
Abstract
The number and diversity of viral sequences that are identified in metagenomic data far exceeds that of experimentally characterized virus isolates. In a recent workshop, a panel of experts discussed the proposal that, with appropriate quality control, viruses that are known only from metagenomic data can, and should be, incorporated into the official classification scheme of the International Committee on Taxonomy of Viruses (ICTV). Although a taxonomy that is based on metagenomic sequence data alone represents a substantial departure from the traditional reliance on phenotypic properties, the development of a robust framework for sequence-based virus taxonomy is indispensable for the comprehensive characterization of the global virome. In this Consensus Statement article, we consider the rationale for why metagenomic sequence data should, and how it can, be incorporated into the ICTV taxonomy, and present proposals that have been endorsed by the Executive Committee of the ICTV.
Similar content being viewed by others

Main
Viruses are obligate intracellular parasites that probably infect all cellular forms of life. Although virologists have traditionally focused on viruses that cause disease in humans, domestic animals and crops, the recent advances in metagenomic sequencing, in particular high-throughput sequencing of environmental samples, have revealed a staggeringly large virome everywhere in the biosphere. At least 1031 virus particles exist globally at any given time in most environments, including marine and freshwater habitats and metazoan gastrointestinal tracts, in which the number of detectable virus particles exceeds the number of cells by 10–100-fold1,2,3,4,5. To help conceptualize the sheer number of viruses in existence, their current biomass has been estimated to equal that of 75 million blue whales (approximately 200 million tonnes) and, if placed end to end, the collective length of their virions would span 65 galaxies6. In addition to their remarkable abundance, viruses are spectacularly diverse in the nature and organization of their genetic material, gene sequences and encoded proteins, replication mechanisms, and interactions with their cellular hosts, whether they are antagonistic, commensal or mutualistic7. Aquatic environments contain particularly diverse forms of viruses, including single-stranded (ss) and double-stranded (ds) DNA and RNA viruses with genomes that range in size from less than 2,000 bases to more than 2 million bases4. Although dsDNA viruses that infect bacteria (bacteriophages) are the best studied to date, recent work suggests that around 50% of marine viruses have ssDNA or RNA genomes8.
Metagenomic data are changing our views on virus diversity and are therefore challenging the way in which we recognize and classify viruses9. Historically, the description and classification of a new virus by the International Committee on Taxonomy of Viruses (ICTV) have required substantial information on host range, replication cycle, and the structure and properties of virus particles, which were then used to define groups of viruses. However, high-throughput sequencing and metagenomic approaches have radically changed virology, with many more viruses now known solely from sequence data than have been characterized experimentally. For example, the family Genomoviridae currently comprises a single classified virus, whereas more than 120 possible members have been sequenced from diverse environments. However, these sequenced viruses lack information about their hosts and other biological properties that would guide their assignment into species and genera in the family10. Indeed, vast numbers of complete, or nearly complete, genome sequences have been assembled and characterized from metagenomic data for viruses with small11,12,13,14, medium15,16,17,18 and even large19,20 genomes. The identification of entirely new groups of viruses from such analyses emphasizes the power of metagenomic approaches in discovering viruses, some of which could have key functions in the regulation of ecosystems, whereas others could coexist with their hosts without causing recognizable disease or may even be mutualists7. However, realistically, few of these viruses are ever likely to receive the same level of experimental characterization as pathogens that cause human disease or influence the global economy.
The question of whether viruses that are identified by metagenomics can, and should, be incorporated into the official ICTV taxonomy scheme on the basis of sequence data alone is pressing. In response to this question, a workshop of invited experts in the field of virus discovery and environmental surveillance, and members of the ICTV Executive Committee, took place in June 2016 to discuss this possibility and to develop a framework for appropriate approaches to virus classification. We present these proposals in this Consensus Statement article, together with an explanation of the rationale for their development. Our proposals have been subsequently endorsed by the ICTV Executive Committee.
Virus diversity
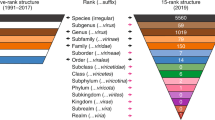
The discrepancy between the number of potential taxa into which viruses in environmental samples could be classified and the number currently recognized by the ICTV is striking. A recent analysis of dsDNA virus sequences that were characterized as part of the Tara Oceans expedition from 43 surface ocean sites worldwide identified 5,476 distinct dsDNA virus populations21, but only 39 of these corresponded to virus groups that have been classified by the ICTV. Most of these populations were both abundant and widely dispersed geographically, but almost all fell outside of established viral taxa (Fig. 1). Early virome studies from different marine habitats hinted at this huge diversity22,23, and, although sequencing technologies at the time precluded direct genome-wide characterization, mathematical modelling predicted several hundred thousand distinct DNA viral genotypes. A recent comprehensive metagenomic analysis of thousands of diverse samples has led to the discovery of approximately 125,000 new viral genomes and a 16-fold increase in the number of identified viral genes24. Similarly, as technology advances, it is becoming clear that ssDNA and RNA viruses in marine and other ecosystems are far more diverse than currently characterized viruses; however, these new viruses remain understudied despite their ecological importance11,25,26,27,28,29,30,31. Many ssDNA viruses identified in metagenomic data encode an evolutionarily conserved replication-associated protein (Rep), whereas the number, orientation and evolutionary origin of other genes are highly variable in these circular Rep-encoding ssDNA viruses (CRESS-DNA viruses)32. Phylogenetic analyses have revealed distinct clustering of some of these viruses into four recognized families, in addition to a vast range of viruses that fall outside of these clusters (Fig. 2). Aside from marine environments, most viruses discovered in wild plants through metagenomics seem to be persistent, and only a tiny proportion of these viruses are species that are recognized by the ICTV33. Highly diverse novel viruses have been similarly reported from insects34,35, and several eukaryotic and prokaryotic viruses have been identified in terrestrial environmental samples24,36.

The 15,222 virus populations that were identified across the Global Ocean Viromes (GOV) dataset69 are shown according to their prevalence (x-axis, number of sampling stations in which the population was detected), average abundance (y-axis, log10 scale, average of normalized coverage across all samples in which the population was detected), and are coloured by the taxonomic affiliation of their host (affiliation is based on best basic local alignment search tool (BLAST) hit of predicted genes; a population was associated to a virus isolate and its host when ≥50% of predicted genes were affiliated to this virus isolate; 512 of the 15,222 populations could be affiliated). Figure courtesy of S. Roux and M.B.S., The Ohio State University, USA.

The replication-associated protein (Rep) sequences of 659 circular Rep-encoding single-stranded DNA (ssDNA) viruses (CRESS-DNA viruses) were compared with 10 representative Repsequences from viruses classified in the families Geminiviridae, Nanoviridae, Circoviridae and Genomoviridae, and a group of alpha satellites that are associated with geminiviruses or nanoviruses. Amino acid sequences were aligned using Multiple Alignment using Fast Fourier Transform (MAFFT; G-INS-i option)70, and a maximum likelihood phylogenetic tree was constructed using Fasttree71. Branches with less than 50% SH (Shimodaira–Hasegawa)-like support were collapsed.
Metagenomic studies have also uncovered astonishingly abundant novel viruses in the human gastrointestinal tract that, despite decades of research, had not been detected previously. For example, the ∼97 kb genome of a dsDNA bacteriophage, named crAssphage, is six-times more abundant in publicly available metagenomic datasets from sewage or wastewater samples than all other known bacteriophages combined. This virus contributes up to 90% of all sequence reads in virus-like particle-derived metagenomes and accounts for ∼1.7% of all human faecal metagenomic sequence reads in public databases17.
Furthermore, numerous viruses are hidden in publicly available microbial genomic datasets. A recently developed tool, VirSorter37,38, identified 12,498 new viral genome sequences in ∼15,000 bacterial and archaeal genomes37, which increased the number of known prokaryotic viruses ∼10-fold and identified viruses that infect 13 prokaryotic phyla37,38. These advances are a striking testimony to the fundamental change in virus discovery: the overwhelming majority of new viral genomes now come from metagenomic data and have never been directly linked to biological agents. Virologists, especially viral taxonomists, have no choice but to work within this new reality.
Current taxonomy of viruses
The framework that is provided by taxonomy enhances our understanding of viruses. It helps communication among virologists, and between virologists and other stakeholders, such as farmers, growers, regulators and potential funders. However, the taxonomy of viruses differs in some fundamental aspects from that of cellular life forms. In particular, viruses lack universal genes that can be used to construct a unified phylogeny into which all viruses can be placed39,40,41,42. Therefore, there is no viral equivalent to the cellular tree of life that has been established through comparisons of ribosomal RNA and (nearly) universal protein-coding genes in bacteria, archaea and eukaryotes (notwithstanding the complications that are caused by horizontal gene transfer)43,44,45.
The ICTV is solely responsible for the classification of viruses into taxa and naming them. Currently, classified viruses are assigned to the hierarchical ranks of family, genus and species, and each taxon has a defined, unique and regulated name. Some families are also divided into subfamilies that each contain separate genera, and a minority of families are also assigned to the higher taxon of order. The ICTV disseminates information on virus taxonomy through the master species list (MSL), which currently lists 7 orders, 112 families, 610 genera and 3,704 species46 (see Virus Taxonomy: 2015 Release), and through periodic publication of ICTV reports that contain additional descriptive material47. The MSL is updated annually based on the submission of taxonomic proposals to the ICTV Executive Committee (see current ICTV Executive Committee webpage), mostly by specialized study groups (see ICTV Study Groups). These proposals are made available to the public and are then scrutinized by the ICTV Executive Committee for compliance with a minimal set of rules that are laid out in the International Code of Virus Classification and Nomenclature (ICVCN; see International Code of Virus Classification and Nomenclature webpage), and for the robustness of the supporting evidence. The new taxonomy is then ratified by voting members of the ICTV and incorporated into the MSL annually.
The lowest taxonomic rank is that of species, which is defined in the ICVCN as “a monophyletic group of viruses whose properties can be distinguished from those of other species by multiple criteria”. Historically, the term “multiple criteria” has been interpreted as referring to attributes such as replication properties in cell culture, virion morphology, serology, nucleic acid sequence, host range, pathogenicity, and epidemiology or epizootiology. However, there is considerable variation in the way in which these criteria have been applied to viruses in different families by the respective Study Groups and approved by the ICTV.
The ICVCN provides greater freedom for specifying the higher taxonomic ranks, with a genus defined as “a group of species sharing certain common characters”, a family defined as “a group of genera (whether or not these are organized into subfamilies) sharing certain common characters” and an order defined as “a group of families sharing certain common characters”. These looser criteria accommodate the substantial variation in the way in which they are applied among the higher ranks. As an approximate guide for vertebrate and plant viruses, members of different genera in a family typically have similar genome organizations with homologous structural and replication-associated genes, but often have non-homologous accessory genes, such as those that are involved in the evasion of host defence and in viral movement in plants. By contrast, between families, viruses often have completely different genome organizations and may lack any detectable genetic relatedness. The presence of homologous, even if not closely similar, RNA-dependent RNA polymerases (RdRps), proteases and helicases in RNA viruses, and Rep-encoding genes in small ssDNA viruses, may, however, enable distant evolutionary relationships between virus families to be identified; such relationships may form a basis for the creation of orders. The process of identifying such distant relationships and assessing their appropriateness for higher rank taxonomic classification is not trivial, and, consequently, the creation of orders requires particularly careful consideration. For example, the existence of a substantial set of shared genes in diverse large or giant dsDNA viruses of eukaryotes has prompted a proposal for the creation of the order 'Megavirales' (Ref. 48), which has thus far not been accepted by the ICTV owing to the lack of consensus in the field. Similarly, the creation of an order for the CRESS-DNA viruses is currently being considered by the relevant ICTV Study Groups.
Virus taxonomy in the age of metagenomics
In the past, the approval of a new species by the ICTV was typically dependent on the availability of data that demonstrate the distinct biological characteristics of the respective virus. This requirement has limited the number of viruses that have been classified and incorporated into the MSL. As most viruses are now discovered by metagenomics and lack direct correlation with biological agents, a workshop was convened to develop a new framework for virus taxonomy in the era of metagenomics (Box 1; Supplementary information S1 (box)). The discussions at the workshop reflected the fact that the challenges that are posed by metagenomic data are not unique to viruses (Box 2).
Sequence assemblies that are derived from environmental samples often contain complete, verified genome sequences of new viruses, but do not directly provide information on biological properties. This perceived limitation has raised the concern that virus classification based on sequence information alone would result in a taxonomy of sequences rather than of viruses49. However, with appropriate precautions (see below), we believe that the detection of a viral sequence in a sample is sufficient evidence to infer the existence of the corresponding virus. Indeed, the concept that a virus can be detected, characterized and classified entirely through analysis of its sequence has gained traction in the burgeoning field of virus discovery. Given that the properties of a virus are largely, or entirely, encoded by its genome, it follows that virus classification based on sequence information alone is not limited primarily by the absence of biological attributes, but by our inability to accurately read such information and robustly infer enzymatic functions, virion structure and other phenotypic attributes.
Sequence data provide a wealth of information that can be used for the purposes of taxonomy, such as evolutionary relationships, overall genome organization (gene content and order, prediction of encoded proteins and the presence of characteristic repeated sequences), features of genome expression, genome replication strategy, the presence or absence of various distinctive motifs (for example, polyprotein cleavage sites, internal ribosome entry sites, terminal sequences, structural folds and host range determinants50), and features of global and local genome composition (for example, GC content, dinucleotide frequencies51 and codon usage). Sequence analyses could thus provide the 'multiple criteria' that are required for classification into species. Indeed, the successful use of sequence information in virus classification has been foreshadowed in the pre-metagenomic era. For example, the bioinformatic characterization of cloned sequences was responsible for the discovery of hepatitis C virus, the prediction of its properties and replication strategy, the characterization of its similarity to members of the family Flaviviridae, and the development of effective diagnostic and screening assays52,53; such advances preceded the visualization of virus particles, the detection of viral proteins in vivo and the achievement of viral growth in cell culture by many years.
However, it is important to recognize that there are several technical problems with using viral genomes that are assembled from metagenomic datasets for taxonomy. Such sequences are often derived from mixed virus populations and, consequently, there is a risk of assembling artificially chimeric genomes. Furthermore, current methodologies are unsuitable for assembling complete genome sequences from viruses that have segmented or multipartite genomes. Another practical problem arises from virus-derived sequences that are integrated into host genomes (for example, endogenous virus-like elements and prophages), many of which are transcribed and hence are present in the RNA pool. To use metagenomic sequences for classification, these problems need to be addressed by robust computational and experimental methods. However, these caveats do not represent fundamental barriers to virus classification, as the technology that is used to create metagenomic sequences is improving continuously, and many of the problems, particularly those that are associated with de novo assembly, will be resolved. These improvements include methods that generate longer sequence reads and those that use template circularization to decrease error rates54.
Proposals
The workshop reached a consensus view on classifying viruses solely on the basis of metagenomic sequence data and, consequently, developed a set of proposals (Box 1; Supplementary information S1 (box)). These proposals are diagrammatically summarized in Fig. 3.

The proposed classification pipeline (red arrows) enables both metagenomic sequence data and conventionally derived virus sequences to be classified. Inferred biological properties that are obtained by bioinformatic analysis of virus sequences together with information on sequence relatedness and gene content, and, optionally, any observed biological properties (dotted line), may all be used as defining criteria for species and higher rank taxonomic assignment in the International Committee on Taxonomy of Viruses (ICTV) taxonomy. This procedure differs from current (green arrows) and previous practice (blue arrows), in which biological data and/or host information and sequence data (current), or biological data alone (1970s–1990s), were required for classification.
Basis of classification. Classifying viruses that are identified only from metagenomic data will advance virus taxonomy, dependent on appropriate checks on data integrity and following the standard procedures of assignment. This is expected to involve the creation of higher rank taxa that consist entirely of viruses that are identified from metagenomic sequence data.
Creating new species. The current ICTV species definition suffices for the classification of viruses based only on sequence information. Virus characteristics that can be inferred from sequence data, including genome organization, replication strategy, presence of homologous genes, and, potentially, host range or type of vector, may serve as additional biological characteristics. These may be used to delineate species in the absence of phenotypic data that have often been relied on for existing species definitions. Such information is best inferred from genomic sequences that comprise the complete coding potential of the respective virus and should be a minimum requirement for classification based on sequences alone.
Assigning new species and genera to existing families. Demarcation procedures vary widely between virus groups and are typically based on parameters that include sequence-based phylogeny and various biological attributes. Although recognizing that direct biological information may form a part of the definition of existing taxa, viruses that are identified from metagenomic data can be classified into additional taxa (species and genera) if their sequence relationships are comparable to those among existing taxa in that family.
Delineating new families and orders. Viruses that have genome sequences that lack close relationships to viruses in existing taxa pose a particular problem, as there is no phenotypically derived standard by which they can be classified. In this situation, assignment of a virus to a new family could be based on limited or absent genetic homology to viruses in recognized families and the existence of major differences in genome organization or inferred replication strategy. Clustering and patterns of variation among more closely related metagenomic sequences might be used to assign viruses hierarchically to lower taxonomic ranks in such groups. However, the creation of a new family, and the assignment of genera and species within it, would require a considerable amount of sequence information and the development of a sound classification framework that is capable of accommodating it. Formalized clustering and network analysis methods that create similarity metrics that are based on the detection of homologous genes and their genetic divergence55,56,57 could be valuable for taxonomic assignments and should be critically evaluated for their effectiveness in the development of a robust classification approach. Frameworks of this kind may have to be tailored to the virus group. For example, bacteriophage taxonomy is typically based on virion sequence and structure58, but these characteristics may not be appropriate for the classification of animal and plant RNA viruses, in which deeper relationships are most often apparent in the gene sequences of the RNA polymerase and other conserved replication-associated proteins59.
Nomenclature of taxa identified only from sequence data. The system that is currently used by the ICTV for taxon nomenclature is readily extendable to additional species, genera and families that are created from metagenomic sequence data. Furthermore, taxa may contain viruses that were identified by various methods. Hence, a species that initially comprises viruses that are characterized solely from sequence data could eventually include viruses that are identified by isolation and that have directly defined biological properties. Thus, metagenomic status belongs to, and would be recoverable from, the sequence record for a particular virus and not to the entire taxon to which it is assigned. Although some virologists have adopted the term 'associated' as part of the nomenclature of viruses that were identified in metagenomics datasets (for example, human stool-associated circular virus (GQ404856 (Ref. 60)); for other examples see Refs 12,13,26,61), it is unnecessary to incorporate this or other such terms that are equivalent to the bacterial term 'Candidatus' into virus taxon names.
Improvement of the procedure for the classification of viruses. The current process of submitting taxonomic proposals to the ICTV suffices, in principle, for dealing with viruses that are known only from sequence data. However, the process could be substantially improved and streamlined through the development of electronic submission methods that incorporate appropriate quality checks for accuracy and completeness of data. In particular, the format could be modified to enable numerous species (possibly many hundreds or thousands) to be proposed in the same submission without the unnecessary repetition of information. In addition, procedures could be developed that shorten the time that is required for processing proposals and updating the MSL.
ICTV endorsement. As an important initial step towards metagenomics-based virus taxonomy, the proposals that were developed during the workshop were presented to, and discussed at, the ICTV Executive Committee meeting from 22–24 August 2016. The proposals were supported by all members of the Executive Committee that were present (one member was unavoidably absent but has since expressed support) and their practical implementation was seen as a matter of high priority for the ICTV. This process will include actively inviting the virology community to submit taxonomic proposals that are based on metagenomic sequences, providing guidelines on data standards (including sequence quality and completeness) and developing more effective data submission tools for large sequence datasets. The ICTV Executive Committee plans to explain and develop these steps in a separate article.
Conclusions
We believe that the time has come to advance the philosophy and practice of virus taxonomy by admitting viruses that are identified only from metagenomics data as being bona fide viruses, dependent on appropriate checks on data integrity and following the standard procedures of taxonomic assignment. We expect that this process will lead to the imminent creation of higher rank taxa that consist entirely of viruses identified by metagenomics.
We believe that the implementation of the proposals outlined here will enable the creation of a vastly expanded formal taxonomy for viruses that will be a major contribution to future research on virus diversity. Only by accepting that sequences that are generated by metagenomic methods truly represent existing viruses and by including them in classification schemes, can we hope to better understand the ecology, history and impact of the global virome.
References
Edwards, R. A. & Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 3, 504–510 (2005).
Mokili, J. L., Rohwer, F. & Dutilh, B. E. Metagenomics and future perspectives in virus discovery. Curr. Opin. Virol. 2, 63–77 (2012).
Rosario, K. & Breitbart, M. Exploring the viral world through metagenomics. Curr. Opin. Virol. 1, 289–297 (2011).
Chow, C. E. & Suttle, C. A. Biogeography of viruses in the sea. Annu. Rev. Virol. 2, 41–66 (2015).
Wigington, C. H. et al. Re-e xamination of the relationship between marine virus and microbial cell abundances. Nat. Microbiol. 1, 15024 (2016).
Suttle, C. A. Viruses: unlocking the greatest biodiversity on Earth. Genome 56, 542–544 (2013).
Roossinck, M. J. Move over, bacteria! Viruses make their mark as mutualistic microbial symbionts. J. Virol. 89, 6532–6535 (2015).
Steward, G. F. et al. Are we missing half of the viruses in the ocean? ISME J. 7, 672–679 (2013).
Simmonds, P. Methods for virus classification and the challenge of incorporating metagenomic sequence data. J. Gen. Virol. 96, 1193–1206 (2015).
Krupovic, M., Ghabrial, S. A., Jiang, D. & Varsani, A. Genomoviridae: a new family of widespread single-stranded DNA viruses. Arch. Virol. 161, 2633–2643 (2016).
Labonte, J. M. & Suttle, C. A. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 7, 2169–2177 (2013).
Dayaram, A. et al. Diverse small circular DNA viruses circulating amongst estuarine molluscs. Infect. Genet. Evol. 31, 284–295 (2015).
Dayaram, A. et al. Diverse circular replication-associated protein encoding viruses circulating in invertebrates within a lake ecosystem. Infect. Genet. Evol. 39, 304–316 (2016).
Rosario, K., Schenck, R. O., Harbeitner, R. C., Lawler, S. N. & Breitbart, M. Novel circular single-stranded DNA viruses identified in marine invertebrates reveal high sequence diversity and consistent predicted intrinsic disorder patterns within putative structural proteins. Front. Microbiol. 6, 696 (2015).
Yutin, N., Shevchenko, S., Kapitonov, V., Krupovic, M. & Koonin, E. V. A novel group of diverse Polinton-like viruses discovered by metagenome analysis. BMC Biol. 13, 95 (2015).
Zhou, J. et al. Diversity of virophages in metagenomic data sets. J. Virol. 87, 4225–4236 (2013).
Dutilh, B. E. et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 5, 4498 (2014).
Yutin, N., Kapitonov, V. V. & Koonin, E. V. A new family of hybrid virophages from an animal gut metagenome. Biol. Direct 10, 19 (2015).
Zhang, W. et al. Four novel algal virus genomes discovered from Yellowstone Lake metagenomes. Sci. Rep. 5, 15131 (2015).
Yau, S. et al. Virophage control of antarctic algal host–virus dynamics. Proc. Natl Acad. Sci. USA 108, 6163–6168 (2011).
Brum, J. R. et al. Ocean plankton. Patterns and ecological drivers of ocean viral communities. Science 348, 1261498 (2015).
Angly, F. E. et al. The marine viromes of four oceanic regions. PLoS Biol. 4, e368 (2006).
Breitbart, M. et al. Diversity and population structure of a near-shore marine-sediment viral community. Proc. Biol. Sci. 271, 565–574 (2004).
Paez-Espino, D. et al. Uncovering Earth's virome. Nature 536, 425–430 (2016).
Culley, A. I. et al. The characterization of RNA viruses in tropical seawater using targeted PCR and metagenomics. mBio 5, e01210–14 (2014).
Kraberger, S. et al. Characterisation of a diverse range of circular replication-associated protein encoding DNA viruses recovered from a sewage treatment oxidation pond. Infect. Genet. Evol. 31, 73–86 (2015).
Roossinck, M. J. Plants, viruses and the environment: ecology and mutualism. Virology 479–480, 271–277 (2015).
Labonte, J. M., Hallam, S. J. & Suttle, C. A. Previously unknown evolutionary groups dominate the ssDNA gokushoviruses in oxic and anoxic waters of a coastal marine environment. Front. Microbiol. 6, 315 (2015).
Szekely, A. J. & Breitbart, M. Single-stranded DNA phages: from early molecular biology tools to recent revolutions in environmental microbiology. FEMS Microbiol. Lett. 363, fnw027 (2016).
Hopkins, M. et al. Diversity of environmental single-stranded DNA phages revealed by PCR amplification of the partial major capsid protein. ISME J. 8, 2093–2103 (2014).
Roux, S., Krupovic, M., Poulet, A., Debroas, D. & Enault, F. Evolution and diversity of the Microviridae viral family through a collection of 81 new complete genomes assembled from virome reads. PLoS ONE 7, e40418 (2012).
Rosario, K., Duffy, S. & Breitbart, M. A field guide to eukaryotic circular single-stranded DNA viruses: insights gained from metagenomics. Arch. Virol. 157, 1851–1871 (2012).
Roossinck, M. J. Plant virus metagenomics: biodiversity and ecology. Annu. Rev. Genet. 46, 359–369 (2012).
Li, C. X. et al. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 4, e05378 (2015).
Shi, M. et al. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J. Virol. 90, 659–669 (2015).
Zablocki, O. et al. High-level diversity of tailed phages, eukaryote-associated viruses, and virophage-like elements in the metaviromes of antarctic soils. Appl. Environ. Microbiol. 80, 6888–6897 (2014).
Roux, S., Hallam, S. J., Woyke, T. & Sullivan, M. B. Viral dark matter and virus–host interactions resolved from publicly available microbial genomes. eLife 4, e08490 (2015).
Bolduc, B., Youens-Clark, K., Roux, S., Hurwitz, B. L. & Sullivan, M. B. iVirus: facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. ISME J. http://dx.doi.org/10.1038/ismej.2016.89 (2016).
Koonin, E. V., Senkevich, T. G. & Dolja, V. V. The ancient Virus World and evolution of cells. Biol. Direct 1, 29 (2006).
Holmes, E. C. What does virus evolution tell us about virus origins? J. Virol. 85, 5247–5251 (2011).
Koonin, E. V. & Dolja, V. V. A virocentric perspective on the evolution of life. Curr. Opin. Virol. 3, 546–557 (2013).
Rohwer, F. & Edwards, R. The Phage Proteomic Tree: a genome-based taxonomy for phage. J. Bacteriol. 184, 4529–4535 (2002).
Woese, C. R. Bacterial evolution. Microbiol. Rev. 51, 221–271 (1987).
O'Malley, M. A. & Koonin, E. V. How stands the Tree of Life a century and a half after The Origin? Biol. Direct 6, 32 (2011).
Pace, N. R., Sapp, J. & Goldenfeld, N. Phylogeny and beyond: scientific, historical, and conceptual significance of the first tree of life. Proc. Natl Acad. Sci. USA 109, 1011–1018 (2012).
Adams, M. J. et al. Ratification vote on taxonomic proposals to the International Committee on Taxonomy of Viruses (2016). Arch. Virol. 161, 2921–2949 (2016).
King, A. M. Q., Adams, M. J., Carstens, E. B. & Lefkowitz, E. J. (eds) Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses (Elsevier, 2011).
Colson, P. et al. “Megavirales”, a proposed new order for eukaryotic nucleocytoplasmic large DNA viruses. Arch. Virol. 158, 2517–2521 (2013).
Van Regenmortel, M. H. V. Classes, taxa and categories in hierarchical virus classification: a review of current debates on definitions and names of virus species. Bionomina 10, 1–21 (2016).
Cai, Y. et al. Nonhuman transferrin receptor 1 is an efficient cell entry receptor for Ocozocoautla de Espinosa virus. J. Virol. 87, 13930–13935 (2013).
Kapoor, A., Simmonds, P., Lipkin, W. I., Zaidi, S. & Delwart, E. Use of nucleotide composition analysis to infer hosts for three novel picorna-like viruses. J. Virol. 84, 10322–10328 (2010).
Choo, Q. L. et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 244, 359–362 (1989).
Kuo, G. et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science 244, 362–364 (1989).
Acevedo, A. & Andino, R. Library preparation for highly accurate population sequencing of RNA viruses. Nat. Protoc. 9, 1760–1769 (2014).
Hurwitz, B. L. & Sullivan, M. B. The Pacific Ocean virome (POV): a marine viral metagenomic dataset and associated protein clusters for quantitative viral ecology. PLoS ONE 8, e57355 (2013).
Lima-Mendez, G., Van Helden, J., Toussaint, A. & Leplae, R. Reticulate representation of evolutionary and functional relationships between phage genomes. Mol. Biol. Evol. 25, 762–777 (2008).
Lauber, C. & Gorbalenya, A. E. Partitioning the genetic diversity of a virus family: approach and evaluation through a case study of picornaviruses. J. Virol. 86, 3890–3904 (2012).
Hatfull, G. F. & Hendrix, R. W. Bacteriophages and their genomes. Curr. Opin. Virol. 1, 298–303 (2011).
Koonin, E. V., Wolf, Y. I., Nagasaki, K. & Dolja, V. V. The Big Bang of picorna-like virus evolution antedates the radiation of eukaryotic supergroups. Nat. Rev. Microbiol. 6, 925–939 (2008).
Li, L. et al. Multiple diverse circoviruses infect farm animals and are commonly found in human and chimpanzee feces. J. Virol. 84, 1674–1682 (2010).
Steel, O. et al. Circular replication-associated protein encoding DNA viruses identified in the faecal matter of various animals in New Zealand. Infect. Genet. Evol. 43, 151–164 (2016).
Parker, C. T., Tindall, B. J. & Garrity, G. M. International Code of Nomenclature of Prokaryotes. Int. J. Syst. Evol. Microbiol. http://dx.doi.org/10.1099/ijsem.0.000778 (2015).
Konstantinidis, K. T. & Rossello-Mora, R. Classifying the uncultivated microbial majority: a place for metagenomic data in the Candidatus proposal. Syst. Appl. Microbiol. 38, 223–230 (2015).
Hedlund, B. P., Dodsworth, J. A. & Staley, J. T. The changing landscape of microbial biodiversity exploration and its implications for systematics. Syst. Appl. Microbiol. 38, 231–236 (2015).
Murray, R. G. & Stackebrandt, E. Taxonomic note: implementation of the provisional status Candidatus for incompletely described procaryotes. Int. J. Syst. Bacteriol. 45, 186–187 (1995).
Hawksworth, D. L. et al. The Amsterdam declaration on fungal nomenclature. IMA Fungus 2, 105–112 (2011).
Schoch, C. L. et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc. Natl Acad. Sci. USA 109, 6241–6246 (2012).
Hibbett, D. S. & Taylor, J. W. Fungal systematics: is a new age of enlightenment at hand? Nat. Rev. Microbiol. 11, 129–133 (2013).
Roux, S. et al. Ecogenomics and potential biogeochemical impacts of globally abundant ocean viruses. Nature 537, 689–693 (2016).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2 — approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Acknowledgements
The workshop and discussions were funded by the Wellcome Trust (grant WT108418AIA). The authors thank the members of the International Committee on Taxonomy of Viruses (ICTV) Executive Committee who did not attend (A. Kropinski, R. Harrison, H. Sanfaçon, A. Mushegian, N. Knowles and S. Siddell) for thoughtful comments and discussion, and S. Roux from the Department of Microbiology at Ohio State University, USA, for the preparation of figure 1. B.H. and M. Benko were supported by the Hungarian Scientific Research Fund (OTKA; grants NN107632 and K100163, respectively). The work of A.E.G. was partially supported by the European Union's Horizon 2020 research and innovation programme (under grant agreement number 653316; the European Virus Archive goes Global, EVAg). R.A.v.d.V. was partly supported by the EU Cooperation in Science and Technology (COST) programme (action FA1407). S.S. acknowledges partial financial support from Mississippi State University (MSU)–Mississippi Agricultural and Forestry Experiment Station (MAFES) Strategic Research Initiative grants. The work of E.D. was supported by the US National Heart, Lung, and Blood Institute (NHLBI; grant R01 HL105770). Research by J.R.B. and E.V.K. was supported, in part, by the Intramural Research Program of the US National Institutes of Health (NIH), US National Library of Medicine. M.Breitbart was supported through the Assembling the Tree of Life Programme of the US National Science Foundation (grant DEB-1239976). M.B.S. was supported by an award from the Gordon and Betty Moore Foundation (number 3790). The involvement of J.H.K. was supported, in part, through the prime contract of the Battelle Memorial Institute with the US National Institute of Allergy and Infectious Diseases (NIAID) under contract number HHSN272200700016I. A subcontractor to the Battelle Memorial Institute who carried out this work is J.H.K., who is an employee of Tunnell Government Services, Inc. The views and conclusions that are contained in this document are those of the authors and should not be interpreted as necessarily representing the official policies, either expressed or implied, of the US Department of Health and Human Services or of the institutions and companies that are affiliated with the authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Supplementary information
Supplementary information S1 (box)
Classification of viruses derived from metagenomic sequences (DOC 45 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Simmonds, P., Adams, M., Benkő, M. et al. Virus taxonomy in the age of metagenomics. Nat Rev Microbiol 15, 161–168 (2017). https://doi.org/10.1038/nrmicro.2016.177
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrmicro.2016.177
This article is cited by
-
Genome sequencing identifies “Limestone Canyon virus” as Montaño virus (Hantaviridae: Orthohantavirus montanoense) circulating in brush deermice in New Mexico
npj Viruses (2024)
-
Double-stranded RNA sequencing reveals distinct riboviruses associated with thermoacidophilic bacteria from hot springs in Japan
Nature Microbiology (2024)
-
Long-read assembled metagenomic approaches improve our understanding on metabolic potentials of microbial community in mangrove sediments
Microbiome (2023)
-
Deciphering the functional diversity of the gut microbiota of the black soldier fly (Hermetia illucens): recent advances and future challenges
Animal Microbiome (2023)
-
Guidelines for public database submission of uncultivated virus genome sequences for taxonomic classification
Nature Biotechnology (2023)
