
Key Points
-
Coronaviruses are positive strand RNA viruses that cause disease in humans, and domestic and companion animals. They are most notorious for causing severe acute respiratory syndrome (SARS) outbreaks in 2002–2003. All coronaviruses follow the same basic strategy of replication.
-
All coronaviruses encode 15 or 16 replicase related proteins, 4 or 5 structural proteins and 1–8 group-specific or accessory proteins. Many of the replicase proteins are assembled into replication machinery in double-membrane vesicles (DMVs) and on a reticular network of membranes that are derived from the endoplasmic reticulum.
-
Coronaviruses are readily transmitted across species. This phenomenon was illustrated when the SARS-coronavirus crossed species from bats to intermediate hosts, such as palm civets, and then to humans. It also explains the large number of species, including humans, that are infected with viruses closely related to bovine coronavirus.
-
In many coronavirus infections, disease severity increases during virus clearance, suggesting that the host immune response is both protective and pathogenic. Furthermore, inhibition of specific aspects of the immune response results in less severe disease and less tissue destruction, without diminishing the kinetics of virus clearance.
-
Like all successful viruses, coronaviruses have evolved both passive and active mechanisms to evade the interferon response. Replication in DMVs may contribute to passive evasion of the innate immune response by making double-stranded RNA inaccessible to cellular sensors.
Abstract
Although coronaviruses were first identified nearly 60 years ago, they only received notoriety in 2003 when one of their members was identified as the aetiological agent of severe acute respiratory syndrome. Previously these viruses were known to be important agents of respiratory and enteric infections of domestic and companion animals and to cause approximately 15% of all cases of the common cold. This Review focuses on recent advances in our understanding of the mechanisms of coronavirus replication, interactions with the host immune response and disease pathogenesis. It also highlights the recent identification of numerous novel coronaviruses and the propensity of this virus family to cross species barriers.
Similar content being viewed by others


Main
Coronaviruses, a genus in the Coronaviridae family (order Nidovirales; Fig. 1), are pleomorphic, enveloped viruses. Coronaviruses gained prominence during the severe acute respiratory syndrome (SARS) outbreaks of 2002–2003 (Ref. 1). The viral membrane contains the transmembrane (M) glycoprotein, the spike (S) glycoprotein and the envelope (E) protein, and surrounds a disordered or flexible, probably helical, nucleocapsid2,3. The viral membrane is unusually thick, probably because the carboxy-terminal region of the M protein forms an extra internal layer, as revealed by cryo-electron tomography2. Coronaviruses are divided into three groups, and further subdivided into subgroups (Table 1), based initially on serologic, and more recently on genetic, analyses. With the identification of more distantly related viruses, the taxonomy of these viruses is likely to undergo further changes.

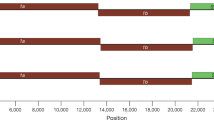
Phylogenetic relationship of viruses in the order Nidoviruses.
Coronaviruses contain a single stranded, 5′-capped, positive strand RNA molecule that ranges from 26–32 kb and that contains at least 6 open reading frames (ORFs). The first ORF (ORF1a/b) comprises approximately two-thirds of the genome and encodes replicase proteins (Fig. 2a). Translation begins in ORF1a and continues in ORF1b after a −1 frameshift signal. The large ORF1a and ORF1ab polypeptides, commonly referred to as pp1a and pp1ab, respectively, are processed primarily by the virally encoded chymotrypsin-like protease 3CLpro (also called Mpro or main protease) with additional cleavage performed by one or two viral papain-like proteases (PLPs), depending on the species of coronavirus4. The majority of the remaining one-third of the genome encodes four structural proteins: S, E, M and nucleocapsid (N) proteins. A subset of group 2 coronaviruses encode an additional haemagglutinin-esterase (HE) protein (Fig. 2a,b). The HE protein, which may be involved in virus entry or egress, is not required for replication, but appears to be important for infection of the natural host5.

a | Schematic diagram of representative genomes from each of the coronavirus groups. Approximately the first two-thirds of the 26–32 Kb, positive-sense RNA genome encodes a large polyprotein (ORF1a/b; green) that is proteolytically cleaved to generate 15 or 16 non-structural proteins (nsps; nsps for severe acute respiratory syndrome coronavirus (SARS-CoV) are illustrated). The 3′-end third of the genome encodes four structural proteins — spike (S), membrane (M), envelope (E) and nucleocapsid (N) (all shown in blue) — along with a set of accessory proteins that are unique to each virus species (shown in red). Some group 2 coronaviruses express an additional structural protein, haemagglutinin-esterase (not shown). b | Schematic diagram of the coronavirus virion. 2′OMT, ribose-2′-O-methyltransferase; ExoN, 3′→5′ exonuclease; Hel, helicase; IBV, infection bronchitis virus; NendoU, uridylate-specific endoribonuclease; RDRP, RNA-dependent RNA polymerase; ssRBP, single-stranded RNA binding protein; ssRNA, single-stranded RNA; TGEV, transmissible gastroenteritis virus.
Receptors for several coronaviruses have been identified (Table 1). The prototypical coronavirus, mouse hepatitis virus (MHV), uses CEACAM1a, a member of the murine carcinoembryonic antigen family, to enter cells. Deletion of this protein makes mice resistant to infection6. Several group 1 coronaviruses use aminopeptidase N to adhere to host cells, consistent with their respiratory and enteric tract tropisms (reviewed in Ref. 7). SARS-CoV, a group 2 coronavirus, enters host cells through an interaction of the S protein with human angiotensin converting enzyme 2 (ACE2)8. Strikingly, human coronavirus-NL63 (HCoV-NL63), which causes mild disease, also uses ACE2, although it binds to a different part of the protein than does SARS-coronavirus (SARS-CoV)9,10. ACE2 is postulated to have a protective role in the inflamed lung, and SARS-CoV S protein binding to ACE2 is thought to contribute to disease severity11,12. As infection with HCoV-NL63 produces mild disease, however, binding to ACE2 by itself cannot be sufficient for this process.
The N protein is important for encapsidation of viral RNA and acts as an interferon (IFN) antagonist (see below). Additionally, it causes upregulation of FGL2, a prothrombinase that contributes to fatal hepatic disease in mice that are infected with MHV-3 (Ref. 13) and that modifies transforming growth factor-β (TGFβ) signalling in SARS-CoV-infected cells14.
The E proteins are small integral membrane proteins with roles in virus morphogenesis, assembly and budding. In the absence of E proteins, virus release is inhibited completely (in the case of transmissible gastroenteritis virus (TGEV)) or partially (in the case of SARS-CoV and MHV)15,16,17. The E protein also possesses ion channel activity, which is required for optimal virus replication18,19.
Interspersed between and in these structural genes are one to eight genes that encode accessory proteins, depending on the virus strain. These show no sequence similarity with other viral or cellular proteins and are not required for virus replication in cultured cells20,21,22. However, they are conserved in virus species isolated at different times and locales (for example, for SARS-CoV23), which suggests that these proteins have an important role in replication in the natural host. Several accessory proteins are virion-associated24,25,26,27 although whether these proteins are truly structural is controversial28.
The genes that encode non-replicase proteins are expressed from a set of 'nested' subgenomic mRNAs that have common 3′ ends and a common leader that is encoded at the 5′ end of genomic RNA. Proteins are produced generally only from the first ORF of subgenomic mRNAs, which are produced during minus strand RNA synthesis. Transcription termination and subsequent acquisition of a leader RNA occurs at transcription regulatory sequences (TRS), located between ORFs. These minus strand subgenomic RNAs serve as templates for the production of subgenomic mRNAs (Fig. 3), an efficient process that results in a high ratio of subgenomic mRNA to minus strand subgenomic RNA29.

Following entry into the cell and uncoating, the positive sense RNA genome is translated to generate replicase proteins from open reading frame 1a/b (ORF1a/b). These proteins use the genome as a template to generate full-length negative sense RNAs, which subsequently serve as templates in generating additional full-length genomes (a). Coronavirus mRNAs all contain a common 5′ leader sequence fused to downstream gene sequences. These leaders are added by a discontinuous synthesis of minus sense subgenomic RNAs using genome RNA as a template (reviewed in Ref. 29). Subgenomic RNAs are initiated at the 3′ end of the genome and proceed until they encounter one of the transcriptional regulatory sequences (TRS; red) that reside upstream of most open-reading frames (b). Through base-pairing interactions, the nascent transcript is transferred to the complementary leader TRS (light red) (c) and transcription continues through the 5′ end of the genome (d). These subgenomic RNAs then serve as templates for viral mRNA production (e).
Coronavirus replication
One consequence of the SARS epidemic was an increase in efforts to understand coronavirus replication and identify additional possible targets for anti-viral therapy. ORF1 of most coronaviruses encodes 16 proteins that are involved in viral replication (Fig. 2a); ORF1 of group 3 coronaviruses lacks nsp1 and thus encodes only 15 proteins. The structure of many of these proteins has been solved by X-ray crystallography or nuclear magnetic resonance, facilitating structure-function studies30,31,32,33,34,35,36,37,38,39,40. Functions were predicted41 and later confirmed for many of these proteins (Table 2), including PL1pro and PL2pro (papain-like proteases) contained in nsp3 and 3CLpro or Mpro contained in nsp5, the RNA-dependent RNA polymerase nsp12 and the helicase nsp13. A second RNA polymerase, nsp8, may function as a primase42. The nsp3 protease has additional roles in the assembly of virus replication structures (see below) and possesses poly(ADP-ribose) binding capabilities, and deubiquitylating activity in its protease domain, although the role of the latter in virus replication is not yet known43.
Nsp7, nsp8, nsp9 and nsp10 are postulated to have a role in subgenomic and genomic RNA replication, and all four proteins are essential for viral replication44. Nsp7 and nsp8 form a hexadecameric structure, with RNA binding activity31. The structure of nsp9 also suggests that it binds RNA45. Mutations in nsp10 inhibit minus strand RNA synthesis, but this effect may be indirect, as studies have showed that nsp10 is required for proper function of the main viral protease (Mpro)46.
Nsp14, a bifunctional protein, is a 3′→5′ exonuclease, with a role in maintaining fidelity of RNA transcription47 and a (guanine-N7)-methyl transferase (N7-MTase), involved in RNA cap formation48. Coronaviruses also encode a novel uridylate-specific endoribonuclease (NendoU), nsp15, that distinguishes nidoviruses in general from other RNA viruses and that is crucial for virus replication49. Cleavage of RNA by NendoU results in 2′-3′ cyclic phosphate ends, but its function in the virus life cycle remains unknown. Nsp16 is an S-adenosyl-L-methionine-dependent RNA (nucleoside-2′O)-methyl transferase (2′O-MTase) and, like nsp14, is involved in cap formation50. Nsp15 has been postulated to function with nsp14 and nsp16 in RNA processing or cap production, but this remains to be proven.
RNA replication is thought to occur on double-membrane vesicles (DMVs)51 (Fig. 4). Newly synthesized genomic RNA is then incorporated into virions on membranes that are located between the endoplasmic reticulum (ER) and the Golgi apparatus (ER-Golgi intermediate compartment (ERGIC); reviewed in Ref. 52). Initial studies suggested that these DMVs assemble using components of the autophagy pathway53, but other studies showed replication proceeded normally and that DMVs were produced in macrophages lacking ATG5, a key component of autophagosomes54. Thus, whether autophagy is involved at all or whether its involvement is cell-specific remains uncertain. In addition, the unfolded protein response (UPR) is induced during coronavirus infections and may contribute to DMV formation55.

Coronavirus infection induces the formation of a reticulovesicular network of modified membranes that are thought to be the sites of virus replication. These modifications, which include double-membrane vesicles (DMVs), vesicle packets (VPs, single-membrane vesicles surrounded by a shared outer membrane) and convoluted membranes (CMs), are all interconnected and contiguous with the rough endoplasmic reticulum (RER). Viral double-stranded RNA is mostly localized to the interior of the DMVs and inner vesicles of the VPs, whereas replicase proteins (that is, nsp3, nsp5 and nsp8) are present on the surrounding CM. Some nsp8 can be detected inside the DMVs. All membranes are bound by ribosomes. (Figure based on data from Refs. 56, 141, 142.)
Recent results show that DMVs are likely to originate from the ER. Using electron tomography of cryo-fixed SARS-CoV-infected Vero E6 cells and three-dimensional reconstruction imaging, Knoops et al. showed that DMVs are not isolated vesicles, but rather are part of a reticulovesicular network of modified ER membranes56. At later times after infections, these networks appear to merge into large single-membrane vesicles. Proteins involved in virus replication (nsp3, nsp5 and nsp8; Table 2) are located mainly outside of DMVs, in adjacent reticular structures. Double-stranded RNA, representing either replicative intermediates or 'dead end' double-stranded RNA, was detected primarily in DMVs and, surprisingly, no obvious connections between the interior of these vesicles and the cytosol were detected56. Thus, it remains unknown how newly synthesized RNA might be transported to sites of virus assembly, assuming that RNA transcription occurs in DMVs.
Formation of DMVs requires membrane curvature, and this may be initiated by insertion of specific viral proteins into membranes. Based on studies of equine arteritis virus57, a non-coronavirus member of the nidovirus order (Fig. 1), nsp3 and nsp4 are probably sufficient for DMV formation. Mutations in nsp4 result in aberrant formation of DMVs, further supporting a role for this protein in establishing sites of virus replication58. Nsp6, like nsp3 and nsp4, also contains multiple transmembrane regions and may be involved in membrane modification59,60,61. Notably, nsp3 and nsp6 encode an odd number of hydrophobic domains, but both the amino and carboxyl termini of these proteins are in the cytoplasm, suggesting that one hydrophobic region does not span the membrane60; whether this region contributes to membrane curvature or has another function requires further investigation.
Coronavirus-mediated diseases
Before the SARS epidemic of 2002–2003, two human coronaviruses, HCoV-OC43 and HCoV-229E, were recognized as important causes of upper respiratory tract infections and were occasionally associated with more severe pulmonary disease in the elderly, newborn and immunocompromised62. SARS-CoV, unlike HCoV-OC43 and HCoV-229E, causes a severe respiratory disease, and nearly 10% mortality was observed in 2002–2003 (Ref. 1). Notable features of the disease were an apparent worsening of symptoms as the virus was cleared (suggesting the disease had an immunopathological basis), and a lack of contagion until lower respiratory tract symptoms were apparent. This latter feature made control of the epidemic by quarantine feasible, as it simplified identification of infected patients. Unlike HCoV-OC43 and HCoV-229E, SARS-CoV also caused systemic disease, with evidence of infection of the gastrointestinal tract, liver, kidney and brain, among other tissues63. Although the virus spread primarily via respiratory droplets, infection of the gastrointestinal tract may have facilitated other routes of spread.
The recognition that SARS was caused by a coronavirus intensified the search for other pathogenic coronaviruses associated with human disease, which led to the identification of HCoV-NL63 and HCoV-HKU1. These viruses were isolated from hospitalized patients, either young children with severe respiratory disease (HCoV-NL63)64,65 or elderly patients with underlying medical problems (HCoV-HKU1)65,66. HCoV-NL63 has infected human populations for centuries, as phylogenetic studies show that it diverged from HCoV-229E nearly 1,000 years ago67. HCoV-NL63 and HCoV-HKU1 have worldwide distributions and generally cause mild upper respiratory tract diseases, with the exception that HCoV-NL63 is also an aetiological agent of croup68. HCoV-NL63 can be propagated in tissue culture cells, and an infectious cDNA clone of this virus was recently engineered, facilitating future studies69. By contrast, HCoV-HKU1 cannot be grown in tissue culture cells, which makes it imperative that an infectious cDNA clone be developed for future studies.
Although the severe disease forming capabilities of human coronaviruses were only recognized because of the SARS epidemic, it was well known that animal coronaviruses could cause life-threatening disease. TGEV, which causes diarrhoea in piglets, infectious bronchitis virus (IBV), a cause of severe upper respiratory tract and kidney disease in chickens, and bovine coronavirus (BCoV), which causes respiratory tract disease and diarrhoea in cattle ('winter dysentery' and 'shipping fever'), are all economically important pathogens. Feline infectious peritonitis virus (FIPV), a virulent feline coronavirus (FCoV), causes an invariably fatal systemic disease in domestic cats and other felines. Unlike most strains of FCoV, which are endemic causes of mild diarrhoea, FIPV arises sporadically, most likely by mutation or deletion in felines persistently infected with enteric strains of FCoV70, and is macrophage-tropic.
Perhaps the most convincing explanation for FIPV-mediated disease was suggested by the observation that progressive waves of virus replication, lymphopenia and ineffectual T cell responses occurred in feline infectious peritonitis (FIP)71. In conjunction with previous studies, these results raised the possibility that FIPV infection of macrophages and dendritic cells caused aberrant cytokine and/or chemokine expression and lymphocyte depletion, resulting in enhanced virus loads and, consequently, a fatal outcome. Although this explanation is appealing, additional work is needed to prove its validity. Notably, anti-FIPV antibody-mediated enhancement has been implicated in pathogenesis, but this has been shown only after immunization with S protein expressing vaccines72; it has not been shown to play a role in a natural feline infection.
Cross-species transmission
A striking feature of the 2002–2003 SARS epidemic was the ability of the SARS-CoV to cross species from Himalayan palm civets (Paguma larvata), raccoon dogs (Nyctereutes procyonoides) and Chinese ferret badgers (Melogale moschata) to infect human populations73 (Fig. 5a). Transmission occurred in live animal retail (wet) markets, where animal handlers became infected. In retrospect, it seems that variants of SARS-CoV related to the epidemic strain infected human populations in the wet markets fairly frequently, as is shown by the high seropositivity rate detected in animal handlers who did not develop SARS-like illnesses73. The epidemic began when a physician who was treating personnel in the wet markets became infected and subsequently infected multiple contacts74.

a | Severe acute respiratory syndrome (SARS)-like bat coronavirus (BtCoV) spread and adapted to wild animals such as the Himalayan palm civet that was sold as food in Chinese wet markets. The virus frequently spread to animal handlers in these markets, but caused minimal or no disease. Further adaptation resulted in strains that replicated efficiently in the human host, caused disease and could spread from person to person. b | Human coronavirus OC43 (HCoV-OC43) and bovine coronavirus (BCoV) are closely related and it is thought that the virus originated in one species and then crossed species. BCoV has also spread to numerous other animals, such as alpaca and wild ruminants. c | Feline coronavirus I (FCoV-I) and canine coronavirus I (CCoV-I) are thought to share a common ancestor. CCoV-I underwent recombination with an unknown coronavirus to give rise to canine coronavirus II (CCoV-II). CCoV-II in turn underwent recombination with FCoV-I (in an unknown host) to give rise to feline coronavirus II (FCoV-II). CCoV-II probably also spread to pigs, resulting in transmissible gastroenteritis virus (TGEV).
Genetic analyses of virus isolates from infected palm civets and humans during the epidemic showed that the virus underwent rapid adaptation in both hosts75,76, primarily in the receptor binding domain (RBD) of the S protein, to allow more efficient infection of human cells77. In particular, mutations K479N and S487T in the RBD of the S protein were key to adaptation to the human receptor (ACE2). These results were recently confirmed using cell lines expressing civet ACE2 or human ACE2 (Ref. 78).
The observation that SARS-CoV could not be detected in either farmed or wild palm civets79, together with evidence of adaptive changes detected in virus isolated from infected animals, suggested that palm civets and other animals in wet markets were not the primary reservoir for the virus. As SARS-like CoV were isolated from Chinese horseshoe bats (Rhinolophus spp.)23,80, which were also present in the live animal markets, the virus may have recently spread from bats to other mammals, such as palm civets, and then to humans (Fig. 5a). Consistent with a recent spread, antibodies to SARS-CoV were detected at extremely low levels (0.008%) in population studies in Hong Kong81. Bat SARS-like CoV cannot replicate in cells that express bat ACE2, although productive infection of cells expressing human ACE2 occurs if the RBD of the bat S protein is replaced with that of a human isolate82,83. Collectively, these observations suggest that virus spread from bats to other species. Host cell entry does not occur via ACE2 in bats, although it does in palm civets and humans.
Besides SARS-CoV, there are other examples of coronavirus cross-species transmission. BCoV and HCoV-OC43 are similar and the virus may have crossed from bovine to human hosts approximately 100 years ago84. BCoV has continued to cross species, as a related virus (99.5% similarity) has been isolated from an alpaca with enteritis and from captive wild ruminants85,86 (Fig. 5b). Furthermore, canine coronavirus (CCoV), feline and porcine viruses show evidence they have recombined with each other, indicating that they were present in the same host. Recombination events between early CCoV and FCoV strains (CCoV-I and FCoV-I) and an unknown coronavirus resulted in two sets of novel viruses — CCoV-II and FCoV-II. Sequence data suggest that TGEV resulted from a cross-species transmission of CCoV-II from an infected canine87 (Fig. 5c).
Molecular surveillance studies have identified at least 60 novel bat coronaviruses in China88, North America50, Europe89,90 and Africa91. These bat CoVs may have originated from a common source and then subsequently diverged as they adapted to growth in different species of bat; they are now only distantly related to other coronaviruses. These studies also identified several novel avian group 3 coronaviruses92 that were related to a novel coronavirus isolated from Asian leopard cats (Prionailurus bengalensis) and Chinese ferret badgers sold in illegal wild animal markets in China93, suggesting that this virus, like SARS-CoV, can cross species. Another novel group 3 coronavirus, isolated from a deceased beluga whale (Delphinapterus leucas), is only distantly related to IBV-like and novel avian coronaviruses, suggesting that it comprises a third subgroup94. Thus group 3 coronaviruses, which formerly included only avian viruses, now consist of at least 3 subgroups and include viruses that infect mammalian hosts.
Immunopathology in coronavirus infections
It is generally accepted that the host response is responsible for many of the disease manifestations in infections caused by coronaviruses95,96. This was shown initially in mice infected with the neurotropic strains of mouse hepatitis virus (the JHMV and MHV-A59 strains). Many attenuated strains of JHMV cause a subacute or persistent infection in the central nervous system, with persistence in glia, especially oligodendrocytes. A consequence of host efforts to clear the virus is myelin destruction (demyelination). However, JHMV infection of mice that lack T or B cells (sublethally irradiated mice or mice with severe combined immunodeficiency or genetically deficient in recombination activating gene 1 (RAG1−/−)) results, eventually, in death in all mice, but without demyelination. Adoptive transfer of CD4+ or CD8+ T splenocytes 7 or 30 days after immunization with JHMV to infected RAG1−/− or SCID mice results in virus clearance and demyelination95,96,97. Myelin destruction is also observed if anti-JHMV antibody is transferred to infected RAG1−/− mice in the absence of T cells98, or if mice are infected with virus expressing the macrophage chemoattractant CCL2 in the absence of other interventions99. In all cases, infiltrating macrophages appear to be crucial for virus clearance and subsequent demyelination; these results suggest that the process of macrophage infiltration can be initiated by T cells, anti-JHMV antibody or overexpression of a single macrophage chemoattractant. These results have been extended to mice with encephalitis caused by virulent strains of JHMV. Although CD4+ and CD8+ T cells are both required for virus clearance100, partial abrogation of the CD4+ T cell response (by mutating the immunodominant CD4+ T cell epitope rJ.MY135Q) results in disease amelioration, and virulence is regained if another CD4+ T cell epitope is reintroduced into the rJ.MY135Q genome101. Thus acute encephalitis, like chronic demyelination, is at least partially mediated by the immune system.
Similar processes may occur in SARS-CoV-infected humans, as pulmonary disease often worsens at 1–2 weeks after onset of respiratory symptoms, concomitant with the onset of virus clearance1. Although worsening clinical disease occurring as a consequence of virus clearance has not been duplicated in any animal model of SARS, the severe disease observed in older patients can be mimicked in SARS-CoV-infected aged mice102,103,104. This has been attributed, in part, to a suboptimal T cell response resulting in delayed kinetics of virus clearance. A suboptimal T cell response, occurring as a consequence of infection of macrophages or dendritic cells, may also be critical for the immunopathological lethal disease that is observed in FIPV-infected felines71. Thus, in many instances, host efforts to clear a coronavirus infection result in some tissue destruction.
Evasion of the innate immune response
Although anti-viral T cells and antibodies are crucial for virus clearance and for the prevention of recrudescence (reviewed in Ref. 96), the efficacy of the innate immune response determines the extent of initial virus replication and thus the load that the host must overcome to clear the infection (Fig. 6a). Coronaviruses, like all other successful viruses, have developed strategies to counter the innate immune response (Fig. 6b-d). IFN expression is a crucial component of this initial response, and coronaviruses have developed 'passive' and 'active' tools to prevent IFN induction and signalling. Interferon is not induced in fibroblasts that are infected with either SARS-CoV or MHV105,106,107. However, in both instances, treatment of cells with polyinosinic:polycytidylic acid or with other IFN-inducing agents, results in activation of IFN regulating factor 3 (IRF3) and IFN induction105,106. Thus, in these cells, viruses appear to be invisible to intracellular viral sensors (such as RIG-I, MDA5 and TLR3), perhaps because double stranded RNA, a potent stimulator of the innate immune system, is buried in a DMV (Fig. 6b).

a | Coronaviruses, as exemplified by severe acute respiratory syndrome coronavirus (SARS-CoV) and mouse hepatitis virus (MHV), induce a type 1 interferon (IFN) response in plasmacytoid dendritic cells (pDC) and macrophages, via TLR7- and MDA5-dependent pathways, respectively. b | IFNα and/or IFNβ is not produced in either SARS-CoV fibroblasts or DCs, partly because coronavirus macromolecules appear to be invisible to immune sensors. Additionally, coronaviruses encode proteins that actively inhibit IFNα and/or IFNβ expression (such as nucleocapsid (N) protein, nsp3, ORF6 and ORF3b) or signalling through the type 1 IFN receptor (such as N, nsp1, ORF6 and ORF3b). c | Consequently, the kinetics of virus clearance is delayed, with subsequent robust T and B cell and cytokine and/or chemokine responses. d | This pro-inflammatory response results in immunopathological disease that occurs during the process of virus clearance. In MHV-infected mice, virus clearance involves recruitment of activated macrophages and microglia to sites of virus infection, leading to demyelination. Similar mechanisms with exuberant cytokine production may function in the lungs of SARS-CoV-infected humans, leading to severe pulmonary disease (adult respiratory distress syndrome, ARDS). AP1, activator protein 1; DMV, double-membrane vesicle; dsRNA, double-stranded RNA; NF-κB, nuclear factor-κB; ssRNA, single-stranded RNA.
Additionally, viral proteins, in particular nsp1, nsp3, N protein and the SARS-CoV accessory proteins ORF6 and ORF3b, also prevent IFN induction108,109,110,111,112,113. The N protein of MHV inhibits activator protein 1 (AP1) signalling and protein kinase R (PKR) function, whereas the N protein of SARS-CoV also inhibits nuclear factor-κB activation108,109,110 when expressed in transfection assays. Whether these inhibitory functions of the N protein are coronavirus or cell-type specific, and whether they occur in infected cells, remains to be determined. The ORF6 protein inhibits IFN signalling by binding to karyopherin-α2, thereby tethering karyopherin-β to cytoplasmic membranes114. This, in turn, prevents nuclear translocation of proteins containing classical nuclear import signals115, including STAT1, a crucial component of IFNα, IFNβ and IFNγ signalling pathways. Of note, deletion of ORF6 does not increase the IFN sensitivity of SARS-CoV116, probably because mechanisms of IFN antagonism are redundant.
SARS-CoV and MHV nsp1 function, at least in part, by degrading host cell mRNA and inhibiting translation111,112,113,117. Nsp1 also inhibits IFN signalling in both SARS-CoV- and MHV-infected cells, in part by inhibiting STAT1 phosphorylation112,113. Mutation of nsp1 attenuates SARS-CoV and MHV growth in mice and tissue culture cells in the presence of an intact IFN system, but not when IFN function is deficient111,112,113. Nsp3 is also an IFN antagonist, and it inhibits phosphorylation and nuclear importation of IRF3 (Ref. 118).
Both MHV and SARS-CoV inhibit IFNα and IFNβ induction and signalling. However, IFNα and/or IFNβ are detected in infected mice and humans119,120 and mice deficient in IFNα and/or IFNβ receptor expression are exquisitely sensitive to MHV infection113,121, showing that IFNα and/or IFNβ has a major role in the anti-virus immune response. Reconciling these disparate results, recent studies showed that IFNα is produced in large amounts in SARS-CoV- and MHV-infected plasmacytoid dendritic cells, via a TLR7-dependent mechanism122. Furthermore, IFNβ is expressed by macrophages and microglia, but not by dendritic cells after MHV infection123. Macrophages, and to a lesser extent dendritic cells, are the major targets for IFNα and/or IFNβ in MHV-infected mice124.
In addition to IFN, multiple chemokines and cytokines are also induced as part of the host response to coronaviruses such as MHV, SARS-CoV and FIPV. Cytokines such as interleukin 1 (IL-1), IL-6 and IL-12 and chemokines such as IL-8, CCL2 and CXCL10 are elevated in SARS patients. Using genomics and proteomics, Cameron et al. found that IFNα and/or IFNβ and IFNγ, as well as chemokines such as CXCL10 and CCL2, are elevated at early times post infection in all patients and diminished in those who recovered, accompanied by a robust anti-virus antibody response119. However, levels of CXCL10, CCL2 and other proinflammatory mediators remained elevated and anti-SARS-CoV antibody titres were low in those patients who developed severe disease. SARS-CoV-infected pulmonary epithelial cells were the source of at least some of the cytokines and/or chemokines, such as CCL2, IL-6, IL-1β and tumour necrosis factor (TNF)125. Others have suggested that a strong TH2 (IL4, IL-5 and IL-10) response correlated with a poor outcome126. It has been postulated that an over-exuberant cytokine response contributed to a poor outcome in patients with SARS in 2002–2003 (reviewed in Refs 95, 127, 128). Collectively, these results do not strongly prove or disprove a role for an exuberant cytokine and chemokine response in severe SARS, in part because virus titres could not be determined concomitantly and also because serum levels, but not pulmonary cytokine or chemokine levels were measured.
Animal models for SARS
As human SARS has disappeared, the role of an exuberant (but perhaps appropriate for the titre of the virus) immune response will need to be addressed using animal models of SARS. Mice, cats, ferrets, macaques and civet cats are all susceptible to SARS-CoV, but none, with the exception of aged mice, develop severe disease (reviewed in Ref. 129). In efforts to develop models that closely mimic human disease, mice that are transgenic for the expression of human ACE2 were developed and infected with SARS-CoV130,131. Although these mice develop more severe pulmonary disease than non-transgenic mice, they also develop an overwhelming neuronal infection, accompanied by high cytokine and/or chemokine expression and minimal cellular infiltration in the brain132. Although the severity of the brain infection observed in human ACE2 transgenic mice is greater than that seen in human patients, infection of this organ has been detected in some studies and patients who survived SARS had a greater incidence of neurological and psychiatric sequelae than anticipated63,133,134. The high susceptibility of these mice to infection with SARS-CoV makes them useful for vaccine and therapeutic trials. Another approach to developing an animal model for SARS was to adapt the virus by passage 10–15 times through the lungs of BALB/c mice or rats103,135,136. Three to six mutations were detected in the adapted viruses, with changes most commonly observed in the S protein and in nsp5 (3CLpro). The adapted virus caused extensive pulmonary infection and disease was most severe in aged animals. These viruses will be useful for studies of pathogenesis and for vaccine and therapeutic trials.
Some models have been tested on the genomic and proteomic level. Studies of SARS-CoV infected macaques showed that several chemokines and/or cytokines, such as IL-6, IL-8, CXCL10 and CCL2, as well as IFNα, IFNβ and IFNγ, were upregulated137. These animals recovered, showing that the same inflammatory mediators that are associated with severe human disease are also produced as part of the inflammatory response in animals that mount an appropriate response. Genomics studies of mice infected with the Urbani strain of SARS-CoV showed continued expression of inflammatory mediators, such as IL-6, TNF, CXCL10 and CCL2, accompanied by slower kinetics of virus clearance and worse outcomes in aged compared to young animals138, paralleling disease patterns in patients with SARS119. These two studies also showed changes in expression of proteins that are involved in cell growth, cycling, cell-to-cell signalling and development and death. It will be important to determine whether these changes are useful as a 'fingerprint' for SARS or whether they represent generalized responses to pulmonary stress.
Future directions
Perhaps the most important insight made over the past several years is that coronaviruses have and will likely continue to cross between species and cause disease in unrelated hosts. This disease may be mild, like the disease caused by the SARS-like CoV that was transmitted to animal handlers in wet markets in China, but it may be severe, as illustrated by the transmission that triggered the SARS epidemic. Further, SARS-CoV appeared to use an entirely new receptor when it crossed species from bats to palm civets and humans. As part of this transmission to a new species, the virus also needed to evolve strategies to evade the innate immune response of the new hosts. One future goal will be to further delineate how the virus evades the immune response and better understand its interaction with the T and B cell responses, both in the original host (bats), in which disease appears to be mild, and in humans and experimentally infected animals.
Although coronaviruses use host proteins as part of their replication strategies, it has also become clear that immune, metabolic, stress, cell cycling and other pathways are activated by infection. Assessing the biological function of these pathways in virus replication and in disease outcome will be critical. Determining the extent to which virus–host interactions are coronavirus-specific and organ-specific will be possible, using genomics and proteomics, as well as new reagents and collaborative cross mice. The collaborative cross, a panel of approximately 1,000 recombinant inbred mouse strains derived from 8 founder strains, will be useful for analyses of complex genetic traits139.
Using sophisticated microscopy and biochemical approaches, details of coronavirus replication in infected cells have been revealed. However, these new results have led to a new set of questions about the relationship between sites of viral RNA replication and virus assembly. Furthermore, although putative functions have been assigned to many of the proteins encoded by the large ORF1 replicase gene, the precise roles of these proteins in virus replication still require additional investigation. Progress in these fields will take advantage of new methodologies that allow detailed observations of both fixed and living cells at high resolution.
Finally, no effective treatments exist for any coronavirus infections, including SARS140; vaccines, even for animal coronaviruses, are not effective; and live attenuated vaccines are prone to recombination with circulating coronaviruses. One future goal will be to translate new information about the structure and function of coronavirus proteins into specific anti-virus therapies. Also, development of live, attenuated, safe vaccines that do not recombine in the wild is another goal, made more feasible as more is learned about basic coronavirus biology. Over the past few years, the development of new technologies has simplified the identification of novel coronaviruses; the next major goals will be to understand viral pathogenesis and to design effective coronavirus vaccines and therapies.
References
Peiris, J. S., Guan, Y. & Yuen, K. Y. Severe acute respiratory syndrome. Nature Med. 10, S88–S97 (2004).
Barcena, M. et al. Cryo-electron tomography of mouse hepatitis virus: insights into the structure of the coronavirion. Proc. Natl Acad. Sci. USA 106, 582–587 (2009). This paper and Ref. 3 use novel methodologies to obtain detailed models of the structure of the intact coronavirion.
Neuman, B. W. et al. Supramolecular architecture of severe acute respiratory syndrome coronavirus revealed by electron cryomicroscopy. J. Virol. 80, 7918–7928 (2006).
Masters, P. S. The molecular biology of coronaviruses. Adv. Virus Res. 66, 193–292 (2006).
Lissenberg, A. et al. Luxury at a cost? Recombinant mouse hepatitis viruses expressing the accessory hemagglutinin esterase protein display reduced fitness in vitro. J. Virol. 79, 15054–15063 (2005).
Hemmila, E. et al. Ceacam1a−/− mice are completely resistant to infection by murine coronavirus mouse hepatitis virus A59. J. Virol. 78, 10156–10165 (2004).
Weiss, S. R. & Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. 69, 635–664 (2005).
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003). This is the first report identifying the SARS-CoV receptor.
Li, W. et al. The S proteins of human coronavirus NL63 and severe acute respiratory syndrome coronavirus bind overlapping regions of ACE2. Virology 367, 367–374 (2007).
Hofmann, H. et al. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl Acad. Sci. USA 102, 7988–7993 (2005).
Kuba, K. et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nature Med. 11, 875–879 (2005). This report suggests that interactions between the SARS-CoV S protein and its receptor may contribute to disease progression, in addition to facilitating virus entry.
Imai, Y. et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436, 112–116 (2005).
Ning, Q. et al. Induction of prothrombinase fgl2 by the nucleocapsid protein of virulent mouse hepatitis virus is dependent on host hepatic nuclear factor-4α. J. Biol. Chem. 278, 15541–15549 (2003).
Zhao, X., Nicholls, J. M. & Chen, Y. G. Severe acute respiratory syndrome-associated coronavirus nucleocapsid protein interacts with Smad3 and modulates transforming growth factor-β signaling. J. Biol. Chem. 283, 3272–3280 (2008).
DeDiego, M. L. et al. A severe acute respiratory syndrome coronavirus that lacks the E gene is attenuated in vitro and in vivo. J. Virol. 81, 1701–1713 (2007).
Kuo, L. & Masters, P. S. The small envelope protein E is not essential for murine coronavirus replication. J. Virol. 77, 4597–4608 (2003).
Ortego, J., Ceriani, J. E., Patino, C., Plana, J. & Enjuanes, L. Absence of E protein arrests transmissible gastroenteritis coronavirus maturation in the secretory pathway. Virology 368, 296–308 (2007).
Madan, V., Garcia Mde, J., Sanz, M. A. & Carrasco, L. Viroporin activity of murine hepatitis virus E protein. FEBS Lett. 579, 3607–3612 (2005).
Wilson, L., McKinlay, C., Gage, P. & Ewart, G. SARS coronavirus E protein forms cation-selective ion channels. Virology 330, 322–331 (2004).
Yount, B. et al. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J. Virol. 79, 14909–14922 (2005).
Ontiveros, E., Kuo, L., Masters, P. S. & Perlman, S. Inactivation of expression of gene 4 of mouse hepatitis virus strain JHM does not affect virulence in the murine CNS. Virology 290, 230–238 (2001).
de Haan, C. A., Masters, P. S., Shen, X., Weiss, S. & Rottier, P. J. The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology 296, 177–189 (2002).
Li, W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679 (2005). This paper and Ref. 80 show that SARS-CoV probably originated in bats.
Fischer, F., Peng, D., Hingley, S. T., Weiss, S. R. & Masters, P. S. The internal open reading frame within the nucleocapsid gene of mouse hepatitis virus encodes a structural protein that is not essential for viral replication. J. Virol. 71, 996–1003 (1997).
Huang, C., Peters, C. J. & Makino, S. Severe acute respiratory syndrome coronavirus accessory protein 6 is a virion-associated protein and is released from 6 protein-expressing cells. J. Virol. 81, 5423–5426 (2007).
Ito, N. et al. Severe acute respiratory syndrome coronavirus 3a protein is a viral structural protein. J. Virol. 79, 3182–3186 (2005).
Schaecher, S. R., Mackenzie, J. M. & Pekosz, A. The ORF7b protein of severe acute respiratory syndrome coronavirus (SARS-CoV) is expressed in virus-infected cells and incorporated into SARS-CoV particles. J. Virol. 81, 718–731 (2007).
Neuman, B. W. et al. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 82, 5279–5294 (2008).
Sawicki, S. G., Sawicki, D. L. & Siddell, S. G. A contemporary view of coronavirus transcription. J. Virol. 81, 20–29 (2007).
Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 300, 1763–1767 (2003).
Zhai, Y. et al. Insights into SARS-CoV transcription and replication from the structure of the nsp7-nsp8 hexadecamer. Nature Struct. Mol. Biol. 12, 980–986 (2005).
Chatterjee, A. et al. Nuclear magnetic resonance structure shows that the severe acute respiratory syndrome coronavirus-unique domain contains a macrodomain fold. J. Virol. 83, 1823–1836 (2009).
Joseph, J. S. et al. Crystal structure of nonstructural protein 10 from the severe acute respiratory syndrome coronavirus reveals a novel fold with two zinc-binding motifs. J. Virol. 80, 7894–7901 (2006).
Joseph, J. S. et al. Crystal structure of a monomeric form of severe acute respiratory syndrome coronavirus endonuclease nsp15 suggests a role for hexamerization as an allosteric switch. J. Virol. 81, 6700–6708 (2007).
Peti, W. et al. Structural genomics of the severe acute respiratory syndrome coronavirus: nuclear magnetic resonance structure of the protein nsP7. J. Virol. 79, 12905–12913 (2005).
Saikatendu, K. S. et al. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1′'-phosphate dephosphorylation by a conserved domain of nsP3. Structure 13, 1665–1675 (2005).
Almeida, M. S., Johnson, M. A., Herrmann, T., Geralt, M. & Wuthrich, K. Novel β -barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. J. Virol. 81, 3151–3161 (2007).
Serrano, P. et al. Nuclear magnetic resonance structure of the N-terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. J. Virol. 81, 12049–12060 (2007).
Ricagno, S. et al. Crystal structure and mechanistic determinants of SARS coronavirus nonstructural protein 15 define an endoribonuclease family. Proc. Natl Acad. Sci. USA 103, 11892–11897 (2006).
Su, D. et al. Dodecamer structure of severe acute respiratory syndrome coronavirus nonstructural protein nsp10. J. Virol. 80, 7902–7908 (2006).
Snijder, E. J. et al. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331, 991–1004 (2003).
Imbert, I. et al. A second, non-canonical RNA-dependent RNA polymerase in SARS coronavirus. EMBO J. 25, 4933–4942 (2006).
Ratia, K. et al. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl Acad. Sci. USA 103, 5717–5722 (2006).
Deming, D. J., Graham, R. L., Denison, M. R. & Baric, R. S. Processing of open reading frame 1a replicase proteins nsp7 to nsp10 in murine hepatitis virus strain A59 replication. J. Virol. 81, 10280–10291 (2007).
Egloff, M. P. et al. The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a single-stranded RNA-binding subunit unique in the RNA virus world. Proc. Natl Acad. Sci. USA 101, 3792–3796 (2004).
Donaldson, E. F., Sims, A. C., Graham, R. L., Denison, M. R. & Baric, R. S. Murine hepatitis virus replicase protein nsp10 is a critical regulator of viral RNA synthesis. J. Virol. 81, 6356–6368 (2007).
Eckerle, L. D., Lu, X., Sperry, S. M., Choi, L. & Denison, M. R. High fidelity of murine hepatitis virus replication is decreased in nsp14 exoribonuclease mutants. J. Virol. 81, 12135–12144 (2007).
Chen, Y. et al. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl Acad. Sci. USA 10 Feb 2009 (doi:10.1073/pnas.0808790106).
Ivanov, K. A. et al. Major genetic marker of nidoviruses encodes a replicative endoribonuclease. Proc. Natl Acad. Sci. USA 101, 12694–12699 (2004).
Decroly, E. et al. Coronavirus nonstructural protein 16 is a cap-0 binding enzyme possessing (nucleoside-2′O)-methyltransferase activity. J. Virol. 82, 8071–8084 (2008).
Snijder, E. J. et al. Ultrastructure and origin of membrane vesicles associated with the severe acute respiratory syndrome coronavirus replication complex. J. Virol. 80, 5927–5940 (2006).
de Haan, C. A. & Rottier, P. J. Molecular interactions in the assembly of coronaviruses. Adv. Virus Res. 64, 165–230 (2005).
Prentice, E., Jerome, W. G., Yoshimori, T., Mizushima, N. & Denison, M. R. Coronavirus replication complex formation utilizes components of cellular autophagy. J. Biol. Chem. 279, 10136–10141 (2004).
Zhao, Z. et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 3, 581–585 (2007).
Bechill, J., Chen, Z., Brewer, J. W. & Baker, S. C. Coronavirus infection modulates the unfolded protein response and mediates sustained translational repression. J. Virol. 82, 4492–4501 (2008).
Knoops, K. et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6, e226 (2008). This is an elegant electron microscopic study that uses 3D imaging of SARS-CoV-infected cells to delineate the relationship between virus replication and virus-induced membranous changes.
Snijder, E. J., van Tol, H., Roos, N. & Pedersen, K. W. Non-structural proteins 2 and 3 interact to modify host cell membranes during the formation of the arterivirus replication complex. J. Gen. Virol. 82, 985–994 (2001).
Clementz, M. A., Kanjanahaluethai, A., O'Brien, T. E. & Baker, S. C. Mutation in murine coronavirus replication protein nsp4 alters assembly of double membrane vesicles. Virology 375, 118–129 (2008).
Kanjanahaluethai, A., Chen, Z., Jukneliene, D. & Baker, S. C. Membrane topology of murine coronavirus replicase nonstructural protein 3. Virology 361, 391–401 (2007).
Oostra, M. et al. Topology and membrane anchoring of the coronavirus replication complex: Not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J. Virol. 82, 12392–12405 (2008).
Oostra, M. et al. Localization and membrane topology of coronavirus nonstructural protein 4: involvement of the early secretory pathway in replication. J. Virol. 81, 12323–12336 (2007).
Garbino, J. et al. A prospective hospital-based study of the clinical impact of non-severe acute respiratory syndrome (Non-SARS)-related human coronavirus infection. Clin. Infect. Dis. 43, 1009–1015 (2006).
Gu, J. et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 202, 415–424 (2005).
Fouchier, R. A. et al. A previously undescribed coronavirus associated with respiratory disease in humans. Proc. Natl Acad. Sci. USA 101, 6212–6216 (2004).
van der Hoek, L. et al. Identification of a new human coronavirus. Nature Med. 10, 368–373 (2004).
Woo, P. C. et al. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 79, 884–895 (2005).
Pyrc, K. et al. Mosaic structure of human coronavirus NL63, one thousand years of evolution. J. Mol. Biol. 364, 964–973 (2006). This manuscript delineates molecular evolution of HCoV-229E and HCoV-NL63.
van der Hoek, L. et al. Croup is associated with the novel coronavirus NL63. PLoS Med. 2, e240 (2005).
Donaldson, E. F. et al. Systematic assembly of a full-length infectious clone of human coronavirus NL63. J. Virol. 82, 11948–11957 (2008).
Rottier, P. J., Nakamura, K., Schellen, P., Volders, H. & Haijema, B. J. Acquisition of macrophage tropism during the pathogenesis of feline infectious peritonitis is determined by mutations in the feline coronavirus spike protein. J. Virol. 79, 14122–14130 (2005).
de Groot-Mijnes, J. D., van Dun, J. M., van der Most, R. G. & de Groot, R. J. Natural history of a recurrent feline coronavirus infection and the role of cellular immunity in survival and disease. J. Virol. 79, 1036–1044 (2005).
Vennema, H. et al. Early death after feline infectious peritonitis virus challenge due to recombinant vaccinia virus immunization. J. Virol. 64, 1407–1409 (1990).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276–278 (2003).
Riley, S. et al. Transmission dynamics of the etiological agent of SARS in Hong Kong: impact of public health interventions. Science 300, 1961–1966 (2003).
Chinese, S. M. E. C. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science 303, 1666–1669 (2004).
Song, H. D. et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl Acad. Sci. USA 102, 2430–2435 (2005).
Li, W. et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 24, 1634–1643 (2005).
Sheahan, T., Rockx, B., Donaldson, E., Corti, D. & Baric, R. Pathways of cross-species transmission of synthetically reconstructed zoonotic severe acute respiratory syndrome coronavirus. J. Virol. 82, 8721–8732 (2008).
Poon, L. L. et al. Identification of a novel coronavirus in bats. J. Virol. 79, 2001–2009 (2005).
Lau, S. K. et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl Acad. Sci. USA 102, 14040–14045 (2005).
Leung, D. T. et al. Extremely low exposure of a community to severe acute respiratory syndrome coronavirus: false seropositivity due to use of bacterially derived antigens. J. Virol. 80, 8920–8928 (2006).
Becker, M. M. et al. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl Acad. Sci. USA 105, 19944–19949 (2008). This manuscript describes the engineering of a non-cultivable bat SARS-like coronavirus using synthetic DNA technology.
Ren, W. et al. Difference in receptor usage between severe acute respiratory syndrome (SARS) coronavirus and SARS-like coronavirus of bat origin. J. Virol. 82, 1899–1907 (2008).
Vijgen, L. et al. Complete genomic sequence of human coronavirus OC43: molecular clock analysis suggests a relatively recent zoonotic coronavirus transmission event. J. Virol. 79, 1595–1604 (2005).
Alekseev, K. P. et al. Bovine-like coronaviruses isolated from four species of captive wild ruminants are homologous to bovine coronaviruses based on complete genomic sequences. J. Virol. 82, 12422–12431 (2008).
Jin, L. et al. Analysis of the genome sequence of an alpaca coronavirus. Virology 365, 198–203 (2007).
Lorusso, A. et al. Gain, preservation, and loss of a group 1a coronavirus accessory glycoprotein. J. Virol. 82, 10312–10317 (2008).
Woo, P. C. et al. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 81, 1574–1585 (2007).
Dominguez, S. R., O'Shea, T. J., Oko, L. M. & Holmes, K. V. Detection of group 1 coronaviruses in bats in North America. Emerg. Infect. Dis. 13, 1295–1300 (2007).
Gloza-Rausch, F. et al. Detection and prevalence patterns of group I coronaviruses in bats, northern Germany. Emerg. Infect. Dis. 14, 626–631 (2008).
Tong, S. et al. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis. 15, 482–485 (2009).
Woo, P. C. et al. Comparative analysis of complete genome sequences of three novel avian coronaviruses reveals a novel group 3c coronavirus. J. Virol. 83, 908–917 (2009).
Dong, B. Q. et al. Detection of a novel and highly divergent coronavirus from asian leopard cats and Chinese ferret badgers in Southern China. J. Virol. 81, 6920–6926 (2007).
Mihindukulasuriya, K. A., Wu, G., St Leger, J., Nordhausen, R. W. & Wang, D. Identification of a novel coronavirus from a beluga whale by using a panviral microarray. J. Virol. 82, 5084–5088 (2008).
Perlman, S. & Dandekar, A. A. Immunopathogenesis of coronavirus infections: implications for SARS. Nature Rev. Immunol. 5, 917–927 (2005).
Bergmann, C. C., Lane, T. E. & Stohlman, S. A. Coronavirus infection of the central nervous system: host–virus stand-off. Nature Rev. Microbiol. 4, 121–132 (2006).
Savarin, C., Bergmann, C. C., Hinton, D. R., Ransohoff, R. M. & Stohlman, S. A. Memory CD4+ T-cell-mediated protection from lethal coronavirus encephalomyelitis. J. Virol. 82, 12432–12440 (2008).
Kim, T. S. & Perlman, S. Virus-specific antibody, in the absence of T cells, mediates demyelination in mice infected with a neurotropic coronavirus. Am. J. Pathol. 166, 801–809 (2005).
Kim, T. S. & Perlman, S. Viral expression of CCL2 is sufficient to induce demyelination in RAG1−/− mice infected with a neurotropic coronavirus. J. Virol. 79, 7113–7120 (2005).
Williamson, J. S. & Stohlman, S. A. Effective clearance of mouse hepatitis virus from the central nervous system requires both CD4+ and CD8+ T cells. J. Virol. 64, 4589–4592 (1990).
Anghelina, D., Pewe, L. & Perlman, S. Pathogenic role for virus-specific CD4 T cells in mice with coronavirus-induced acute encephalitis. Am. J. Pathol. 169, 209–222 (2006).
Deming, D. et al. Vaccine efficacy in senescent mice challenged with recombinant SARS-CoV bearing epidemic and zoonotic spike variants. PLoS Med. 3, e525 (2006).
Roberts, A. et al. A mouse-adapted SARS-Coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 3, e5 (2007). This is the first report showing that a mouse-adapted SARS-CoV causes a SARS-like illness in mice.
Roberts, A. et al. Aged BALB/c mice as a model for increased severity of severe acute respiratory syndrome in elderly humans. J. Virol. 79, 5833–5838 (2005).
Versteeg, G. A., Bredenbeek, P. J., van den Worm, S. H. & Spaan, W. J. Group 2 coronaviruses prevent immediate early interferon induction by protection of viral RNA from host cell recognition. Virology 361, 18–26 (2007). This report and Ref. 106 are the first to suggest that coronaviruses are invisible to the host innate immune response in some cells.
Zhou, H. & Perlman, S. Mouse hepatitis virus does not induce beta interferon synthesis and does not inhibit its induction by double-stranded RNA. J. Virol. 81, 568–574 (2007).
Spiegel, M. et al. Inhibition of beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. J. Virol. 79, 2079–2086 (2005).
He, R. et al. Activation of AP-1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem. Biophys. Res. Commun. 311, 870–876 (2003).
Kopecky-Bromberg, S. A., Martinez-Sobrido, L., Frieman, M., Baric, R. A. & Palese, P. SARS coronavirus proteins Orf 3b, Orf 6, and nucleocapsid function as interferon antagonists. J. Virol. 81, 548–557 (2006).
Ye, Y., Hauns, K., Langland, J. O., Jacobs, B. L. & Hogue, B. G. Mouse hepatitis coronavirus A59 nucleocapsid protein is a type I interferon antagonist. J. Virol. 81, 2554–2563 (2007).
Narayanan, K. et al. Severe acute respiratory syndrome coronavirus nsp1 suppresses host gene expression, including that of type I interferon, in infected cells. J. Virol. 82, 4471–4479 (2008).
Wathelet, M. G., Orr, M., Frieman, M. B. & Baric, R. S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 81, 11620–11633 (2007).
Zust, R. et al. Coronavirus non-structural protein 1 is a major pathogenicity factor: implications for the rational design of coronavirus vaccines. PLoS Pathog. 3, e109 (2007).
Frieman, M. et al. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 81, 9812–9824 (2007).
Hussain, S., Perlman, S. & Gallagher, T. M. Severe acute respiratory syndrome coronavirus protein 6 accelerates murine hepatitis virus infections by more than one mechanism. J. Virol. 82, 7212–7222 (2008).
Zhao, J. et al. Severe acute respiratory syndrome-CoV protein 6 is required for optimal replication. J. Virol. 83, 2368–2373 (2008).
Kamitani, W. et al. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proc. Natl Acad. Sci. USA 103, 12885–12890 (2006). Describes a novel mechanism of viral protein-induced inhibition of host gene expression.
Devaraj, S. G. et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 282, 32208–32221 (2007).
Cameron, M. J. et al. Interferon-mediated immunopathological events are associated with atypical innate and adaptive immune responses in patients with severe acute respiratory syndrome. J. Virol. 81, 8692–8706 (2007). This report provides a careful description of the changes in cytokine and chemokine expression that occurred in patients during the 2002–2003 epidemic.
Rempel, J. D., Murray, S. J., Meisner, J. & Buchmeier, M. J. Differential regulation of innate and adaptive immune responses in viral encephalitis. Virology 318, 381–392 (2004).
Ireland, D. D., Stohlman, S. A., Hinton, D. R., Atkinson, R. & Bergmann, C. C. Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J. Virol. 82, 300–310 (2008).
Cervantes-Barragan, L. et al. Control of coronavirus infection through plasmacytoid dendritic cell-derived type I interferon. Blood 109, 1131–1137 (2006).
Roth-Cross, J. K., Bender, S. J. & Weiss, S. R. Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J. Virol. 82, 9829–9838 (2008).
Cervantes-Barragan, L. et al. Type I IFN-mediated protection of macrophages and dendritic cells secures control of murine coronavirus infection. J. Immunol. 182, 1099–1106 (2009).
He, L. et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: relation to the acute lung injury and pathogenesis of SARS. J. Pathol. 210, 288–297 (2006).
Li, C. K. et al. T cell responses to whole SARS coronavirus in humans. J. Immunol. 181, 5490–5500 (2008).
Gu, J. & Korteweg, C. Pathology and pathogenesis of severe acute respiratory syndrome. Am. J. Pathol. 170, 1136–1147 (2007).
Chen, J. & Subbarao, K. The immunobiology of SARS. Annu. Rev. Immunol. 25, 443–472 (2007).
Subbarao, K. & Roberts, A. Is there an ideal animal model for SARS? Trends Microbiol. 14, 299–303 (2006).
McCray, P. B. Jr et al. Lethal infection in K18-hACE2 mice infected with SARS-CoV. J. Virol. 81, 813–821 (2006). This paper and REF. 131 show that mice transgenic for the human SARS-CoV develop a lethal infection characterized by extensive infection of the brain.
Tseng, C. T. et al. SARS coronavirus infection of mice transgenic for the human angiotensin-converting enzyme 2 (hACE2) virus receptor. J. Virol. 81, 1162–1173 (2006).
Netland, J., Meyerholz, D. K., Moore, S., Cassell, M. & Perlman, S. Severe acute respiratory syndrome coronavirus infection causes neuronal death in the absence of encephalitis in mice transgenic for human ACE2. J. Virol. 82, 7264–7275 (2008).
Lee, D. T. et al. Factors associated with psychosis among patients with severe acute respiratory syndrome: a case-control study. Clin. Infect. Dis. 39, 1247–1249 (2004).
Xu, J. et al. Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine mig in pathogenesis. Clin. Infect. Dis. 41, 1089–1096 (2005).
Nagata, N. et al. Mouse-passaged severe acute respiratory syndrome-associated coronavirus leads to lethal pulmonary edema and diffuse alveolar damage in adult but not young mice. Am. J. Pathol. 172, 1625–1637 (2008).
Nagata, N. et al. Participation of both host and virus factors in induction of severe acute respiratory syndrome (SARS) in F344 rats infected with SARS coronavirus. J. Virol. 81, 1848–1857 (2007).
de Lang, A. et al. Functional genomics highlights differential induction of antiviral pathways in the lungs of SARS-CoV-infected macaques. PLoS Pathog. 3, e112 (2007).
Baas, T. et al. Genomic analysis reveals age-dependent innate immune responses to severe acute respiratory syndrome coronavirus. J. Virol. 82, 9465–9476 (2008).
Morahan, G., Balmer, L. & Monley, D. Establishment of “The Gene Mine”: a resource for rapid identification of complex trait genes. Mamm. Genome 19, 390–393 (2008).
Stockman, L. J., Bellamy, R. & Garner, P. SARS: systematic review of treatment effects. PLoS Med. 3, e343 (2006).
Gosert, R., Kanjanahaluethai, A., Egger, D., Bienz, K. & Baker, S. C. RNA replication of mouse hepatitis virus takes place at double-membrane vesicles. J. Virol. 76, 3697–3708 (2002).
Sims, A. C., Ostermann, J. & Denison, M. R. Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes. J. Virol. 74, 5647–5654 (2000).
Acknowledgements
Supported in part by research (PO1 AI060699 and RO1 NS36592) and training (T32 AI007533) grants from the National Institutes of Health (USA).
Author information
Authors and Affiliations
Corresponding author
Related links
Glossary
- Prothrombinase
-
Molecule that cleaves thrombin, thereby initiating the coagulation cascade.
- Primase
-
In the case of nsp8, an RNA-dependent RNA polymerase that produces RNA primers that are required for initiation of RNA synthesis by the main viral RNA polymerase, nsp12.
- Double-membrane vesicle
-
A structure that is observed in electron micrographs of infected cells and that is thought to be the site of virus replication.
- Collaborative cross mice
-
A panel of 1,000 recombinant inbred mouse strains derived from 8 genetically diverse founder strains. The crosses were designed for complex trait analysis and will be useful for identifying and establishing the role of host genes in SARS pathogenesis.
Rights and permissions
About this article
Cite this article
Perlman, S., Netland, J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7, 439–450 (2009). https://doi.org/10.1038/nrmicro2147
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro2147
This article is cited by
-
SARS-CoV-2 egress from Vero cells: a morphological approach
Histochemistry and Cell Biology (2024)
-
Emerging SARS-CoV-2 variants of concern potentially expand host range to chickens: insights from AXL, NRP1 and ACE2 receptors
Virology Journal (2023)
-
Prevalence and outcome of chronic hepatitis C patients admitted with COVID-19 to intensive care units: a blessing in disguise
Ain-Shams Journal of Anesthesiology (2023)
-
Secondary zoonotic dog-to-human transmission of SARS-CoV-2 suggested by timeline but refuted by viral genome sequencing
Infection (2023)
-
Covid-19 and its relation to the human eye: transmission, infection, and ocular manifestations
Graefe's Archive for Clinical and Experimental Ophthalmology (2023)
