
Key Points
-
The term 'killer strains' is used to describe yeast and fungal species that produce secreted toxins — known as 'killer toxins' — that have antimycotic activity. This killer phenotype can be associated with double-stranded (ds)RNA mycoviruses and linear dsDNA plasmids or can be chromosomally encoded. This article focuses on the killer phenotype associated with dsRNA viruses, with the emphasis on Saccharomyces cerevisiae.
-
In S. cerevisiae, three killer viruses have been identified — ScV-M1, ScV-M2 and ScV-M28 — each of which produces a specific killer toxin (K1, K2 and K28, respectively). In each case, the killer phenotype requires the presence of two different dsRNA viruses: an L-A helper virus and the toxin-coding killer virus. Besides K1, the K28 killer toxin is the most well studied.
-
The K28 toxin is initially translated in the yeast cytoplasm as a preprotoxin. Following processing and protoxin folding in the endoplasmic reticulum (ER) and late-Golgi compartment, the biologically active protein is secreted into the culture medium as an α/β heterodimer. The β-subunit of the K28 toxin precursor contains a classical ER-retention signal (HDEL) that is initially masked by a C-terminal arginine residue, allowing the toxin precursor to pass through the secretory pathway from the ER to the Golgi. The terminal arginine is removed by a processing reaction in the late-Golgi compartment and the HDEL sequence is then accessible for interaction with the HDEL receptor in the target cell.
-
The secreted mature toxin interacts with receptors in the cell wall and cytoplasmic membrane of sensitive yeast strains. To date, the membrane receptor for K28 has not been definitively identified, although current evidence suggests that it may be the cellular HDEL receptor Erd2p. The membrane receptor for toxin K1 has been identified — Kre1p, an O-glycosylated yeast cell-surface protein that is involved in β-1,6-glucan biosynthesis.
-
After endocytotic uptake and retrograde transport through the Golgi and ER to the cytosol, the K28 β-subunit is ubiquitinated and proteasomally degraded, and the α-subunit exerts its lethal effect in the nucleus, blocking DNA synthesis, causing cell-cycle arrest in early S phase. By contrast, ionophoric virus toxins, such as S. cerevisiae K1 and the Zygosaccharomyces bailii toxin zygocin, disrupt cytoplasmic membrane function by forming cation-selective ion channels.
-
A characteristic of the yeast-killer phenomenon is that toxin-producing yeast strains are immune to the toxin that they produce. A model is presented for K28 functional immunity, which suggests that mature, secreted K28 toxin — either self-produced or produced by other K28 killer cells — is taken up and transported through the Golgi and ER. Once in the cytosol, the mature toxin forms a complex with preprotoxin molecules that have not yet been imported into the ER, which can be ubiquitinated and then proteasomally degraded, protecting the cell from the lethal effects of the α-subunit.
Abstract
Since the discovery of toxin-secreting killer yeasts more than 40 years ago, research into this phenomenon has provided insights into eukaryotic cell biology and virus–host-cell interactions. This review focuses on the most recent advances in our understanding of the basic biology of virus-carrying killer yeasts, in particular the toxin-encoding killer viruses, and the intracellular processing, maturation and toxicity of the viral protein toxins. The strategy of using eukaryotic viral toxins to effectively penetrate and eventually kill a eukaryotic target cell will be discussed, and the cellular mechanisms of self-defence and protective immunity will also be addressed.
Similar content being viewed by others

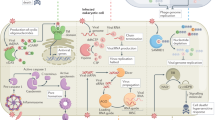

Main
The discovery of toxin-secreting strains of the yeast Saccharomyces cerevisiae , and their phenotypic association with the presence of cytoplasmically inherited double-stranded RNA (dsRNA) viruses, marked the beginning of research into yeast virology in the early 1970s1,2,3. At that time, it was shown that certain yeast strains secrete protein toxins that are lethal to sensitive strains. The toxin-secreting strains were designated 'killer yeasts', and the term 'killer toxin' was used to describe the secreted proteins. Shortly after this discovery, it became evident that toxin-producing killer strains have remarkable antimycotic activity and are not restricted to S. cerevisiae but are frequently found in other yeast and fungal species and genera4,5,6, including Zygosaccharomyces bailii, Hanseniaspora uvarum and Ustilago maydis 7,8,9,10,11,12. With the exception of toxin-secreting strains of Z. bailii, killer-toxin production in yeast is usually associated with specific immunity to this toxin (reviewed in Refs 13, 14).
The killer phenotype is not exclusively associated with dsRNA viruses and is also encoded by linear dsDNA plasmids (in Kluyveromyces lactis and Pichia acaciae)14 and chromosomally encoded (in Williopsis californica and Pichia farinosa )15,16. This article will deal only with the killer phenotype associated with dsRNA viruses, with the emphasis on S. cerevisiae.
The virus genomes
In S. cerevisiae, the killer phenotype is caused by an infection with cytoplasmic-persisting dsRNA viruses of the Totiviridae family, a member of the constantly growing class of mycoviruses, which are widely distributed among yeast and higher fungi17,18. So far, in S. cerevisiae three major killer viruses have been discovered (ScV-M1, ScV-M2 and ScV-M28), with each virus encoding a specific killer toxin (K1, K2 and K28, respectively) and a self-protective immunity component19,20,21,22,23. In each case, the killer phenotype requires the presence of two different dsRNA viruses: an L-A helper virus and the toxin-coding (M) killer virus. In vivo, both dsRNA genomes are separately encapsidated into virus-like particles (VLPs) that stably persist in the cytoplasm of the infected yeast cell. The L-A virus alone does not confer a phenotype upon its host nor does it lead to cell lysis or slow cell growth. All known fungal viruses spread horizontally by cell–cell mating or heterokaryon formation.
As summarized in Table 1, the linear dsRNA genome of the L-A helper virus contains two open reading frames (ORFs) on its positive strand: ORF1 encodes the major capsid protein, Gag, which is necessary for encapsidation and viral particle structure, and ORF2 encodes the RNA-dependent RNA polymerase Pol, which is expressed as a Gag–Pol fusion protein by a −1 ribosomal frameshift event24,25,26,27. In contrast to L-A, each of the three known S. cerevisiae M virus dsRNA genomes contains a single ORF coding for a preprotoxin (pptox), the unprocessed precursor of the mature secreted killer toxin, which also confers functional immunity (Table 1). As each toxin-coding M virus depends on the coexistence of an L-A helper virus for stable maintenance and replication, M viruses resemble classical satellite viruses of L-A. The presence of all three killer virions in a single yeast cell does not occur in vivo, as the toxin-coding M genomes exclude each other at the replicative level; the underlying mechanism of this exclusion is still unknown. However, this limitation can be bypassed in vitro by introducing cDNA copies of the K2 and K28 pptox genes into a natural K1 killer, resulting in stable 'triple-killer' strains that produce three different killer toxins and simultaneously express triple toxin immunity28,29,30.
Viral replication cycle
Intensive studies, mainly in the labs of Reed Wickner and Jeremy Bruenn, have shown that L-A virions are non-infectious icosahedral particles with a diameter of 39 nm that show many striking similarities to mammalian reoviruses and rotaviruses31,32,33,34. Each L-A virion consists of a single copy of the 4.6-kb L-A dsRNA genome, which is encapsidated by 60 asymmetric dimers of the 76-kDa coat protein Gag and two copies of the 171-kDa Gag–Pol fusion protein. During the conservative replication cycle of L-A, a single-stranded positive-strand RNA ((+)ssRNA) is transcribed and subsequently extruded from the virion into the yeast-cell cytoplasm35,36,37. On the one hand, this (+)ssRNA serves as an mRNA for translation into the viral proteins Gag and Gag–Pol; on the other hand, it serves as an RNA template that is packaged into new viral particles (Fig. 1). Once viral coat assembly is completed, Gag–Pol functions as a replicase, synthesizing a new negative strand and generating the dsRNA genome of the mature virus, completing the viral replication cycle. The replication cycle of the toxin-coding M viruses resembles that of L-A, with the exception that each M virion can accept two copies of the smaller M dsRNA genome before the toxin-coding transcripts are extruded into the cytoplasm, a phenomenon that has been described as 'headful packaging'38,39 in analogy to some DNA bacteriophages.

The killer virus (M) and the helper virus (L-A) are both double-stranded RNA (dsRNA) viruses. They compete for the L-A-encoded viral proteins Gag and Gag–Pol, which are essential for (a) single-stranded RNA (ssRNA) encapsidation, (b) virion assembly, (c) negative-strand RNA synthesis (replication), (d) positive-strand RNA synthesis (transcription) and extrusion from the particles into the cytosol, (e) ssRNA translation and (f) ssRNA binding.
Precursor processing and toxin secretion
In yeast harbouring a killer virus, the toxin-encoding (+)ssRNA transcript is translated in the cytoplasm into a pptox that subsequently enters the secretory pathway for further processing, maturation and toxin secretion. In a eukaryotic cell, the secretory pathway is an essential pathway for newly synthesized proteins that are destined for the extracellular space, the plasma membrane or endocytic compartments including endosomes, vacuoles and lysosomes. Entry into this pathway is mediated by a hydrophobic signal sequence at the N terminus of the protein that directs co- and/or post-translational import into the endoplasmic reticulum (ER), where the environment is optimized for protein folding and maturation.
In the case of a yeast killer toxin, the unprocessed toxin precursor consists of an N-terminal signal sequence, which is necessary for pptox import into the lumen of the ER, followed by the α- and β-subunits of the mature toxin separated from each other by a potentially N-glycosylated γ-sequence (Fig. 2). During passage through the yeast secretory pathway, the toxin precursor is enzymatically processed to the biologically active α/β heterodimer in a way that is homologous to prohormone conversion in mammalian cells40,41. In a late-Golgi compartment, the N-glycosylated γ-sequence is removed by the action of the furin-like endopeptidase Kex2p (Ref. 42), the C terminus of the β-subunit is trimmed by the carboxypeptidase Kex1p, and the biologically active protein is secreted into the culture medium as an α/β heterodimer in which the subunits are covalently linked by one or more disulphide bonds (Fig. 2). In the case of the K28 toxin precursor, the C terminus of the β-subunit contains a four-amino-acid epitope that represents a classical ER-retention signal (HDELR), which is normally found on soluble proteins resident in the ER lumen. As this signal is initially masked by a carboxy-terminal arginine residue, ER retention of the toxin precursor is effectively prevented until the protoxin enters the late-Golgi compartment and Kex1p cleavage uncovers the retention signal43.

After in vivo translation of the preprotoxin-coding killer virus transcript, the toxin precursor is post-translationally imported into the lumen of the endoplasmic reticulum (ER) with the help of cytosolic chaperones (Ssa1p–Ssa4p). Following import into the ER through the Sec61p pore complex, signal peptidase (SP) cleavage removes the N-terminal secretion signal (pre-region), and protoxin folding is initiated and catalysed by the action of the Hsp70 chaperones BiP (Kar2p) and calnexin (Cne1p). The intervening γ-sequence is core-N-glycosylated, and a single disulphide bond between α and β is generated by the action of protein disulphide isomerase, Pdi1p. In a late-Golgi compartment, the Kex2p endopeptidase removes both the pro-region and the intramolecular γ-sequence, whereas carboxypeptidase Kex1p cleavage trims the C termini of both subunits, leading to the secretion of mature α/β toxin, the β-C-terminal HDEL motif of which is uncovered and, therefore, accessible for interaction with the HDEL receptor of the target cell.
Uptake of K28 toxin and retrograde transport
To date, the yeast K28 toxin is the sole example of a killer toxin that is taken up by endocytosis after binding to the surface of a sensitive cell44. Yeast mutants that are blocked in the early steps of both fluid-phase endocytosis and receptor-mediated endocytosis are toxin resistant, as the toxin cannot enter the cell and reach its final target compartment. Although the K28 membrane receptor has not yet been identified, there is growing evidence that it might be the cellular HDEL receptor Erd2p, which co-localizes — in low copy number — to the cytoplasmic membrane (J. Spindler, S. Heiligenstein and M.J.S., unpublished data). In accordance with this assumption, K28-toxin-treated cells that lack the HDEL receptor are K28 resistant, accumulate the toxin at the plasma membrane and are incapable of internalizing the toxin44. Interestingly, this phenotype is reflected in a mutated K28 derivative that lacks the β-C-terminal HDEL sequence; this toxin is inactive and unable to enter cells43.
Once the toxin has been internalized and targeted to an early endosomal compartment, it travels the secretion pathway in reverse and enters the yeast-cell cytoplasm. This retrograde transport is mediated by the coat protein COPI and the HDEL ER-targeting motif at the C terminus of the β-subunit, which is exposed after Kex1p cleavage, as discussed above. In yeast and higher eukaryotes, H/KDEL-carrying proteins resemble resident proteins of the ER lumen that are recognized and subsequently recycled from an early Golgi compartment back to the ER by an ER-membrane-bound H/KDEL receptor45,46. In the case of the K28 virus toxin, this sequence allows retrograde transport through the Golgi and ER, and ensures that the toxin can enter the cytoplasm and subsequently transduce its lethal signal into the nucleus. The disulphide bond that covalently links the two subunits of the heterodimeric toxin is believed to be important in ensuring accessibility of the β-C-terminal ER-targeting signal to the HDEL target-cell receptor43.
The strategy of using endocytosis and retrograde transport is a common phenomenon for many bacterial protein toxins47. Pseudomonas exotoxin A, for instance, has also been shown to be internalized by receptormediated endocytosis, followed by reverse secretion through the Golgi and ER. As many other microbial A/B toxins, such as the Escherichia coli toxins heat-labile enterotoxin and Shiga toxin, contain putative ER-retention signals at their C termini, H/KDEL-dependent mechanisms seem to have general importance for toxin entry into eukaryotic target cells48,49,50. In this respect, the main difference between the virally encoded K28 killer toxin and most bacterial A/B toxins is that K28 itself is produced and secreted by a eukaryotic cell, and therefore the ER-targeting signal at the β-C-terminus is initially masked by a terminal arginine residue. This strategy ensures that the viral toxin can successfully pass through the early secretory pathway. Once it has reached a late Golgi compartment, the terminal arginine residue is no longer needed for intracellular toxin transport and is cleaved by Kex1p. Consequently, expression of a truncated pptox in which the carboxy-terminal arginine has been deleted results in a loss of toxicity as the toxin is retained in the ER43,44.
Retrotranslocation of K28 from the ER. Once the toxin has reached the ER, it is retrotranslocated to the cytosol, from where it transduces the toxic signal into the nucleus (Fig. 3). ER-to-cytosol export of K28 is mediated by the Sec61p complex (termed the translocon), a major transport channel in the ER membrane of yeast and higher eukaryotes44,51. In yeast, each translocon contains a core heterotrimeric complex consisting of the transmembrane protein Sec61p and two smaller subunits, Sbh1p and Sss1p (Ref. 52). In addition to being the main channel for co-translational and post-translational protein import into the ER, Sec61p is also involved in the export and removal of misfolded proteins from the secretory pathway and their subsequent proteasomal degradation in the cytosol53,54,55. This mechanism is an important quality-control system in the ER of eukaryotes which ensures that only correctly folded proteins reach their final destination. Native proteins that successfully passed this 'proof-reading' checkpoint are further transported through the secretory pathway, whereas non-native proteins and incompletely assembled conformers are eventually sorted for ER export and proteasomal degradation in a process called ER-associated degradation (ERAD)56. Sec61p as a central component of ER export and ERAD has been shown to be responsible for ER retrotranslocation of plant and microbial A/B toxins such as ricin57, cholera toxin58 and Pseudomonas exotoxin A59, as well as the yeast K28 virus toxin44. However, in contrast to most H/KDEL-carrying microbial toxins, ER-to-cytosol translocation of K28 does not depend on ERAD and, consequently, yeast mutants defective in 'classical' ERAD components show wild-type sensitivity6.

After endocytotic uptake and retrograde transport through the Golgi and endoplasmic reticulum (ER), the toxin is gated through the major export channel (Sec61p) with the help of the lumenal ER chaperone Kar2p. In the cytosol, the β-subunit is ubiquitinated and proteasomally degraded, whereas the α-subunit enters the nucleus. Through the toxin's interaction with essential cellular proteins that are normally involved in eukaryotic cell-cycle control, the toxin kills the host cell by irreversibly blocking DNA synthesis. Cellular components of the ER quality-control pathway ERAD (such as Cue1p, Ubc7p, Der3p/Hrd1p, Ubc6p and Der1p) are not involved in ER-to-cytosol export of the toxin (ERAD components are shown in grey).
Yeast cells carrying a mutated Sec61p translocon, and yeast sec63 mutants, are toxin resistant owing to a block in toxin retrotranslocation to the cytosol44. Recently, it was shown that this Sec61p/Sec63p-mediated ER-to-cytosol export depends on the action of the lumenal ER chaperone Kar2p (binding protein or BiP), to ensure that the heterodimeric toxin is competent for translocation44. However, neither well-known ERAD components nor components of the ubiquitin/proteasome degradation machinery are involved in K28 translocation to the cytosol6,60 (Fig. 3). So far, the component(s) near the ER membrane that are required, or sufficient, for toxin exit through the Sec61p complex are still unknown, and it is not known whether toxin ubiquitination and proteasomal degradation generates the driving force for retrograde transport through the ER membrane. In this respect, it is interesting to note that a pptox variant that lacks all internal lysine residues is still processed in vivo to a biologically active α/β toxin, indicating that toxin retrotranslocation from the ER might be independent of ubiquitination (F.B. et al., unpublished data). Once in the cytosol (at the cytosolic face of the ER), the β-subunit is subsequently ubiquitinated and targeted for proteasomal degradation, whereas the α-subunit enters the nucleus and causes cell death (see below).
Mode of killer-toxin action
Viral killer toxins kill sensitive yeast in a receptor-mediated process by interacting with receptors in the yeast cell wall and cytoplasmic membrane. The initial step involves fast, energy-independent binding to a primary toxin receptor, R1, in the mannoprotein or β-1,6-glucan fraction of the cell wall61,62. It has been speculated that toxin binding to R1 either concentrates the toxin at the level of the cell wall or mediates close contact between the toxin and the target cell membrane. Susceptible strains can become toxin resistant by chromosomal mutations in a set of genes that encode proteins involved in the structure and assembly of the yeast cell wall63. The second, energy-dependent step involves toxin translocation to the cytoplasmic membrane and interaction with a secondary membrane receptor, R2. To date, only the membrane receptor for toxin K1 has been identified — Kre1p, an O-glycosylated yeast cell-surface protein which is initially anchored to the plasma membrane through a glycosylphosphatidylinositol (GPI) anchor and is involved in both β-1,6-glucan biosynthesis and assembly of the K1 cell-wall receptor64,65. After reaching the plasma membrane, ionophoric virus toxins such as K1 and zygocin (the latter toxin is produced by Z. bailii) disrupt cytoplasmic membrane function by forming cation-selective ion channels, whereas K28 toxins block DNA synthesis and arrest cells in early S phase of the cell cycle, creating a medium-sized bud and a single, pre-replicated nucleus in the mother cell66,67,68. Both of these mechanisms are discussed in more detail below.
Ionophoric virus toxins
K1 toxin. K1-toxin-induced ion-channel formation in yeast membranes was initially reported using patch-clamping techniques, and was thought to be a result of direct toxin action68. However, this observation is inconsistent with the resistance found in immune spheroplasts, and no receptor-independent channels were observed in a recent study in which it was shown that K1 activates Tok1p potassium channels in yeast membranes as well as in Tok1p-expressing Xenopus oocytes69. These channels were postulated to be required for the lethal interaction of K1 with the plasma membrane and, furthermore, to be potential K1 membrane receptors. It was assumed that exposure of Tok1p channels to K1 at the cytosolic face of the plasma membrane would prevent channel activation from the external face, and this was postulated to be the mechanism of K1 toxin immunity70. However, several studies have subsequently shown that Tok1p channels are neither the K1 membrane receptor nor the primary toxin target, nor are they responsible for K1 immunity64,71,72. Any effect on Tok1p channels is, therefore, likely to be a late secondary event and is probably not the physiologically relevant lethal mechanism of the K1 virus toxin. So, the primary target of K1 and its molecular mode of cell killing remain obscure.
Zygocin. Zygocin is a monomeric antifungal protein toxin that is secreted by virus-infected strains of the osmotolerant yeast Z. bailii. Its broad killing spectrum encompasses phytopathogenic and human-pathogenic yeast and fungi, including Candida albicans , Candida glabrata , Candida tropicalis , Sporothrix schenckii and the filamentous fungi Fusarium oxysporum and Colletotrichum graminicola73. Similar to the ionophoric activity of K1, zygocin negatively affects plasma-membrane permeability in vivo, although the kinetics of zygocin-mediated membrane permeabilization are much faster, and equivalent molar amounts of K1 require a significantly prolonged time span to achieve a comparable reduction in cell viability68,73. The ionophoric mode of zygocin action is reinforced by in silico sequence analysis74, which shows the presence of a stretch of potential α-helical conformation that forms an amphipathic structure characteristic of various membrane-disturbing antimicrobial peptides such as alamethicin, melittin and dermaseptin. Zygocin also contains a transmembrane helix at the C terminus that is predicted to favour a membrane-permeabilizing potential, not by activating native ion channels but instead by establishing pores itself after toxin oligomerization (Fig. 4). It has been postulated, therefore, that the hydrophobic region in the amphipathic α-helix of zygocin is responsible for toxin binding to the yeast-cell surface. Initial toxin adsorption would only be limited by the toxin's ability to overcome the cell-wall barrier or by additional physicochemical factors that affect zygocin's hydrophilic–hydrophobic transition from the aqueous medium to the cytoplasmic membrane. Therefore, the postulated model of zygocin action (Fig. 4) resembles the mechanism of toxicity of certain α-defensins of human origin75. Although non-specific toxin binding seems to be essential, it is not sufficient to initiate cell killing, as zygocin-producing cells can bind toxin at the plasma-membrane level without being sensitive to the toxin76. In analogy to alamethicin, the toxic effect of zygocin could be mediated by incorporation of its transmembrane helix into the plasma membrane, a process solely driven by the natural transmembrane potential of the energized yeast plasma membrane77,78.

This model is based on experimental data, in silico structure prediction and analogy to antimicrobial peptides of higher eukaryotes. (a) Shows the domain structure of zygocin; (b), unspecific adsorption of the non-polar side of the α-helix (D2) after overcoming the cell-wall barrier by binding to the cell-wall mannoprotein R1 receptor (through domain D1) and recognition of a putative membrane receptor or docking protein; (c), membrane insertion of domain D3 driven by the transmembrane potential of the target cell and formation of a transmembrane helix; (d), toxin oligomerization by self-assembly and subsequent plasma-membrane permeabilization.
This mode of action, along with its rapid energy-dependent toxicity, shows striking similarities to membrane-disturbing peptides produced by virtually all higher eukaryotes79,80. There are few mechanisms of resistance against antimicrobial peptides, and these are mostly limited to changes in the composition of the cytoplasmic membrane. In contrast to higher eukaryotic cells, the outer leaflet of microbial membranes is enriched in negatively charged lipids. Owing to the cationic net charge of antimicrobial peptides (including zygocin), there might be an affinity for these lipids, facilitating toxin adsorption to the target membrane. So it was rather surprising when species of Morganella and Serratia were discovered that showed a resistant phenotype to antimicrobial peptides that was triggered by a reduction in negatively charged lipids in the plasma membrane81. In agreement with this observation, mutants of Staphylococcus aureus that have a more negatively charged membrane than the wild-type are more susceptible to antimicrobial peptides, reflecting significantly improved membrane binding of cationic antimicrobial peptides82.
The recent finding that deletions in the chromosomal yeast genes PDR16 and PDR17 cause a dramatic decrease in zygocin sensitivity at the plasma-membrane level will be extremely valuable in further elucidating its molecular mode of action (F. Weiler and M.J.S., unpublished data). The corresponding gene products affect plasma-membrane lipid composition which, in turn, causes a significant decrease in the sensitivity of yeast Δpdr16 and Δpdr17 mutants to various toxic substances83. Owing to a dramatic reduction in membrane sterol concentration, yeast pdr16 mutants are significantly more sensitive to azole antimycotics, which interfere with ergosterol biosynthesis84. However, whereas the proportion of negatively charged lipids is severely decreased in a Δpdr16 knock-out mutant, the opposite effect can be seen in a yeast Δpdr17 mutant83. As the same studies indicated that Pdr16p/Pdr17p activity is limited to the composition of the cytoplasmic membrane, it can be assumed that changes in plasma-membrane lipid composition are exclusively responsible for the reduced zygocin sensitivity of yeast pdr16/pdr17 mutants. In contrast to K1, a zygocin-specific membrane receptor has not yet been identified, and the effect of a pdr16/pdr17 mutation cannot be attributed to the absence of a particular membrane lipid or a single membrane-docking protein. However, membrane-permeabilizing proteins do not necessarily require a specific secondary membrane receptor or docking protein, as has recently been shown for the antimicrobial polypeptide DmAMP1 produced by Dahlia merckii. The cytocidal effect of this antimicrobial protein, which belongs to the diverse group of defensins, depends on sphingolipid-containing membranes85. Therefore, knowledge of the molecular structure of the zygocin protein is required to gain deeper insight into its native conformation and in vivo toxicity.
K28 toxin. As the cytotoxic α-subunit of K28 is only 10.5 kDa, it can enter the nucleus by passive diffusion without the need for an active nuclear import machinery67. However, if the α-subunit is additionally modified and extended by a classical nuclear-localization sequence (NLS), the in vivo toxicity is significantly enhanced owing to faster and more efficient nuclear import mediated by α/β importins in the host cell (F. Reiter and M.J.S., unpublished data). Once in the nucleus, K28-α specifically interacts with host proteins that are essential in eukaryotic cell-cycle control and progression as well as in initiating DNA synthesis in early S phase. Therefore, because K28 targets essential and evolutionarily conserved host proteins with basic cellular functions, resistance mechanisms based on mutations in essential chromosomal genes rarely occur in vivo, and so the K28 virus toxin has developed an 'intelligent' strategy to effectively penetrate and kill its target cell.
Toxin-induced apoptosis
In yeast, apoptotic markers such as DNA fragmentation, chromatin condensation, exposure of phosphatidylserine on the outer surface of the plasma membrane, accumulation of reactive oxygen species (ROS) and phenotypic changes can be induced by various factors such as H2O2, cell ageing, acetic acid and α-factor pheromone treatment86,87,88,89. Interestingly, active cell death can also be induced by treating yeast with low doses of viral killer toxins, whereas high toxin concentrations prevent apoptosis and cause necrotic cell killing90. Phenotypic analysis of the pronounced toxin hypersensitivity in glutathione-deficient yeast mutants further confirmed that ROS accumulation is the actual trigger of apoptosis in toxin-treated yeast, as it is known that glutathione acts as a redox buffer that protects cells from damage by reducing the amount of ROS91. Although apoptosis is clearly not the primary lethal effect of viral killer toxins, a chromosomal deletion in the yeast caspase 1 gene YCA1 — which encodes a protein that is required for apoptotic host-cell responses92 — results in markedly reduced toxin sensitivity; therefore, mutations in YCA1 can rescue cells from K28-mediated toxicity. Based on all these observations, it can be concluded that in the natural environment of killer yeast, where the toxin concentration is usually low, the induction of apoptosis has a crucial, if not essential, role in efficient toxin-mediated cell killing90.
Toxin immunity
In addition to the fact that the primary toxin target has yet to be identified, the question of how immunity is realized in vivo has still to be answered and remains the most intriguing aspect of the killer phenomenon. In killer yeast, functional immunity is essential for cell survival, as the toxins exclusively target and inhibit eukaryotic cell functions. This is in contrast to bacterial protein toxins such as cholera toxin and Shiga toxin, which selectively kill eukaryotes, therefore making immunity and self-defence in a prokaryotic host dispensable.
Immunity to K1. It has been speculated that K1-toxin immunity might be conferred by the toxin precursor itself acting as a competitive inhibitor of the mature toxin by saturating or eliminating the plasma-membrane receptor that normally mediates toxicity. It was also shown that in vivo expression of a toxin-coding cDNA in a Δkex2 null mutant that is deficient in pptox processing and, therefore, unable to release the α- and β-subunits from the intervening γ-sequence results in immune non-killers93,94,95,96. Based on these observations, a model for K1 immunity was proposed in which either loss or modification of the toxin's secondary plasma-membrane receptor causes immunity. When it was shown that the expression of a cDNA copy of the pptox was sufficient to confer immunity, it was postulated that the γ-component of the toxin precursor might not only act as an intramolecular chaperone, ensuring proper pptox processing in the secretory pathway, but might also provide some sort of 'masking' function by protecting membranes of toxin-producing cells against damage by the hydrophobic α-subunit97.
Later, a more plausible model was proposed, in which it was speculated that immunity in a K1 killer cell results from an interaction of the toxin R2 receptor (Kre1p) with the K1 protoxin during secretion, leading to diversion of the receptor–protoxin complex to the vacuole95. This model was further strengthened by phenotypic analyses of various mutant pptox derivatives, which clearly indicated that the α-toxin was the lethal component and that its secretion in the mature form caused severe growth inhibition, whereas secretion of the α-toxin fused to an N-terminal fragment of the γ-subunit was sufficient to confer immunity98. The dependence of immunity on diversion of the putative membrane receptor to the vacuole is consistent with the defect in immunity observed in many vps mutants, which are known to be blocked in various steps of vacuolar protein sorting99.
The membrane receptor for K1 has been identified as Kre1p, a cell-surface protein involved in the synthesis of the cell-wall component β-1,6-glucan64. Immunity apparently does not involve loss of the membrane receptor but affects a step downstream of binding to Kre1p. Given that the recently proposed immunity mechanism in which K1 leads to an internal blockage of the potassium channel Tok1p (Ref. 70) is unlikely to be of physiological relevance, the precise mechanism of K1 immunity still remains obscure.
Immunity to K28. The terminal phenotype of K28-treated cells (inhibition of DNA synthesis and cell-cycle arrest) indicates that the lethal effect begins in the nucleus. Nevertheless, toxin-producing cells are effectively protected against the toxin. Recently, the mechanism of K28 immunity has been elucidated at the molecular level100. Taking into account the fact that toxicity involves endocytotic uptake and retrograde toxin transport, K28 immunity could involve interaction of external, internalized toxin with toxin that is being secreted to the cell surface at the same time in the secretory pathway of an immune cell. This is not the case, however, as immune cells can still take up external toxin and translocate it back to the cytosol, indicating that immunity manifests in the cytosol. The simultaneous presence of both the re-internalized α/β toxin and its pptox precursor in the cytosol of a killer yeast indicates that an interaction between K28 and pptox might be the key step in toxin immunity. In fact, such a complex has recently been purified in vivo100. According to the model postulated for K28 immunity (Fig. 5), the K28 pptox is initially translated from the killer-virus transcript on free ribosomes. Thereafter, it is post-translationally translocated into the secretory pathway, where it is processed into the α/β heterodimeric toxin, which is secreted into the culture medium. Consistent with the important role of post-translational pptox import into the ER, K28 immunity is negatively affected and severely impaired when the natural ER-import signal in K28 pptox is replaced by the co-translational signal sequence of K1 pptox100.

In the cytosol of a K28-producing killer yeast, the unprocessed preprotoxin — encoded by the M28 double-stranded RNA killer virus — complexes with the mature α/β toxin that has been re-internalized and transported in a retrograde manner through the secretory pathway. In the in vivo generated complex of the toxin precursor and the mature α/β toxin, the β-subunits in the complex are (poly)ubiquitinated (Ubx) and rapidly degraded by the proteasome. Part of the toxin precursor escapes from being ubiquitinated and degraded, and can therefore serve either as template for preprotoxin import into the endoplasmic reticulum (ER) and toxin secretion or as an immunity component to form a complex with the re-internalized α/β toxin.
In addition to pptox import into the secretory pathway and K28 toxin secretion, the killer cell can also take up external toxin (either self-produced or produced by other K28 killer cells) and transport it in a retrograde fashion from the Golgi to the ER. After reaching the ER, the toxin translocates to the cytosol and rapidly forms a complex with pptox molecules that have not yet been imported into the ER. In this complex, the K28 heterodimer is selectively ubiquitinated and proteasomally degraded. In this way, at least part of the pptox moiety of the complex is released and can either be imported into the ER or complexed with a newly internalized K28 heterodimer. Interestingly, selective blockage of proteasomal degradation causes a dramatic decrease in toxin secretion, as effective degradation of the generated K28–pptox complex is prevented, less pptox is available for ER import, and the remaining pptox is unable to fully complex internalized, heterodimeric toxin. Therefore, proteasomal mutants become partially sensitive to K28. Interestingly, the amount of cytosolic ubiquitin has a crucial role in toxin immunity, and increasing free ubiquitin (by overexpression of mutated ubiquitin unable to form polyubiquitin chains) results in a significant decrease in toxin secretion. Under such conditions, cytosolic pptox is primarily monoubiquitinated and incompetent for either ER import or complex formation with the re-internalized heterodimeric toxin100. Such a scenario generates a killer yeast that is no longer protected against its own toxin. Vice versa, decreasing cytosolic ubiquitin by blocking protein deubiquitination (as in a Δdoa4 mutant) causes an increase in toxin secretion and immunity is not impaired, as sufficient pptox is available for K28 complex formation. This simple mechanism ensures that a toxin-producing killer yeast is effectively protected against the lethal action of its own toxin.
Concluding remarks
Over the years, much has been learned about eukaryotic cell biology by studying virus-infected killer yeasts and dissecting toxin maturation and secretion. In the case of the K28 virus toxin, various important cellular processes have been studied in detail — including retrograde protein transport and retrotranslocation, ubiquitination and proteasomal degradation, and even apoptotic host-cell responses — that not only are fundamental to eukaryotic cell biology but also relevant to human disease. However, there are still many open and unresolved questions, and future research is needed to adequately address each aspect. Based on the strategy of microbial A/B toxins to enter and penetrate a eukaryotic target cell, it might be possible in future to specifically design chimaeric toxin variants that function as novel vehicles for protein import and retrograde transport in eukaryotic cells.
References
Bevan, E. A. & Makower, M. The physiological basis of the killer-character in yeast. in Genetics Today, XIth International Congress of Genetics Vol. 1 (ed. Geerts, S. J.) 202–203 (Pergamon Press, Oxford, 1963). The discovery of toxin-secreting yeast.
Bevan, E. A., Herring, A. J. & Mitchell, D. J. Preliminary characterisation of two species of dsRNA in yeast and their relationship to the “killer” character. Nature 245, 81–86 (1973). First report that links yeast dsRNA genomes to killer phenotype expression.
Herring, A. J. & Bevan, E. A. Virus-like particles associated with double-stranded RNA species found in killer and sensitive strains of the yeast Saccharomyces cerevisiae. J. Gen. Virol. 22, 387–394 (1974).
Magliani, W., Conti, S., Gerloni, M., Bertolotti, D. & Polonelli, L. Yeast killer systems. Clin. Microbiol. Rev. 10, 369–400 (1997).
Bruenn, J. A. The double-stranded RNA viruses of Ustilago maydis and their killer toxins. in dsRNA Genetic Elements: Concepts and Applications in Agriculture, Forestry, and Medicine (ed. Tavantzis, S. M.) 109–124 (CRC Press, Boca Raton, 2002).
Schmitt, M. J. & Breinig, F. The viral killer system in yeast: from molecular biology to application. FEMS Microbiol. Rev. 26, 257–276 (2002).
Schmitt, M. J. & Neuhausen, F. Killer toxin-secreting double-stranded RNA mycoviruses in the yeasts Hanseniaspora uvarum and Zygosaccharomyces bailii. J. Virol. 68, 1765–1772 (1994).
Schmitt, M. J., Poravou, O., Trenz, K. & Rehfeldt, K. Unique double-stranded RNAs responsible for the anti-Candida activity of the yeast Hanseniaspora uvarum. J. Virol. 71, 8852–8855 (1997).
Zorg, J., Kilian, S. & Radler, F. Killer toxin producing strains of the yeasts Hanseniaspora uvarum and Pichia kluyveri. Arch. Microbiol. 149, 261–267 (1988).
Radler, F., Herzberger, S., Schonig, I. & Schwarz, P. Investigation of a killer strain of Zygosaccharomyces bailii. J. Gen. Microbiol. 139, 495–500 (1993).
Park, C. M., Banerjee, N., Koltin, Y. & Bruenn, J. A. The Ustilago maydis virally encoded KP1 killer toxin. Mol. Microbiol. 20, 957–963 (1996).
Dawe, A. L. & Nuss, D. L. Hypoviruses and chestnut blight: exploiting viruses to understand and modulate fungal pathogenesis. Annu. Rev. Genet. 35, 1–29 (2001).
Bussey, H. K1 killer toxin, a pore-forming protein from yeast. Mol. Microbiol. 5, 2339–2243 (1991).
Schaffrath, R. & Meinhardt, F. Kluyveromyces lactis zymocin and related plasmid-encoded yeast killer toxins. in Topics in Current Genetics: Microbial Protein Toxins (eds Schmitt, M. J. & Schaffrath, R.) 133–155 (Springer-Verlag, Berlin Heidelberg, 2005).
Theisen, S., Molkenau, E. & Schmitt, M. J. Wicaltin, a new protein toxin secreted by the yeast Williopsis californica and its broad-spectrum antimycotic potential. J. Microbiol. Biotechnol. 10, 547–550 (2000).
Suzuki, C. Acidiphilic structure and killing mechanism of the Pichia farinosa killer toxin SMTK. in Topics in Current Genetics: Microbial Protein Toxins (eds Schmitt, M. J. & Schaffrath, R.) 189–214 (Springer-Verlag, Berlin Heidelberg, 2005).
Buck, K. W. Fungal Virology (CRC Press, Boca Raton, 1986).
Bruenn, J. A. Virus-like particles of yeast. Annu. Rev. Microbiol. 34, 49–68 (1980).
Hanes, S. D., Burn, V. E., Sturley, S. L., Tipper, D. J. & Bostian, K. A. Expression of a cDNA derived from the yeast killer preprotoxin gene: implications for processing and immunity. Proc. Natl Acad. Sci. USA 83, 1675–1679 (1986).
Dignard, D., Whiteway, M., Germain, D., Tessier, D. & Thomas, D. Y. Expression in yeast of a cDNA copy of the K2 killer toxin gene. Mol. Gen. Genet. 227, 127–136 (1991).
Schmitt, M. J. & Tipper, D. J. K28, a unique double-stranded RNA killer virus of Saccharomyces cerevisiae. Mol. Cell. Biol. 10, 4807–4815 (1990).
Schmitt, M. J. Cloning and expression of a cDNA copy of the viral K28 killer toxin gene in yeast. Mol. Gen. Genet. 246, 236–246 (1995).
Wickner, R. B. Double-stranded and single-stranded RNA viruses of Saccharomyces cerevisiae. Annu. Rev. Microbiol. 46, 347–375 (1992). Still an excellent review on yeast dsRNA virology.
Icho, T. & Wickner, R. B. The double-stranded RNA genome of yeast virus L-A encodes its own putative RNA polymerase by fusing two open reading frames. J. Biol. Chem. 264, 6716–6723 (1989).
Dinman, J. D., Icho, T. & Wickner, R. B. A-1 ribosomal frameshift in a double-stranded RNA virus of yeast forms a gag–pol fusion protein. Proc. Natl Acad. Sci. USA 88, 174–178 (1991).
Fujimura, T., Ribas, J. C., Makhov, A. M. & Wickner, R. B. Pol of gag–pol fusion protein required for encapsidation of viral RNA of yeast L-A virus. Nature 359, 746–749 (1992).
Dinman, J. D. & Wickner, R. B. Translational maintenance of frame: mutants of Saccharomyces cerevisiae with altered-1 ribosomal frameshifting efficiencies. Genetics 136, 75–86 (1994).
Schmitt, M. J. & Tipper, D. J. Genetic analysis of maintenance and expression of L and M double-stranded RNAs from yeast killer virus K28. Yeast 8, 373–384 (1992).
Bussey, H., Vernet, T. & Sdicu, A. M. Mutual antagonism among killer yeasts: competition between K1 and K2 killers and a novel cDNA-based K1–K2 killer strain of Saccharomyces cerevisiae. Can. J. Microbiol. 34, 38–44 (1988).
Schmitt, M. J. & Schernikau, G. Construction of a cDNA-based K1/K2/K28 triple killer strain of Saccharomyces cerevisiae. Food Technol. Biotechnol. 35, 281–285 (1997).
Bruenn, J. A. Relationships among the positive strand and double-strand RNA viruses as viewed through their RNA-dependent RNA polymerases. Nucleic Acids Res. 19, 217–226 (1991).
Cheng, R. H. et al. Fungal virus capsids, cytoplasmic compartments for the replication of double-stranded RNA, formed as icosahedral shells of asymmetric Gag dimers. J. Mol. Biol. 244, 255–258 (1994).
Naitow, H., Tang, J., Canady, M., Wickner, R. B. & Johnson, J. E. L-A virus at 3.4 Å resolution reveals particle architecture and mRNA decapping mechanism. Nature Struct. Biol. 9, 725–728 (2002).
Tang, J. et al. The structural basis of recognition and removal of cellular mRNA 7-methyl G caps by a viral capsid protein: a unique viral response to host defense. J. Mol. Recognit. 18, 158–168 (2005).
Wickner, R. B. Double-stranded RNA viruses of Saccharomyces cerevisiae. Microbiol. Rev. 60, 250–265 (1996).
Wickner, R. B., Bussey, H., Fujimura, T. & Esteban, R. Viral RNA and the killer phenomenon of Saccharomyces. in The Mycota Vol. 2 (ed. Kück, U.) 211–226 (Springer, Berlin Heidelberg, 1995).
Caston, J. R. et al. Structure of L-A virus: a specialized compartment for the transcription and replication of double-stranded RNA. J. Cell Biol. 138, 975–985 (1997).
Esteban, R. & Wickner, R. B. Three different M1 RNA-containing viruslike particle types in Saccharomyces cerevisiae: in vitro M1 double-stranded RNA synthesis. Mol. Cell. Biol. 6, 1552–1561 (1986).
Bostian, K. A., Sturgeon, J. A. & Tipper, D. J. Encapsidation of yeast killer double-stranded ribonucleic acids: dependence of M on L. J. Bacteriol. 143, 463–470 (1980).
Schmitt, M. J. & Tipper, D. J. Sequence of the M28 dsRNA: preprotoxin is processed to an α/β heterodimeric protein toxin. Virology 213, 341–351 (1995).
Fuller, R. S., Brake, A. & Thorner, J. Yeast prohormone processing enzyme (KEX2 gene product) is a Ca2+-dependent serine protease. Proc. Natl Acad. Sci. USA 86, 1434–1438 (1989).
Rockwell, N. C. & Thorner, J. W. The kindest cuts of all: crystal structures of Kex2 and furin reveal secrets of precursor processing. Trends Biochem. Sci. 29, 80–87 (2004).
Riffer, F., Eisfeld, K., Breinig, F. & Schmitt, M. J. Mutational analysis of K28 preprotoxin processing in the yeast Saccharomyces cerevisiae. Microbiology 148, 1317–1328 (2002).
Eisfeld, K., Riffer, F., Mentges, J. & Schmitt, M. J. Endocytotic uptake and retrograde transport of a virally encoded killer toxin in yeast. Mol. Microbiol. 37, 926–940 (2000). First description of endocytosis and retrograde transport of a yeast killer toxin.
Townsley, F. M., Frigerio, G. & Pelham, H. R. Retrieval of HDEL proteins is required for growth of yeast cells. J. Cell Biol. 127, 21–28 (1994).
Kreitman, R. J. & Pastan, I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem. J. 307, 29–37 (1995).
Lord, J. M. & Roberts, L. M. Toxin entry: retrograde transport through the secretory pathway. J. Cell Biol. 2, 733–736 (1998).
Pelham, H. R. B., Roberts, L. M. & Lord, J. M. Toxin entry: how reversible is the secretory pathway? Trends Cell Biol. 2, 183–185 (1992).
Lord, J. M. et al. Retrograde transport of toxins accross the endoplasmic reticulum membrane. Biochem. Soc. Trans. 31, 1260–1262 (2003).
Lencer, W. I. & Tsai, B. The intracellular voyage of cholera toxin: going retro. Trends Biochem. Sci. 28, 639–645 (2003).
Snapp, E. L., Reinhart, G. A., Bogert, B. A., Lippincott-Schwartz, J. & Hedge, S. The organization of engaged and quiescent translocons in the endoplasmic reticulum of mammalian cells. J. Cell Biol. 164, 997–1007 (2004).
Beswick, V. et al. Expression, purification, and characterization of Sss1p, an essential component of the yeast Sec61p protein translocation complex. Protein Expr. Purif. 13, 423–432 (1998).
Pilon, M., Schekman, R. & Romisch, K. Sec61p mediates export of a misfolded secretory protein from the endoplasmic reticulum to the cytosol for degradation. EMBO J. 16, 4540–4548 (1997).
Plemper, R. K., Bohmler, S., Bordallo, J., Sommer, T. & Wolf, D. H. Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 388, 891–895 (1997).
Tsai, B., Ye, Y. & Rapoport, T. A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nature Rev. Mol. Cell. Biol. 3, 246–255 (2002).
Ellgaard, L. & Helenius, A. Quality control in the endoplasmic reticulum. Nature Rev. Mol. Cell. Biol. 4, 181–191 (2003).
Wesche, J., Rapak, A. & Olsnes, S. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 274, 34443–34449 (1999).
Schmitz, A., Herrgen, H., Winkeler, A. & Herzog, V. Cholera toxin is exported from microsomes by the Sec61p complex. J. Biol. Chem. 148, 1203–1212 (2000).
Koopman, J. et al. Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity 13, 117–127 (2000).
Sommer, T. & Wolf, D. H. Endoplasmic reticulum degradation: reverse protein flow of no return. FASEB J. 11, 1227–1233 (1997).
Schmitt, M. & Radler, F. Molecular structure of the cell wall receptor for killer toxin KT28 in Saccharomyces cerevisiae. J. Bacteriol. 170, 2192–2196 (1988).
Hutchins, K. & Bussey, H. Cell wall receptor for yeast killer toxin: involvement of 1,6-β-D-glucan. J. Bacteriol. 154, 161–169 (1983). First identification of the killer toxin K1 cell-wall receptor.
Boone, C., Sommer, S. S., Hensel, A. & Bussey, H. Yeast KRE genes provide evidence for a pathway of cell wall β-glucan assembly. J. Cell Biol. 110, 1833–1843 (1990).
Breinig, F., Tipper, D. J. & Schmitt, M. J. Kre1p, the plasma membrane receptor for the yeast K1 viral toxin. Cell 108, 395–405 (2002). First paper that identifies the K1 cytoplasmic membrane receptor.
Breinig, F., Schleinkofer, K. & Schmitt, M. J. Yeast Kre1p is GPI-anchored and involved in both cell wall assembly and architecture. Microbiology 150, 3209–3218 (2004).
Schmitt, M., Brendel, M., Schwarz, R. & Radler, F. Inhibition of DNA synthesis in Saccharomyces cerevisiae by yeast toxin K28. J. Gen. Microbiol. 135, 1529–1535 (1989).
Schmitt, M. J., Klavehn, P., Wang, J., Schonig, I. & Tipper, D. J. Cell cycle studies on the mode of action of yeast K28 killer toxin. Microbiology 142, 2655–2662 (1996).
Martinac, B. et al. Yeast K1 killer toxin forms ion channels in sensitive yeast spheroplasts and in artificial liposomes. Proc. Natl Acad. Sci. USA 87, 6228–6232 (1990). Important study on the ionophoric mode of action of K1 toxin.
Ahmed, A. et al. A molecular target for viral killer toxin: TOK1 potassium channels. Cell 99, 283–291 (1999).
Sesti, F., Shih, T. M., Nikolaeva, N. & Goldstein, S. A. Immunity to K1 killer toxin: internal TOK1 blockade. Cell 105, 637–644 (2001).
Bertl, A. et al. Characterization of potassium transport in wild-type and isogenic yeast strains carrying all combinations of trk1, trk2 and tok1 null mutations. Mol. Microbiol. 47, 767–780 (2003).
Page, N. et al. A Saccharomyces cerevisiae genome-wide mutant screen for altered sensitivity to K1 killer toxin. Genetics 163, 875–894 (2003).
Weiler, F. & Schmitt, M. J. Zygocin, a secreted antifungal toxin of the yeast Zygosaccharomyces bailii, and its effect on sensitive fungal cells. FEMS Yeast. Res. 3, 69–76 (2003).
La Rocca, P., Biggin, P., Tieleman, D. P. & Sansom, M. S. Simulation studies of the interaction of antimicrobial peptides and lipid bilayers. Biochim. Biophys. Acta 1462, 185–200 (1999).
Tossi, A., Sandri, L. & Giangaspero, A. Amphiphatic, α-helical antimicrobial peptides. Biopolymers 55, 4–30 (2000).
Weiler, F., Rehfeldt, K., Bautz, F. & Schmitt, M. J. The Zygosaccharomyces bailii antifungal virus toxin zygocin: cloning and expression in a heterologous fungal host. Mol. Microbiol. 46, 1095–1105 (2002).
Sansom, M. S. Alamethicin and related peptaibols — model ion channels. Eur. Biophys. J. 22, 105–124 (1993).
Cafiso, D. S. Alamethicin: a peptide model for voltage gating and protein–membrane interactions. Annu. Rev. Biophys. Biomol. Struct. 23, 141–165 (1994).
Thevissen, K. et al. Fungal membrane responses induced by plant defensins and thionins. J. Biol. Chem. 271, 15018–15025 (1996).
Thevissen, K., Osborn, R. W., Acland, D. P. & Broekaert, W. F. Specific, high affinity binding sites for an antifungal plant defensin on Neurospora crassa hyphae and microsomal membranes. J. Biol. Chem. 272, 32176–32181 (1997).
Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 415, 389–395 (2002).
Peschel, A. et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor MprF is based on modification of membrane lipids with L-lysine. J. Exp. Med. 193, 1067–1076 (2001).
van den Hazel, H. B. et al. PDR16 and PDR17, two homologous genes of Saccharomyces cerevisiae, affect lipid biosynthesis and resistance to multiple drugs. J. Biol. Chem. 274, 1934–1941 (1999).
Li, X. et al. Identification of a novel family of nonclassic yeast phosphatidylinositol transfer proteins whose function modulates phospholipase D activity and Sec14p-independent cell growth. Mol. Biol. Cell 11, 1989–2005 (2000).
Thevissen, K. et al. A gene encoding a sphingolipid biosynthesis enzyme determines the sensitivity of Saccharomyces cerevisiae to an antifungal plant defensin from dahlia (Dahlia merckii). Proc. Natl Acad. Sci. USA 97, 9531–9536 (2000).
Del Carratore, R. et al. Cell cycle and morphological alterations as indicative of apoptosis promoted by UV irradiation in S. cerevisiae. Mutat. Res. 513, 183–191 (2002).
Laun, P. et al. Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis. Mol. Microbiol. 39, 1166–1173 (2001).
Ludovico, P., Sousa, M. J., Silva, M. T., Leao, C. & Corte-Real, M. Saccharomyces cerevisiae commits to a programmed cell death process in response to acetic acid. Microbiology 147, 2409–2415 (2001).
Madeo, F. et al. Oxygen stress: a regulator of apoptosis in yeast. J. Cell Biol. 145, 757–767 (1999).
Reiter, J., Herker, E., Madeo, F. & Schmitt, M. J. Viral killer toxins induce caspase-mediated apoptosis in yeast. J. Cell Biol. 168, 353–358 (2005).
Carmody, R. J. & Cotter, T. G. Signalling apoptosis: a radical approach. Redox Rep. 6, 77–90 (2001).
Madeo, F. et al. A caspase-related protease regulates apoptosis in yeast. Mol. Cell 9, 911–917 (2002).
Bussey, H., Sacks, W., Galley, D. & Saville, D. Yeast killer plasmid mutations affecting toxin secretion and activity and toxin immunity function. Mol. Cell. Biol. 2, 346–354 (1982).
Bussey, H., Saville, D., Greene, D., Tipper, D. J. & Bostian, K. A. Secretion of Saccharomyces cerevisiae killer toxin: processing of the glycosylated precursor. Mol. Cell. Biol. 3, 1362–1370 (1983).
Sturley, S. L., Elliot, Q., LeVitre, J., Tipper, D. J. & Bostian, K. A. Mapping of functional domains within the Saccharomyces cerevisiae type 1 killer preprotoxin. EMBO J. 5, 3381–3389 (1986).
Boone, C., Bussey, H., Greene, D., Thomas, D. Y. & Vernet, T. Yeast killer toxin: site-directed mutations implicate the precursor protein as the immunity component. Cell 46, 105–113 (1986).
Bostian, K. A. et al. Sequence of the preprotoxin dsRNA gene of type I killer yeast: multiple processing events produce a two-component toxin. Cell 36, 741–751 (1984). First detailed analysis on pptox processing and maturation.
Zhu, Y. S., Kane, J., Zhang, X. Y., Zhang, M. & Tipper, D. J. Role of the γ component of preprotoxin in expression of the yeast K1 killer phenotype. Yeast 9, 251–266 (1993).
Douglas, C. M., Sturley, S. L. & Bostian, K. A. Role of protein processing, intracellular trafficking and endocytosis in production of and immunity to yeast killer toxin. Eur. J. Epidemiol. 4, 400–408 (1988).
Breinig, F., Sendzik, T., Eisfeld, K. & Schmitt, M. J. Dissecting toxin immunity in virus-infected killer yeast uncovers an intrinsic strategy of self protection. Proc. Natl Acad. Sci. USA (in the press).
Acknowledgements
The authors thank all past and present members of the Schmitt laboratory for the many contributions over the years, and apologize to authors whose work could not be cited owing to space limitations. Work in the authors' laboratory that contributed to this review was continuously supported by various grants from the Deutsche Forschungsgemeinschaft.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Related links
DATABASES
Entrez Genome
Entrez Genome Project
FURTHER INFORMATION
Glossary
- Virus-like particles
-
Yeast and fungal viruses that are transmitted in vivo by cell-to-cell passage and lack a natural extracellular route of infection.
- Heterokaryon
-
Coexistence of two or more genetically different nuclei in a common cytoplasm.
- Ribosomal frameshift event
-
A process used by many viral mRNAs that makes translating ribosomes change their reading frame by slipping one base in either the 5′ or 3′ direction. The frequency of ribosomal frameshift events is crucial for virus propagation and assembly, as it determines the stoichiometry of viral structural and enzymatic proteins.
- Satellite viruses
-
Encapsidated viruses or virus-like particles composed of structural and enzymatic proteins that are encoded by a helper virus.
- Conservative replication
-
Parental RNA strands remain associated, and the codogenic positive-strand RNA is made first, followed by negative-strand synthesis on the positive-strand RNA template.
- Fluid-phase endocytosis
-
A receptor-independent process by which eukaryotic cells internalize portions of their cell surface to remove cargo such as proteins, lipids or solutes from the external environment. In yeast, accumulation of Lucifer yellow in the vacuole is used as a marker for fluid-phase endocytosis.
- Coat proteins
-
Molecules that form a proteinaceous coating around vesicles that are involved in endoplasmic-reticulum and Golgi trafficking.
- ER-associated degradation
-
A cellular quality-control system that ensures removal of misfolded and/or unassembled proteins from the endoplasmic-reticulum lumen and their subsequent elimination by the cytoplasmic ubiquitin–proteasome system.
- Patch clamping
-
Technique whereby a small electrode tip is sealed onto a patch of cell membrane, making it possible to record the flow of current through individual ion channels or pores in the patch.
- Spheroplasts
-
Yeast cells the cell wall of which has been enzymatically removed to increase the efficiencies of DNA transformation or virus-like-particle transfection.
- Ergosterol
-
The main sterol in the cell membranes of yeast that is responsible, and essential, for structural and regulatory membrane features such as fluidity and permeability (equivalent to cholesterol in mammalian cells).
- Nuclear-localization sequence
-
A short sequence in a protein, rich in basic residues, which acts as a signal for localization of the protein in the nucleus.
- Importins
-
A family of proteins that transport macromolecules into the nucleus.
- α-factor
-
One of two peptide hormones in Saccharomyces cerevisiae that are responsible for synchronized mating between yeast cells of opposite mating type.
Rights and permissions
About this article
Cite this article
Schmitt, M., Breinig, F. Yeast viral killer toxins: lethality and self-protection. Nat Rev Microbiol 4, 212–221 (2006). https://doi.org/10.1038/nrmicro1347
Issue Date:
DOI: https://doi.org/10.1038/nrmicro1347
This article is cited by
-
A virus from Aspergillus cibarius with features of alpha- and betachrysoviruses
Virus Genes (2024)
-
Chemical characteristics and anti-Escherichia coli mechanism of water-soluble extracts from yeast cell walls
Beni-Suef University Journal of Basic and Applied Sciences (2023)
-
An Overview of Mycoviral Curing Strategies Used in Evaluating Fungal Host Fitness
Molecular Biotechnology (2023)
-
The revenge of Zygosaccharomyces yeasts in food biotechnology and applied microbiology
World Journal of Microbiology and Biotechnology (2021)
-
Chance and necessity in the pleiotropic consequences of adaptation for budding yeast
Nature Ecology & Evolution (2020)
