabolites. As well as having different abilities in terms of activating PRRs, each population of bacteria carries out specific metabolic processes. Correspondingly, the composition of the microbiota has been shown to correlate with both faecal and urinary metabolites38,39. Thus, antibiotic treatment results in changes in the metabolic profiles of intestinal contents and faeces, as well as those of the blood and urine (following absorption of metabolites from the gut). Metabolic profiles have been studied in mice under various antibiotic regimens40,41. A common feature of the metabolic profiles of antibiotic-treated mice and humans is a reduced production of short-chain fatty acids (SCFAs)40,41,42,43. Two independent metabolomic studies of enrofloxacin- or vancomycin-treated mice showed decreased concentrations of amino acids and SCFAs in faeces from treated mice compared with concentrations in faeces from untreated mice, whereas oligosaccharide levels were increased in faeces from treated mice, indicating that microbial fermentation of carbohydrates was disrupted41. Although vancomycin had a dramatic effect on Clostridium leptum and Clostridium coccoides clusters (important butyrate-producing bacteria), these populations recovered 2–3 days post-treatment, coinciding with a recovery of fatty acid levels by day 7. In agreement with observations that some populations of bacteria do not recover after antibiotic treatment, levels of some metabolites had not recovered weeks after the cessation of antibiotic treatment41.
The depletion of SCFAs following antibiotic treatment is important with regard to intestinal health and immunity. SCFAs are rapidly absorbed in the colon, providing a preferred energy source for colonocytes and regulating cell proliferation, differentiation, growth and apoptosis44,45,46. Butyrate is the SCFA with the strongest effect on the cell cycle and has also been implicated in regulating many aspects of intestinal immunity. Effects on mucosal defence include improved tight junction assembly, antimicrobial secretion and mucin expression47,48,49. Most studies have described butyrate as anti-inflammatory in the gut; for example, it has been shown to inhibit the constitutive expression of monocyte chemotactic protein 1 (MCP1; also known as CCL2) in a dose-dependent manner, both in the absence and in the presence of LPS50, and to reduce neutrophil production of reactive oxygen species51. These anti-inflammatory properties are generally considered to be beneficial, preventing overt inflammation, oxidative stress and carcinogenesis52. Butyrate's anti-inflammatory effects on neutrophils may contribute to the increased infiltration of neutrophils that is observed in mice that are treated with streptomycin and then infected with S. Typhimurium, and could result in an increased inflammatory state in neutrophil-associated diseases. One mechanism by which butyrate acts is to induce peripheral blood mononuclear cells to produce prostaglandin E2, a prostanoid that reduces the LPS-mediated induction of tumour necrosis factor (TNF) expression53. Intriguingly, reduced concentrations of prostaglandin E2 metabolites have been observed in patients with Crohn's disease, along with a disrupted microbiota, reduced butyrate production and uncontrolled intestinal inflammation39,54. However, the effects of butyrate can be cell specific; therefore, caution should be used when interpreting the results of such studies. For example, the effect of butyrate on cathelicidin expression and antimicrobial activity is specific to colonocytes, as the molecule does not have the same effect on keratinocytes or monocytes48. Butyrate has also been shown to increase the expression of NOD2, resulting in an increased induction of interleukin-8 (IL-8) following NOD2 activation by peptidoglycan in intestinal epithelial cells55. As well as playing an important part in regulating mucosal immunity, SCFAs also regulate T cell anergy, identifying these cells as targets for systemic and obesity-associated inflammation56. Taken together, the data indicate an important anti-inflammatory role for butyrate, suggesting that the reduced butyrate levels seen during antibiotic treatment may contribute to increased inflammation following pathogen insult.
Butyrate is certainly not the only microbial metabolite that affects the host. However, in general, the metabolites produced by the microbiota are poorly understood. In a study examining metabolites in the faeces of patients with Crohn's disease, most (∼90%) of the metabolites that correlated with disease could not be assigned to any metabolic pathway, indicating that the established databases are incomplete39. Many of these unknown metabolites probably have important roles in regulating host physiology and mucosal immune homeostasis.
Loss of specific bacterial signals. The loss of specific bacteria also means the loss of specific signals that are associated with those bacteria. For example, although the process is not yet well understood, the population of segmented filamentous bacteria (SFB) is particularly adapted to induce the differentiation of T helper 17 (TH17) cells6. Other specific microbial signals are involved in inhibiting inflammation. For example, B. thetaiotaomicron limited the inflammation induced by Salmonella enterica subsp. enterica serovar Enteritidis by activating nuclear export of nuclear factor-κB (NF-κB) through a peroxisome proliferator-activated receptor-γ (PPARγ)-dependent pathway57. Saccharomyces boulardii reduced host inflammatory responses by secreting a heat-stable, water-soluble factor that blocked the degradation of NF-κB inhibitor-α (IκBα), thus reducing NF-κB binding to DNA and subsequent IL-8 expression58. Polysaccharide A, a component of Bacteroides fragilis, is able to induce FOXP3+ regulatory T cells to produce IL-10, preventing inflammation in an experimental model of infectious colitis59. It is expected that many more microorganism-specific signals remain uncharacterized.
Antibiotic-related changes in immunity
The microbiota has many key roles in preparing the mucosal immune system for battle against infection. Germ-free animals have poorly developed lymphoid tissue, reduced and imbalanced T cell populations, increased intestinal permeability, and reduced expression of antimicrobial peptides (AMPs) and secretory immunoglobulin A60. Although germ-free models have provided interesting contributions to our understanding of the role of the microbiota in immune development, these models do not address how changes in the microbiota later in the life of the host may affect host immunity. Studies using antibiotics have revealed that the microbiota not only contributes to development, but also is required for maintenance of the immune system (Table 2).
Many studies report reduced expression and secretion of AMPs on antibiotic treatment. AMPs, including C-type lectins, defensins and cathelicidins, are the first line of defence to incoming pathogens, as they are secreted into and maintained in the mucus layer61. AMPs have direct antimicrobial activity and are variably expressed between intestinal cell types; however, enterocytes, goblet cells and particularly Paneth cells all contribute to their production. Treatment with a combination of vancomycin, neomycin and metronidazole resulted in reduced expression of the gene encoding REG3γ, a C-type lectin that targets Gram-positive bacteria62. The expression of this gene could be corrected by administering LPS from Gram-negative bacteria, but not LTA from Gram-positive bacteria5. Conversely, treatment of mice with streptomycin has been shown to affect transcription of the mRNA encoding REG3β37. The depletion of this transcript, and of the protein itself, seemed to be caused by reduced microbial activation of TLR2, as adding a TLR2 agonist resulted in recovery of expression37.
Global gene analysis revealed that amoxicillin treatment also resulted in decreased expression of genes involved in antimicrobial defence, including those encoding α-defensins, matrilysin and phospholipase A2 (Ref. 63). Although treatment with amoxicillin had no effect on total bacterial numbers in the small intestine, it nearly eradicated the lactobacilli. It should be noted that host responses differ depending on the location examined. Of the hundreds of genes affected by amoxicillin treatment, very few showed overlapping changes in the proximal small intestine, distal small intestine and large intestine. The only genes that were globally downregulated were those involved in antigen presentation, including those encoding major histocompatibility complex (MHC) class II and MHC class Ib complexes. Importantly, MHC class II complexes are required for tolerization to luminal antigens64.
Although amoxicillin treatment reduced the expression of genes related to antigen presentation and the secretion of defence molecules, this treatment increased the expression of mast cell proteases. In fact, increased numbers of mucosal mast cells have been associated with the administration of other antibiotics65. Mast cells play a part in host defence against pathogens66, but they also have a key role in chronic intestinal inflammation67. Treatment with antibiotics alone did not result in increased signs of inflammation; however, increased mast cell number and activity may predispose the host to exacerbated inflammatory reactions following insult with a pathogen.
Depleting specific subsets of the microbial population with antibiotics has provided clues as to which bacteria are responsible for regulating T cell differentiation and activation. The treatment of mice with vancomycin or ampicillin, which target Gram-positive bacteria, resulted in a depletion of TH17 cells in the intestine, whereas treatment with metronidazole and neomycin sulphate, which target strict anaerobes and Gram-negative bacteria, respectively, had no effect on the TH17 cell population4. It was this preliminary evidence that identified Gram-positive bacteria, and more recently SFB, as major contributors to this T cell phenotype6. It has also been shown that specific populations of mutualistic bacteria — not including those obligate anaerobes that are affected by metronidazole treatment — are required for maintaining IL-17 production by γδ T cells68.
The reduced frequency of CD4+ T cells expressing interferon-γ (IFNγ) or IL-17A following a 10-day antibiotic treatment period (using a combination of ampicillin, gentamicin, metronidazole, neomycin and vancomycin) supports the role of continued microbial signalling in maintaining effector T cell populations in the gut69. As well as affecting T cell activity, this combination of antibiotics was found to reduce the production of resistin-like molecule-β (RELMβ), another important AMP produced by goblet cells69. The changes that were seen in the microbial population with this antibiotic treatment were extensive, but in general the species most strongly affected were members of the phylum Firmicutes, whereas certain members of the phyla Bacteroidetes and Proteobacteria were able to persist69.
As well as influencing antimicrobial defences, antibiotics can affect the activity of immune cells. The eradication of enteric bacteria from 8-week-old mice after 3 weeks of antibiotic administration has recently been shown to reduce the expression of TLR2 and TLR4 in peritoneal macrophages70. This reduced expression of TLRs resulted in a markedly impaired response to challenge with LPS, indicating that continued mucosal stimulation by enteric bacteria is required for the priming of macrophages in preparation for invasion70. How this phenomenon is affected by short-term antibiotic therapy or more clinical doses has yet to be shown.
Mucosal imbalance leads to susceptibility
Changes in immunity, such as those that occur as a consequence of antibiotic treatment, result in increased susceptibility to infection by pathogens (Fig. 3). Antimicrobial secretions have been shown to be very important for impeding colonization of enteric pathogens5,71. The disruption of mucus layer integrity, as is seen in mucin 2 (Muc2)-knockout mice, allows bacteria to contact the epithelium and, indeed, results in increased attachment of the enteric pathogen Citrobacter rodentium72,73. Treatment with metronidazole — an antibiotic that targets anaerobic bacteria — affects the expression of MUC2, reduces the integrity of the mucous layer and accelerates mucosal attachment of C. rodentium74. Intriguingly, streptomycin treatment did not result in reduced MUC2 expression, thus implying that bacteria which are obligate anaerobes (which are targeted by metronidazole but not streptomycin) are involved in triggering MUC2 expression. Treatment of rats with metronidazole also resulted in modifications in the mucus layer, resulting in greater microbial colonization of intestinal crypts75.

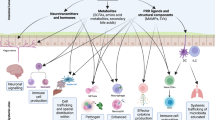
a | In the absence of antibiotics, epithelial homeostasis is maintained. Microorganism-associated molecular patterns (MAMPs) from the microbiota are sensed intracellularly and extracellularly by pattern recognition receptors (PRRs) (that is, Toll-like receptors and NOD-like receptors). PRRs that recognize MAMPs from the microbiota instruct intestinal epithelial cells (IECs) called Paneth cells to secrete antimicrobial peptides (AMPs; for example, C-type lectins, defensins and cathelicidins) into the adjacent mucus layer. Secretory immunoglobulin A (IgA) and AMPs are important for controlling bacterial numbers and maintaining epithelial homeostasis. Tight junctions and a thick mucus layer prevent incoming pathogens from damaging the mucosal barrier. b | Antibiotic exposure disrupts epithelial homeostasis. Subsets of the normal microbiota are lost, thus reducing epithelial exposure to microbiota-derived MAMPs. Fewer MAMPs result in decreased host–microbiota signalling (that is, decreased PRR stimulation), which leads to decreased AMP production by IECs. Some antibiotics cause thinning of the mucus layer and disruption of tight junctions, rendering the host more susceptible to damage to the mucosal barrier, caused by incoming pathogens. The loss of AMP production and of the integrity of the epithelial cell barrier facilitates successful invasion by enteric pathogens.
Vancomycin-resistant Enterococcus (VRE) was also able to colonize more effectively in mice treated with antibiotics. Increased colonization was not a result of reduced competition, but was instead due to a reduced production of the antimicrobial peptide REG3γ, as a consequence of depleted microbial signals5. A recent study of patients undergoing allogeneic haematopoietic stem cell transplantation also revealed that antibiotic treatment enabled VRE strains to dominate the microbiota, an event shown to precede bacterial invasion into the bloodstream76. It is likely that parallels can be drawn between infection with VRE and with C. difficile. Antibiotic-induced susceptibility to C. difficile has been reproduced in a mouse model, providing a means of identifying how changes in mucosal immunity correlate with disease77. However, it has yet to be determined whether host resistance to colonization by C. difficile is a product of changes in host immunity, or due to direct inhibition or competition from the microbiota. Certainly, immunological changes would be expected, as demonstrated in certain cases of AAD: changes in the composition of the microbiota in individuals with AAD have revealed a marked decrease in the prevalence of butyrate-producing bacteria78.
It has recently been shown that S. Typhimurium uses gut inflammation to its competitive advantage when colonizing the gut79. Specifically, reactive oxygen species produced on initial pathogen insult react with luminal sulphur compounds to form tetrathionate, a respiratory electron acceptor that is used by S. Typhimurium to gain a competitive advantage80. Interestingly, the inflammation that is activated on initial infection and required for S. Typhimurium to expand and cause colitis does not occur in mice unless they are pretreated with antibiotics10. Probiotics have been shown to prevent colitis by inhibiting P-selectin upregulation and, thus, reducing the interactions between endothelial cells and both leukocytes and platelets81. Therefore, although this has not yet been shown experimentally, one might speculate that the loss of microbiota-produced anti-inflammatory signals that is known to occur on antibiotic treatment (as discussed above for butyrate) is involved in the increased inflammation that is seen on infection with S. Typhimurium. The high-dose streptomycin pretreatment model used in S. Typhimurium-induced colitis suggests that the microbiota as a whole was inhibiting S. Typhimurium colonization in the gut82. However, low-dose administration of streptomycin and vancomycin also enabled colonization by S. Typhimurium10, suggesting that different subsets of the microbiota are involved in modulating susceptibility to S. Typhimurium infection in the intestine. High-dose streptomycin treatment was also found to cause alterations in the microbiota that persisted for several weeks and resulted in continued susceptibility to S. Typhimurium-induced gastroenteritis83.
Antibiotic-mediated depletion of bacteria has also been shown to affect the development of an adaptive immune response. Reducing the amount of CpG DNA (a bacterial MAMP) resulted in suppressed immune responses to oral vaccination with an antigen and to oral infection with a protozoan parasite84. Combined treatment with amoxicillin and cluvulanic acid also resulted in reduced serum IgG levels, although IgA concentrations were unaffected85, suggesting that systemic effects occurred.
Systemic effects
We are now learning that germ-free mice also have several extra-intestinal deficits, indicating that the effects of the microbiota on the host — which were previously believed to be restricted to the gut — have systemic implications. Antibiotic-induced perturbation of the microbiota demonstrated that NOD1-mediated recognition of peptidoglycan from Gram-negative bacteria is required for the generation of adaptive lymphoid tissues86. Microbiota-derived peptidoglycan has been shown to interact with NOD1 (Ref. 3) and to therefore systemically prime the innate immune system. This priming occurs through a mechanism that involves peptidoglycan from the microbiota entering the circulation, binding NOD1 receptors in bone marrow-derived neutrophils and initiating a signalling cascade that enhances the ability of these neutrophils to kill microorganisms87. The concept of the microbiota affecting systemic immunity is not new: over 10 years ago, antibiotics were used to show that antigens from the microbiota were involved in regulating cell-mediated responses by priming CD4+ T cells to secrete IL-4 (in other words, to become TH2 polarized)88. This was observed even after challenge with Leishmania major, a pathogen requiring a TH1-type response for clearance.
Antibiotics have also been used to investigate the role of extra-intestinal diseases that are caused by gut-specific microorganisms. Over the past few decades, there has been an increase in the prevalence of immunological disorders (both of autoimmune and of atopic origin) in the developed world. These observations, in combination with causal relationships established by animal models and clinical studies, have provided increased support for the hygiene hypothesis89,90. Recent epidemiological studies have associated early antibiotic use with an increased risk of atopy91,92,93, encouraging an extension of the hygiene hypothesis, coined the 'microflora hypothesis'. This is the theory that reduced microbial exposure during childhood inhibits normal maturation of the intestinal microbiota, altering immune development in a way that enhances the symptoms of allergic hypersensitivity94. Although these studies remain correlative, antibiotic exposure in animal models of allergic-airways disease11,95 and atopic dermatitis96 support these findings; however, most of the mechanisms involved in these models remain undefined.
Although no studies have linked antibiotic exposure to autoimmunity in a clinical setting, experiments in animal models have provided some exciting insights. Oral antibiotic treatment has demonstrated the substantial impact of the microbiota on experimental autoimmune encephalomyelitis (EAE), the experimental model of multiple sclerosis. Depleting the intestinal microbiota with a cocktail of antibiotics impaired the development of EAE. Protection was accompanied by diminished pro-inflammatory responses and an accumulation of FOXP3+ regulatory T cells in the peripheral lymph nodes97. Interestingly, antibiotic-treated mice reconstituted with polysaccharide A-producing B. fragilis remained protected from EAE, whereas colonization of mice with a polysaccharide A-deficient mutant of B. fragilis restored EAE susceptibility. Recent data suggest that polysaccharide A enhances the conversion of naive CD4+ T cells into IL-10-producing FOXP3+ regulatory T cells through induction by tolerogenic CD103+ DCs, an effect that protects mice against EAE induction9,98 (Fig. 4).

Following antibiotic-mediated disruption of the intestinal microbiota, wild-type and polysaccharide A (PSA)-deficient Bacteroides fragilis were administered to mice. The wild-type bacteria protected against experimental autoimmune encephalopathy (EAE), but the PSA-deficient mutant did not9. The suggested mechanism of protection involves transport of the PSA antigen across the epithelial cell barrier, induction of tolerogenic dendritic cells (DCs) in the underlying lamina propria in the presence of epithelial cell-secreted cytokines, and subsequent transport of the tolerogenic DCs in the circulation (through an unknown mechanism) to cervical lymph nodes. In the cervical lymph nodes, tolerogenic DCs promote the induction of FOXP3+ regulatory T cells. The expansion of FOXP3+ regulatory T cells in the cervical lymph node corresponds with protection against demyelination of the central nervous system in EAE. TSLP, thymic stromal lymphopoetin; TGFβ, transforming growth factor-β; RA, retinoic acid.
Antibiotic studies have helped to identify a role for the intestinal microbiota in several complex multifactorial disorders, such as metabolic syndrome and autism. Interestingly, there is evidence to suggest that certain antibiotic regimens may actually improve insulin sensitivity in both animals8,99 and humans100. Associations between extensive antibiotic use and late-onset autism (which develops at 18–24 months of age) suggest that a reassembly of the microbiota may promote the overgrowth of microorganisms that trigger autistic behavioural patterns101. According to one study, oral vancomycin treatment actually reduces autistic symptoms in some patients, suggesting that certain gut microorganisms may influence the onset of this disorder102.
Challenges
Many interactions between host and microorganism require a functional immune system; therefore, an antibiotic approach is, in some ways, better than a gnotobiotic approach for understanding host–microbial interactions, as immune function in gnotobiotic animals is drastically different from that in conventional animals. Although this is an important limitation of the gnotobiotic system, using antibiotics to understand host–microbial interactions has many of its own limitations.
One of the major challenges associated with using antibiotics to study host–microbial interactions is the direct effect that antibiotics can have on the immune system. Macrolide antibiotics in particular have been described as having many non-antimicrobial effects on host physiology103. This class of antibiotics typically decreases the production of pro-inflammatory cytokines, including IL-1β, IL-8 and macrophage inflammatory protein, and decreases neutrophil infiltration104,105. The aminoglycoside gentamicin has been shown to have direct inhibitory effects on the production of TNF and MCP1 in proximal tubular cells, despite an increased accumulation of the mRNAs encoding these proteins106. Similarly, although not demonstrated as a direct effect in the gut, LPS induced greater transcription of TNF in mice treated with gentamicin. Concanamycin B has also been found to inhibit the acidification of endosomes and lysosomes in macrophages107. Certainly, this could affect the ability of macrophages to clear intracellular pathogens.
Reproducing antibiotic-induced alterations in the microbiota is also an important challenge. How the microbiota responds to antibiotic administration is largely dependent on the length of exposure and the number of repeated exposures108. It has been well documented that the number of antibiotic-resistance genes in the microbiota is dramatically increased following antibiotic exposure, causing a reorganization of the phylogenetic hierarchy of the community17,18. It is thought that repeated exposure to the same antibiotic fails to reproduce the same compositional shifts owing to the overgrowth of these resistant strains. There is also considerable variability in the composition of the microbiota among individuals, particularly at the genus and species levels109,110. As such, antibiotic treatment results in individualized responses to perturbation of the microbiota16. Similarly, different mouse strains111, or even the same mouse strain housed in a different animal facility, respond differently to the same disease model.
Because even narrow-spectrum antibiotics do not target a single species or even a single genus, identifying those changes in the microbiota that are associated with a subsequent downstream effect (that is, an immune response, or an altered susceptibility to a pathogen or disease) is challenging. Many antibiotic studies remain correlative because no direct cause-and-effect relationship has been identified.
Transplantation is an alternative to antibiotic methods of microbiota manipulation. However, reintroducing specific organisms into a host is challenging owing to the lack of cultured organisms available. Transfer models are being developed in which faeces or caecal contents can be used to inoculate a host with a particular community112; however, this involves the transfer of a complete microbiota rather than single organisms. Achieving efficient transfer is difficult, which is why many transplants are performed in germ-free mice. More effective, targeted approaches are needed to supplant the correlative data that has been accumulated using antibiotics.
Where do we go from here?
Antibiotics create stable, lasting alterations in the gut microbiota, weakening the fidelity of this community by disrupting the host–microorganism associations that have co-evolved to maximize mutual benefit. The loss of these mutualistic traits affects the remaining community members and their hosts across generations, shifting homeostatic mechanisms toward potentially unfavourable outcomes108. Although this Review focuses on the role of antibiotics in disrupting the homeostatic balance of immunity, it must be noted that antibiotics can also restore a homeostatic state in cases in which the host is unable to regulate the microbiota. A lack of immunoglobulin genes in the host results in a 100-fold expansion of the microbiota, resulting in hyperplasia of lymphoid follicles; however, antibiotic treatment corrects this response113.
With an increasing number of studies demonstrating that antibiotics have detrimental effects on the immediate and long-term health of the host, there is a push to develop treatments that are better targeted to specific infecting pathogens; the challenges are not limited to the development of these drugs, but also include methods for rapid, accurate diagnoses in the clinic15. Because eliminating the need for antibiotics is unrealistic at this time, a better understanding of how antibiotics impact the microbiota and host immunity is needed, so that strategies to mitigate these effects can be developed. Bacteriotherapy is rapidly becoming a recognized treatment for antibiotic-induced C. difficile-associated diarrhoea13. As some microorganisms do not return in the microbiota after antibiotic therapy, it might be prudent to collect faecal samples before treatment, to be used as a source of 'individualized' healthy microorganisms that could be administered post-treatment. Moreover, probiotic intervention strategies are being implemented following antibiotic therapy in infants with necrotizing enterocolitis (NEC)114, as well as in individuals being treated for AAD115 and bacterial vaginosis116. Activation of TLR4 seems to be one of the mechanisms involved in bacteria-mediated protection from diseases, including NEC117, suggesting that TLR agonists could be administered during antibiotic therapy to help restore immune homeostasis. Similarly, systemic administration of flagellin has been used to experimentally limit infection with VRE; through activation of TLR5, systemically administered flagellin restored antibiotic-impaired immune defences (specifically, REG3γ expression), drastically reducing colonization by VRE14. Although most of these approaches are still experimental, they hold substantial promise as adjunct therapies to antibiotics.
References
Hooper, L. V. Bacterial contributions to mammalian gut development. Trends Microbiol. 12, 129–134 (2004).
De La Cochetière, M. F. et al. Effect of antibiotic therapy on human fecal microbiota and the relation to the development of Clostridium difficile. Microb. Ecol. 56, 395–402 (2008).
Clarke, T. B. et al. Recognition of peptidoglycan from the microbiota by Nod1 enhances systemic innate immunity. Nature Med. 16, 228–231 (2010). This article is the first to provide a detailed mechanism of how antigens from the microbiota specifically direct immune cells at systemic sites.
Ivanov, II. et al. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349 (2008). This paper presents important evidence that the composition of the microbiota is critical for the induction of T H 17 cells.
Brandl, K. et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 455, 804–807 (2008). This work provides the first evidence that an increased susceptibility to enteric pathogens is the result of a reduced innate immune defence.
Ivanov, II. et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009).
Finegold, S. M. Therapy and epidemiology of autism–clostridial spores as key elements. Med. Hypotheses 70, 508–511 (2008).
Cani, P. D. et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481 (2008).
Ochoa-Reparaz, J. et al. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J. Immunol. 185, 4101–4108 (2010). This article is the latest and the most mechanistically detailed in a series of manuscripts detailing the involvement of microbial antigens in the development of EAE.
Sekirov, I. et al. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect. Immun. 76, 4726–4736 (2008).
Noverr, M. C., Falkowski, N. R., McDonald, R. A., McKenzie, A. N. & Huffnagle, G. B. Development of allergic airway disease in mice following antibiotic therapy and fungal microbiota increase: role of host genetics, antigen, and interleukin-13. Infect. Immun. 73, 30–38 (2005).
Stecher, B. & Hardt, W. D. Mechanisms controlling pathogen colonization of the gut. Curr. Opin. Microbiol. 14, 82–91 (2011).
Khoruts, A., Dicksved, J., Jansson, J. K. & Sadowsky, M. J. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J. Clin. Gastroenterol. 44, 354–360 (2010).
Kinnebrew, M. A. et al. Bacterial flagellin stimulates Toll-like receptor 5-dependent defence against vancomycin-resistant enterococcus infection. J. Infect. Dis. 201, 534–543 (2010). This paper presents a novel approach for mitigating the antibiotic-mediated defects in mucosal immunity using systemic delivery of bacterial flagellin.
Nathan, C. Antibiotics at the crossroads. Nature 431, 899–902 (2004).
Jakobsson, H. E. et al. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS ONE 5, e9836 (2010).
Jernberg, C., Lofmark, S., Edlund, C. & Jansson, J. K. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 1, 56–66 (2007).
Lofmark, S., Jernberg, C., Jansson, J. K. & Edlund, C. Clindamycin-induced enrichment and long-term persistence of resistant Bacteroides spp. and resistance genes. J. Antimicrob. Chemother. 58, 1160–1167 (2006).
Dethlefsen, L., Huse, S., Sogin, M. L. & Relman, D. A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6, e280 (2008).
De La Cochetière, M. F. et al. Resilience of the dominant human fecal microbiota upon short-course antibiotic challenge. J. Clin. Microbiol. 43, 5588–5592 (2005).
Samuel, B. S. & Gordon, J. I. A humanized gnotobiotic mouse model of host–archaeal–bacterial mutualism. Proc. Natl Acad. Sci. USA 103, 10011–10016 (2006).
Belenguer, A. et al. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl. Environ. Microbiol. 72, 3593–3599 (2006).
Robinson, C. J. & Young, V. B. Antibiotic administration alters the community structure of the gastrointestinal microbiota. Gut Microbes 1, 279–284 (2010).
Antonopoulos, D. A. et al. Reproducible community dynamics of the gastrointestinal microbiota following antibiotic perturbation. Infect. Immun. 77, 2367–2375 (2009).
Smith, D. L., Harris, A. D., Johnson, J. A., Silbergeld, E. K. & Morris, J. G. Jr. Animal antibiotic use has an early but important impact on the emergence of antibiotic resistance in human commensal bacteria. Proc. Natl Acad. Sci. USA 99, 6434–6439 (2002).
Janczyk, P. et al. Parenteral long-acting amoxicillin reduces intestinal bacterial community diversity in piglets even 5 weeks after the administration. ISME J. 1, 180–183 (2007).
Tedesco, F. J., Barton, R. W. & Alpers, D. H. Clindamycin-associated colitis. A prospective study. Ann. Intern. Med. 81, 429–433 (1974).
Sobel, J. D. Vulvovaginal candidosis. Lancet 369, 1961–1971 (2007).
Yaguchi, Y. et al. Influences of long-term antibiotic administration on Peyer's patch lymphocytes and mucosal immunoglobulin A levels in a mouse model. J. Parenter. Enteral Nutr. 30, 395–399 (2006).
Giuliano, M., Barza, M., Jacobus, N. V. & Gorbach, S. L. Effect of broad-spectrum parenteral antibiotics on composition of intestinal microflora of humans. Antimicrob. Agents Chemother. 31, 202–206 (1987).
Vanogtrop, M. L., Guiot, H. F. L., Mattie, H. & Vanfurth, R. Modulation of the intestinal flora of mice by parenteral treatment with broad-spectrum cephalosporins. Antimicrob. Agents Chemother. 35, 976–982 (1991).
Atarashi, K. et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341 (2011).
Comelli, E. M. et al. Multifaceted transcriptional regulation of the murine intestinal mucus layer by endogenous microbiota. Genomics 91, 70–77 (2008).
Wells, J. M., Loonen, L. M. & Karczewski, J. M. The role of innate signaling in the homeostasis of tolerance and immunity in the intestine. Int. J. Med. Microbiol. 300, 41–48 (2010).
Abreu, M. T. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nature Rev. Immunol. 10, 131–144 (2010).
Netea, M. G., van Deuren, M., Kullberg, B. J., Cavaillon, J. M. & Van der Meer, J. W. Does the shape of lipid A determine the interaction of LPS with Toll-like receptors? Trends Immunol. 23, 135–139 (2002).
Dessein, R. et al. Toll-like receptor 2 is critical for induction of Reg3β expression and intestinal clearance of Yersinia pseudotuberculosis. Gut 58, 771–776 (2009).
Li, M. et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl Acad. Sci. USA 105, 2117–2122 (2008).
Jansson, J. et al. Metabolomics reveals metabolic biomarkers of Crohn's disease. PLoS ONE 4, e6386 (2009).
Romick-Rosendale, L. E. et al. NMR-based metabonomics analysis of mouse urine and fecal extracts following oral treatment with the broad-spectrum antibiotic enrofloxacin (Baytril). Magn. Reson. Chem. 47, S36–S46 (2009).
Yap, I. K. et al. Metabonomic and microbiological analysis of the dynamic effect of vancomycin-induced gut microbiota modification in the mouse. J. Proteome Res. 7, 3718–3728 (2008).
Woodmansey, E. J., McMurdo, M. E. T., Macfarlane, G. T. & Macfarlane, S. Comparison of compositions and metabolic activities of fecal microbiotas in young adults and in antibiotic-treated and non-antibiotic-treated elderly subjects. Appl. Environ. Microbiol. 70, 6113–6122 (2004).
Hoverstad, T. et al. Influence of ampicillin, clindamycin, and metronidazole on faecal excretion of short-chain fatty acids in healthy subjects. Scand. J. Gastroenterol. 21, 621–626 (1986).
Millard, A. L. et al. Butyrate affects differentiation, maturation and function of human monocyte-derived dendritic cells and macrophages. Clin. Exp. Immunol. 130, 245–255 (2002).
Flavia, L. A. R. et al. Apoptosis induced by butyrate is independent of Jak/STAT signaling in a fibrosarcoma cell line. Biochem. Biophys. Res. Commun. 301, 968–973 (2003).
Hossain, Z., Konishi, M., Hosokawa, M. & Takahashi, K. Effect of polyunsaturated fatty acid-enriched phosphatidylcholine and phosphatidylserine on butyrate-induced growth inhibition, differentiation and apoptosis in Caco-2 cells. Cell Biochem. Funct. 24, 159–165 (2006).
Peng, L., Li, Z. R., Green, R. S., Holzman, I. R. & Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 139, 1619–1625 (2009).
Schauber, J., Dorschner, R. A., Yamasaki, K., Brouha, B. & Gallo, R. L. Control of the innate epithelial antimicrobial response is cell-type specific and dependent on relevant microenvironmentat stimuli. Immunology 118, 509–519 (2006).
Gaudier, E., Rival, M., Buisine, M. P., Robineau, I. & Hoebler, C. Butyrate enemas upregulate Muc genes expression but decrease adherent mucus thickness in mice colon. Physiol. Res. 58, 111–119 (2009).
Cox, M. A. et al. Short-chain fatty acids act as antiinflammatory mediators by regulating prostaglandin E-2 and cytokines. World J. Gastroenterol. 15, 5549–5557 (2009).
Vinolo, M. A. R., Hatanaka, E., Lambertucci, R. H., Newsholme, P. & Curi, R. Effects of short chain fatty acids on effector mechanisms of neutrophils. Cell Biochem. Funct. 27, 48–55 (2009).
Hamer, H. M. et al. Review article: the role of butyrate on colonic function. Aliment. Pharmacol. Ther. 27, 104–119 (2008).
Usami, M. et al. Butyrate and trichostatin A attenuate nuclear factor κB activation and tumor necrosis factor α secretion and increase prostaglandin E2 secretion in human peripheral blood mononuclear cells. Nutr. Res. 28, 321–328 (2008).
Marchesi, J. R. et al. Rapid and noninvasive metabonomic characterization of inflammatory bowel disease. J. Proteome Res. 6, 546–551 (2007).
Leung, C. H., Lam, W., Ma, D. L., Gullen, E. A. & Cheng, Y. C. Butyrate mediates nucleotide-binding and oligomerisation domain (NOD) 2-dependent mucosal immune responses against peptidoglycan. Eur. J. Immunol. 39, 3529–3537 (2009).
Meijer, K., de Vos, P. & Priebe, M. G. Butyrate and other short-chain fatty acids as modulators of immunity: what relevance for health? Curr. Opin. Clin. Nutr. Metab. Care 13, 715–721 (2010).
Kelly, D. et al. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-γ and RelA. Nature Immunol. 5, 104–112 (2004).
Sougioultzis, S. et al. Saccharomyces boulardii produces a soluble anti-inflammatory factor that inhibits NF-κB-mediated IL-8 gene expression. Biochem. Biophys. Res. Commun. 343, 69–76 (2006).
Round, J. L. & Mazmanian, S. K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl Acad. Sci. USA 107, 12204–12209 (2010).
Round, J. L. & Mazmanian, S. K. The gut microbiota shapes intestinal immune responses during health and disease. Nature Rev. Immunol. 9, 313–323 (2009).
Meyer-Hoffert, U. et al. Secreted enteric antimicrobial activity localises to the mucus surface layer. Gut 57, 764–771 (2008).
Cash, H. L., Whitham, C. V., Behrendt, C. L. & Hooper, L. V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 313, 1126–1130 (2006).
Schumann, A. et al. Neonatal antibiotic treatment alters gastrointestinal tract developmental gene expression and intestinal barrier transcriptome. Physiol. Genomics 23, 235–245 (2005).
Desvignes, C., Bour, H., Nicolas, J. F. & Kaiserlian, D. Lack of oral tolerance but oral priming for contact sensitivity to dinitrofluorobenzene in major histocompatibility complex class II-deficient mice and in CD4+ T cell-depleted mice. Eur. J. Immunol. 26, 1756–1761 (1996).
Enerback, L., Lowhagen, G., Lowhagen, O. & Wingren, U. The effect of polymyxin B and some mast-cell constituents on mucosal mast cells in the duodenum of the rat. Cell Tissue Res. 214, 239–246 (1981).
Malaviya, R., Ikeda, T., Ross, E. & Abraham, S. N. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-α. Nature 381, 77–80 (1996).
Xu, X. et al. Mast cells involvement in the inflammation and fibrosis development of the TNBS-induced rat model of colitis. Scand. J. Gastroenterol. 37, 330–337 (2002).
Duan, J., Chung, H., Troy, E. & Kasper, D. L. Microbial colonization drives expansion of IL-1 receptor 1-expressing and IL-17-producing γ/δ T cells. Cell Host Microbe 7, 140–150 (2010).
Hill, D. A. et al. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 3, 148–158 (2010).
Umenai, T. et al. Eradication of the commensal intestinal microflora by oral antimicrobials interferes with the host response to lipopolysaccharide. Eur. J. Clin. Microbiol Infect. Dis. 29, 633–641 (2010). This study demonstrates that peritoneal macrophages are dependent on continued stimulation by intestinal microorganisms for their proper function.
Iimura, M. et al. Cathelicidin mediates innate intestinal defence against colonization with epithelial adherent bacterial pathogens. J. Immunol. 174, 4901–4907 (2005).
Johansson, M. E. V. et al. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl Acad. Sci. USA 105, 15064–15069 (2008).
Bergstrom, K. S. B. et al. Muc2 protects against lethal infectious colitis by disassociating pathogenic and commensal bacteria from the colonic mucosa. PLoS Pathog. 6, e1000902 (2010).
Wlodarska, M., Willing, B., Keeney, K. M., Menendez, A., Bergstrom, K. S., Gill, N., Russell, S. L., Vallance, B. A. & Finlay, B. B. Antibiotic treatment alters the colonic mucus layer and predisposes the host to exacerbated Citrobacter rodentium-induced colitis. Infect. Immun. 14 Feb 2011 (doi:10.1128/IAI.01104-10).
Pelissier, M. A. et al. Metronidazole effects on microbiota and mucus layer thickness in the rat gut. FEMS Microbiol. Ecol. 73, 601–610 (2010).
Ubeda, C. et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J. Clin. Invest. 120, 4332–4341 (2010).
Chen, X. et al. A mouse model of Clostridium difficile-associated disease. Gastroenterology 135, 1984–1992 (2008).
Young, V. B. & Schmidt, T. M. Antibiotic-associated diarrhea accompanied by large-scale alterations in the composition of the fecal microbiota. J. Clin. Microbiol. 42, 1203–1206 (2004).
Stecher, B. et al. Salmonella enterica serovar Typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS Biol. 5, 2177–2189 (2007).
Winter, S. E. et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429 (2010).
Schreiber, O. et al. Lactobacillus reuteri prevents colitis by reducing P-selectin-associated leukocyte- and platelet-endothelial cell interactions. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G534–G542 (2009).
Barthel, M. et al. Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect. Immun. 71, 2839–2858 (2003).
Croswell, A., Amir, E., Teggatz, P., Barman, M. & Salzman, N. H. Prolonged impact of antibiotics on intestinal microbial ecology and susceptibility to enteric Salmonella infection. Infect. Immun. 77, 2741–2753 (2009).
Hall, J. A. et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune responses. Immunity 29, 637–649 (2008).
Dufour, V. et al. Effects of a short-course of amoxicillin/clavulanic acid on systemic and mucosal immunity in healthy adult humans. Int. Immunopharmacol. 5, 917–928 (2005).
Bouskra, D. et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 456, 507–510 (2008).
Philpott, D. J. & Girardin, S. E. Gut microbes extend reach to systemic innate immunity. Nature Med. 16, 160–161 (2010).
Julia, V. et al. Priming by microbial antigens from the intestinal flora determines the ability of CD4+ T cells to rapidly secrete IL-4 in BALB/c mice infected with Leishmania major. J. Immunol. 165, 5637–5645 (2000).
Wills-Karp, M., Santeliz, J. & Karp, C. L. The germless theory of allergic disease: revisiting the hygiene hypothesis. Nature Rev. Immunol. 1, 69–75 (2001).
Guarner, F. et al. Mechanisms of disease: the hygiene hypothesis revisited. Nature Clin. Pract Gastroenterol. Hepatol 3, 275–284 (2006).
Marra, F. et al. Antibiotic use in children is associated with increased risk of asthma. Pediatrics 123, 1003–1010 (2009).
Verhulst, S. L. et al. A longitudinal analysis on the association between antibiotic use, intestinal microflora, and wheezing during the first year of life. J. Asthma 45, 828–832 (2008).
Kummeling, I. et al. Early life exposure to antibiotics and the subsequent development of eczema, wheeze, and allergic sensitization in the first 2 years of life: the KOALA birth cohort study. Pediatrics 119, e225–e231 (2007).
Noverr, M. C. & Huffnagle, G. B. The 'microflora hypothesis' of allergic diseases. Clin. Exp. Allergy 35, 1511–1520 (2005).
Noverr, M. C., Noggle, R. M., Toews, G. B. & Huffnagle, G. B. Role of antibiotics and fungal microbiota in driving pulmonary allergic responses. Infect. Immun. 72, 4996–5003 (2004).
Watanabe, J., Fujiwara, R., Sasajima, N., Ito, S. & Sonoyama, K. Administration of antibiotics during infancy promoted the development of atopic dermatitis-like skin lesions in NC/Nga mice. Biosci. Biotechnol. Biochem. 74, 358–363 (2010).
Ochoa-Reparaz, J. et al. Role of gut commensal microflora in the development of experimental autoimmune encephalomyelitis. J. Immunol. 183, 6041–6050 (2009).
Ochoa-Reparaz, J. et al. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 3, 487–495 (2010).
Brugman, S. et al. Antibiotic treatment partially protects against type 1 diabetes in the Bio-Breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia 49, 2105–2108 (2006).
Gen, R., Demir, M. & Ataseven, H. Effect of Helicobacter pylori eradication on insulin resistance, serum lipids and low-grade inflammation. South Med. J. 103, 190–196 (2010).
Finegold, S. M. et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 35, S6–S16 (2002).
Sandler, R. H. et al. Short-term benefit from oral vancomycin treatment of regressive-onset autism. J. Child. Neurol. 15, 429–435 (2000).
Kanoh, S. & Rubin, B. K. Mechanisms of action and clinical application of macrolides as immunomodulatory medications. Clin. Microbiol. Rev. 23, 590–615 (2010).
Sakito, O. et al. Interleukin 1β, tumor necrosis factor alpha, and interleukin 8 in bronchoalveolar lavage fluid of patients with diffuse panbronchiolitis: A potential mechanism of macrolide therapy. Respiration 63, 42–48 (1996).
Schultz, M. J. et al. Intravenous infusion of erythromycin inhibits CXC chemokine production, but augments neutrophil degranulation in whole blood stimulated with Streptococcus pneumoniae. J. Antimicrob. Chemother. 46, 235–240 (2000).
Zager, R. A., Johnson, A. C. M. & Geballe, A. Gentamicin suppresses endotoxin-driven TNF-α production in human and mouse proximal tubule cells. Am. J. Physiol. Renal Physiol. 293, F1373–F1380 (2007).
Woo, J. T., Shinohara, C., Sakai, K., Hasumi, K. & Endo, A. Inhibition of the acidification of endosomes and lysosomes by the antibiotic concanamycin-B in macrophage J774. Eur. J. Biochem. 207, 383–389 (1992).
Dethlefsen, L. & Relman, D. A. Microbes and Health Sackler Colloquium: Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl Acad. Sci. USA. 16 Sep 2010 (doi: 10.1073/pnas.1000087107). This work furthers our understanding of the impacts of antibiotics on the microbiota, addressing important issues such as repeat exposure to antibiotics, individualized responses and the loss of mutualistic traits from changing microbial communities.
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005).
Gill, S. R. et al. Metagenomic analysis of the human distal gut microbiome. Science 312, 1355–1359 (2006).
Vallance, B. A., Deng, W., Jacobson, K. & Finlay, B. B. Host susceptibility to the attaching and effacing bacterial pathogen Citrobacter rodentium. Infect. Immun. 71, 3443–3453 (2003).
Manichanh, C. et al. Reshaping the gut microbiome with bacterial transplantation and antibiotic intake. Genome Res. 20, 1411–1419 (2010).
Fagarasan, S. et al. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science 298, 1424–1427 (2002).
Deshpande, G., Rao, S., Patole, S. & Bulsara, M. Updated meta-analysis of probiotics for preventing necrotizing enterocolitis in preterm neonates. Pediatrics 125, 921–930 (2010).
Kale-Pradhan, P. B., Jassal, H. K. & Wilhelm, S. M. Role of Lactobacillus in the prevention of antibiotic-associated diarrhea: a meta-analysis. Pharmacotherapy 30, 119–126 (2010).
Marcone, V., Calzolari, E. & Bertini, M. Effectiveness of vaginal administration of Lactobacillus rhamnosus following conventional metronidazole therapy: how to lower the rate of bacterial vaginosis recurrences. New Microbiol. 31, 429–433 (2008).
Tatum, P. M. Jr, Harmon, C. M., Lorenz, R. G. & Dimmitt, R. A. Toll-like receptor 4 is protective against neonatal murine ischemia-reperfusion intestinal injury. J. Pediatr. Surg. 45, 1246–1255 (2010).
McFarland, L. V. Update on the changing epidemiology of Clostridium difficile-associated disease. Nature Clin. Pract. Gastroenterol. Hepatol. 5, 40–48 (2008).
Kozyrskyj, A. L., Ernst, P. & Becker, A. B. Increased risk of childhood asthma from antibiotic use in early life. Chest 131, 1753–1759 (2007).
Freter, R. The fatal enteric cholera infection in the guinea pig, achieved by inhibition of normal enteric flora. J. Infect. Dis. 97, 57–65 (1955).
Acknowledgements
S.L.R. is funded by the Michael Smith Foundation for Health Research. B.B.F. is a US Howard Hughes Medical Institute International Research Scholar and The University of British Columbia Peter Wall Distinguished Professor; work in his laboratory is funded by operating grants from the Canadian Institutes of Health Research and the Canadian Crohn's and Colitis Foundation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Glossary
- Microbiota
-
The consortium of microorganisms that inhabit the body.
- Mutualist
-
Pertaining to a member of the gut microbiota: a species that provides a service to the host while benefiting from the environment afforded by the host.
- Extra-intestinal disease
-
A disease that manifests outside of the gastrointestinal tract but is influenced by the gut microbiota.
- Bacteriotherapy
-
The introduction of the microbiota from a healthy donor into a patient with an imbalanced microbiota.
- Sub-therapeutic
-
Below the dosage levels used to treat diseases.
- Gut homeostasis
-
The dichotomy between preventing infection by luminal microorganisms and avoiding overt intestinal inflammation.
- Co-evolved
-
Resulting from the evolution of interdependent species, such that each organism adapts to changes in the other.
- Microorganism-associated molecular pattern
-
A molecule that is found as part of a microorganism and is recognized by cells of the host innate immune system.
- Pattern recognition receptor
-
A protein that is expressed as part of the innate immune system and recognizes a microorganism-associated molecular pattern; when activated, these receptors elicit a downstream immune response.
- Short-chain fatty acid
-
A fatty acid that is produced by the microbiota through the fermentation of host dietary fibre.
- Metabolomic
-
Pertaining to the study of the collective metabolites in an environment.
- Anergy
-
Reduced responsiveness to an antigen.
- Antimicrobial peptide
-
A peptide that is secreted as part of the innate immune response and has broad-spectrum antibiotic activity.
- Tolerization
-
The development of tolerance to innocuous antigens, including food and resident microbiota, thus contributing to intestinal homeostasis.
- Hygiene hypothesis
-
The hypothesis that insufficient exposure to microorganisms, parasites and infectious agents alters natural immune development, leading to an increased susceptibility to allergic diseases.
- Atopy
-
An allergic reaction that is characterized by immunoglobulin E-mediated inflammation.
Rights and permissions
About this article
Cite this article
Willing, B., Russell, S. & Finlay, B. Shifting the balance: antibiotic effects on host–microbiota mutualism. Nat Rev Microbiol 9, 233–243 (2011). https://doi.org/10.1038/nrmicro2536
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro2536
This article is cited by
-
Impact of an oligosaccharide-based polymer on the metabolic profiles and microbial ecology of weanling pigs experimentally infected with a pathogenic E. coli
Journal of Animal Science and Biotechnology (2024)
-
Ocular mucosal homeostasis of teleost fish provides insight into the coevolution between microbiome and mucosal immunity
Microbiome (2024)
-
Effects of probiotics (Bacillus coagulans) supplementation after antibiotic administration on growth, immunity, and intestinal microflora in turbot Scophthalmus maximus
Aquaculture International (2024)
-
Over supplementation with vitamin B12 alters microbe-host interactions in the gut leading to accelerated Citrobacter rodentium colonization and pathogenesis in mice
Microbiome (2023)
-
Phage therapy minimally affects the water microbiota in an Atlantic salmon (Salmo salar) rearing system while still preventing infection
Scientific Reports (2023)
