
Key Points
-
The respiratory tract is a major portal of entry for viruses into the body. Infection of the respiratory tract can, if severe, induce life-threatening damage to the lungs. Various strategies to control virus replication and to limit immune-mediated inflammation and tissue injury have evolved in the respiratory tract.
-
Multiple innate immune cell types, particularly dendritic cells (DCs), within the pulmonary interstitium and between airway epithelial cells are strategically poised to recognize and sample airway particulates, such as viruses.
-
In response to respiratory virus infection, several distinct DC subsets are stimulated to migrate from the site of infection in the lungs to the draining lymph nodes. Here, these migrant DCs have a crucial role in initiating the antivirus adaptive immune response to the invading viruses.
-
After entering the infected lungs, effector T cells that were generated in the lymph nodes undergo further modifications that are shaped by the inflammatory milieu.
-
Co-stimulatory receptor–ligand interactions between effector T cells and various cell types presenting viral antigens in the infected lungs modulate the host adaptive immune response in situ.
-
Effector T cells that produce pro-inflammatory mediators are also the major producers of regulatory (anti-inflammatory) cytokines, providing a fine-tuning mechanism of self-control by effector T cells responding to viruses in the inflamed tissue.
-
The immune mechanisms that control virus replication and/or excessive inflammation in the virus-infected lungs can also predispose the individual recovering from a virus infection to bacterial superinfection. Therapeutic strategies should consider balancing the need to inhibit virus replication and excessive inflammation with the need to optimize the antibacterial functions of innate immune phagocytes, which are crucial for clearing the bacteria from the lungs.
Abstract
Recent years have seen several advances in our understanding of immunity to virus infection of the lower respiratory tract, including to influenza virus infection. Here, we review the cellular targets of viruses and the features of the host immune response that are unique to the lungs. We describe the interplay between innate and adaptive immune cells in the induction, expression and control of antiviral immunity, and discuss the impact of the infected lung milieu on moulding the response of antiviral effector T cells. Recent findings on the mechanisms that underlie the increased frequency of severe pulmonary bacterial infections following respiratory virus infection are also discussed.
Similar content being viewed by others


Main
Viruses are highly evolved and ubiquitous pathogens. Although infection with many viruses results in a mild, self-limited disease, other viruses can produce severe and frequently fatal infections. The respiratory tract is a major portal through which viruses enter to initiate infection. Whereas some viruses can initiate infection in the respiratory tract and then disseminate to other sites in the body, other viruses typically remain localized to the respiratory tract, where they replicate and induce tissue injury (see Supplementary information S1 (table)). Various defence mechanisms have evolved in the respiratory tract to prevent and control infection by viruses and other pathogens.
The respiratory tract can be divided anatomically into the upper respiratory tract (including the nose, mouth and pharynx) and the lower respiratory tract (which consists of the trachea, bronchi and lungs), with the lymphoid tissue of Waldeyer's ring (including the pharyngeal and palatine tonsils) representing the line of demarcation. Because of the crucial role of the lower respiratory tract in respiration, we limit the focus of this Review to recent advances in our understanding of the host immune response to virus infection at this site.
Stromal cells (typically CD45− non-haematopoietic cells) and haematopoietic cells (CD45+ bone marrow-derived cells) comprise the cellular network of the lower respiratory tract. Although all cell types presumably have important roles during respiratory virus infection, type I and type II alveolar epithelial cells and conducting airway epithelial cells are of particular importance among the CD45− stromal cells, as infection of these cells by viruses and the subsequent host immune response can compromise respiratory function and can result in acute respiratory distress syndrome (ARDS). Type I and type II alveolar epithelial cells are important cellular targets for infection by severe acute respiratory syndrome (SARS) coronavirus1,2 and certain type A influenza virus strains, most notably the highly pathogenic avian H5N1 viruses3,4,5. Viruses can also target epithelial cells of the conducting airways (Supplementary information S1 (table)).
The severe lung inflammation associated with respiratory infection by certain viruses poses a unique challenge to the immune system: not only must the virus be rapidly eliminated by the immune system, but tissue inflammation must also be controlled to prevent acute respiratory failure. Furthermore, during the recovery or resolution phase of infection, the immune system must help to orchestrate tissue repair to restore normal lung architecture and function and prevent permanent defects in respiratory function.
The contributions of physical and chemical barriers to infection and of the various innate immune mechanisms and cell types that mediate host responses to virus infections have been recently reviewed6,7,8,9 and are not discussed in detail here (although they are summarized in Supplementary information S2 (box)). Likewise, the function of B cells in immunity to respiratory viruses has been reviewed elsewhere10. Here, we primarily focus on recent results regarding the induction of T cell responses to virus infection in the respiratory tract andthe role of specific respiratory dendritic cell (DC) subsets and macrophages in controlling this process. In addition, we describe recent findings on the regulation of immunity, pulmonary inflammation and injury during virus infection. Finally, we briefly consider the potential link between respiratory virus infection and chronic respiratory diseases and discuss in more detail the recent findings relating to the effect of respiratory tract virus infection on the susceptibility to concurrent or subsequent bacterial infection (known as bacterial superinfection).
Respiratory APC activation and emigration
The lungs are bombarded by a plethora of innocuous inhaled antigens and presumably by commensal microorganisms. A unique challenge for lung-resident antigen-presenting cells (APCs) is to discriminate between invading pathogens and innocuous particles and to deliver only relevant microbial antigens to T cells. The mechanisms of this discrimination in the lungs are poorly understood but ultimately result in the activation of respiratory DCs and their migration to draining lymph nodes.
Alveolar macrophages. If intracellular innate immune mechanisms in primary target cells (such as epithelial cells) fail to adequately control early virus replication, and replication proceeds, then released virions and the contents of dying or dead infected cells can be taken up by a variety of APCs9 (Table 1). The first respiratory tract CD45+ immune cell type to encounter viral antigens is the alveolar macrophage. Not only are alveolar macrophages superb phagocytes that are capable of rapidly eliminating large numbers of microorganisms from the lung, but they also have a crucial role in dampening immune responses at this site11. It is widely believed that alveolar macrophages are non-migratory and do not contribute to the induction of adaptive immune responses. However, recent evidence indicates that alveolar macrophages can migrate from the lung to the draining lymph nodes under homeostatic conditions and can serve as antigen carriers following bacterial infection12, although their contribution to the induction of adaptive immune responses in the draining lymph nodes is currently undefined.
Dendritic cells. DCs reside below the airway epithelial cells within the pulmonary interstitium,and they can extend processes between airway epithelial cells, making them strategically poised to sample airway particulates, such as viruses. Several phenotypically and functionally distinct subsets of respiratory DCs have been studied extensively in rodents13. These populations include two main resident mature (MHC class IIhiCD11chi) respiratory DC subsets, which are characterized as CD103+ and CD11bhiCD103− respiratory DCs and are crucial for the induction of adaptive immune responses, at least in mice14,15,16,17. CD103+ respiratory DCs localize at the mucosal surface, adjacent to the respiratory epithelium, and surround adjoining vasculature. By contrast, the more abundant CD11bhi respiratory DC subset is found within the lung interstitium. CD103+ respiratory DCs express the mannose-binding C-type lectin langerin (also known as CLEC4K)16,18 and require expression of specific transcriptional factors (such as BATF3) for their normal development in the lung and at other mucosal surfaces19,20,21. Activated CD103+ respiratory DCs produce modest levels of pro-inflammatory mediators, whereas CD11bhi respiratory DCs are major chemokine producers in response to pulmonary inflammation22.
Monocyte-derived DCs, which are phenotypically immature (MHC class IIlowCD11clow), are more abundant than the mature DC subsets and localize within the interstitium. They can take up particulates in the inflamed lung and migrate to the draining lymph nodes. However, they were found to be poor activators of naive T cells when analysed during experimental virus infections14. They may, however, serve as precursors for mature DCs23 (Fig. 1). Distinct from these conventional respiratory DCs are plasmacytoid DCs (pDCs). pDCs are recognized as major producers of type I interferons (IFNs) during infection, and can also transport antigens from the infected lung to the draining lymph nodes. However, the low-level expression of co-stimulatory ligands on pDCs renders them weak activators of naive T cells.

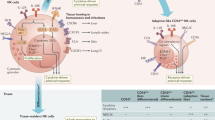
Virus infection of respiratory epithelial cells is first detected by cytosolic and/or endosomal innate sensors in the infected epithelial cells (not shown). Recognition of the invading virus by these innate immune receptors leads to the production of pro-inflammatory cytokines such as interleukin-1β (IL-1β) and IL-18, and chemokines such as CC-chemokine ligand 2 (CCL2). These soluble mediators that are released by infected cells activate adjacent CD45− parenchymal cells, such as fibroblasts and epithelial cells, and neighbouring innate immune cells. Following activation, these cells convert latent transforming growth factor-β (TGFβ) to an active form, resulting in increased secretion of the chemokines CCL2 and CCL20 by parenchymal stromal cells and of cytokines such as IL-12 and tumour necrosis factor (TNF) by inflammatory cells. This enhanced production of chemokines and cytokines facilitates the maturation of tissue-resident CD103+ and CD11b+ dendritic cells (DCs) and the recruitment and maturation of monocyte-derived DCs. Antigen acquisition and activation of immature antigen-bearing respiratory DCs results in their mobilization and migration out of the infected lungs along chemokine gradients of CCL21 and sphingosine-1-phosphate (S1P) to the lymph nodes draining the infected lung. Once in the lymph nodes, these DCs participate in initiating adaptive immune responses to the respiratory virus (not shown).
Macrophages. In addition to alveolar macrophages, CD11b+ (CD11c−) tissue-resident macrophages — which originate from circulating CC-chemokine receptor 2 (CCR2)+ monocytic subsets in the blood — are found within the non-inflamed pulmonary interstitium9,24,25. These cells have the capacity to transport antigens from the lungs to the draining lymph nodes during virus infection14, although their contribution to the induction of adaptive immune responses is unclear. The number of these macrophages can increase dramatically in response to virus infection, and they may serve as APCs for activated effector T cells in the infected lungs24,25 (see below). Recent evidence suggests that the increase in the number of macrophages during infection may result both from an influx of blood monocyte precursors and from local cell proliferation26.
Migration of respiratory DCs. Respiratory virus infection (and other inflammatory stimuli) triggers antigen uptake by and activation of most types of respiratory DC, resulting in their mobilization and migration out of the infected lungs9,27. Although this process is not fully understood, multiple mediators — including chemokines such as CC-chemokine ligand 2 (CCL2), CCL5, CCL20 and CCL21 — probably work in concert to orchestrate respiratory DC activation and mobilization. These chemokines are produced by respiratory epithelial cells and vascular and lymphatic endothelial cells, and then engage their corresponding receptors on respiratory DCs28,29. Additional pro-inflammatory mediators (such as interleukin-1β (IL-1β) and IL-18) that are released by infected epithelial cells can activate adjacent CD45− parenchymal cells, such as fibroblasts28. This may increase the expression of αVβ8 integrin on the fibroblastsand subsequent integrin-dependent activation of transforming growth factor-β (TGFβ), resulting in increased secretion of the TGFβ-dependent chemokines CCL2 and CCL20 (Ref. 28). Similarly, Toll-like receptor 4 (TLR4) signalling in epithelial cells in response to viral proteins upregulates CCL2 and CCL20 expression29, which may facilitate the activation and mobilization of DCs within the lung, as well as the recruitment of monocyte progenitors into the lung from the circulation (Fig. 1).
The infection-induced crosstalk between CD45− lung parenchymal cells and mature resident DCs in the inflamed lung leads to the upregulation on the DCs of several chemotactic receptors, most notably CCR7 and sphingosine-1-phosphate receptors. These receptors are crucial for transendothelial migration of activated respiratory DCs and their egress to the draining lymph nodes14,15,30,31. Unlike CD103+ and CD11bhi respiratory DCs, other potential APCs — such as alveolar macrophages, pDCs and monocyte-derived DCs — use as yet unidentified pathways that are independent of CCR7 to enter the draining lymph nodes. Activated respiratory DCs also upregulate their expression of co-stimulatory and adhesion molecules (such as CD40, CD80, CD86 and ICAM1) and of antigen-presenting molecules, and this renders the migrating respiratory DCs competent to serve as potent APCs for the activation of naive virus-specific T cells in the draining lymph nodes.
T cell activation and differentiation
The migration of respiratory DCs bearing viral antigens to the draining lymph nodes serves to concentrate viral antigens at a site where extremely rare virus-specific adaptive immune cells, such as naive32,33 and memory antiviral T cells34,35, can encounter their cognate antigens. Although the transfer of viral antigens from migrant respiratory DCs (and macrophages) to lymph node-resident CD8α+ DCs has been documented in a respiratory virus infection36, several lines of evidence suggest that it is the antigen-bearing migratory DCs that most efficiently present viral antigens to naive CD4+ and CD8+ T cells during respiratory virus infections14,17,37,38,39,40,41 (Table 1). However, the relative contributions of migratory respiratory DCs and lymph node-resident CD8α+ DCs to the activation of naive T cells during respiratory virus infection remain to be fully explored and are probably pathogen dependent.
Migrant CD103+ respiratory DCs reach maximum numbers in the draining lymph nodes early (2–4 days) following respiratory virus infection and serve as the most potent APCs for naive virus-specific CD8+ T cells early in the response to an active infection14,41. Furthermore, CD103+ respiratory DCs can efficiently cross-present antigens (such as products of infected cells) that were initially taken up in the lungs, possibly via efferocytosis42. The limited data currently available do not allow a definitive determination of whether infected CD103+ respiratory DCs are more potent APCs for naive CD8+ T cells than CD103+ respiratory DCs that cross-present acquired viral antigens14,41. Recently, the superiority of CD103+ respiratory DCs over CD11bhi respiratory DCs in supporting antiviral CD8+ T cell activation during influenza virus infection was linked with their enhanced capacity to process and load viral antigens onto MHC class I molecules for presentation to CD8+ T cells43.
CD11bhi respiratory DCs reach peak numbers in the draining lymph nodes at later time points (5–7 days) following influenza virus infection14,41 and may act to expand the pool size of previously activated effector CD8+ T cells in the draining lymph nodes through engagement of the co-stimulatory receptor CD27 (Ref. 40). The speed and magnitude of migratory DC accumulation in the lymph nodes are probably regulated by virus-intrinsic factors, such as replication potential, target cell type and cytotoxic effects, as well as by host-intrinsic factors, such as underlying genetic susceptibility.
Both major migratory respiratory DC subsets also efficiently activate naive virus-specific CD4+ T cells14. Furthermore, the influx of inflammatory DCs into the draining lymph nodes from the circulation has an important role in activating potent T helper 1 (TH1) cell responses to respiratory virus infection44. Following the resolution of acute infection, CD103+ and CD11b+ respiratory DCs continue to ferry and present residual antigens to memory T cells34,35, and as a consequence they influence the trafficking and recall responses of antiviral memory T cells45,46,47.
T cell-mediated antiviral responses
Following their encounter with APCs in the draining lymph nodes, naive (and memory) virus-specific T cells undergo a stepwise process of activation, proliferation and differentiation to become effector T cells that can migrate to the site of infection and mediate antiviral immune responses.
Effector mechanisms. The effector mechanisms used by antiviral T cells can be categorized into three groups. First, there are T cell-associated lytic mechanisms that promote the lysis of infected cells following the exocytosis of perforin- and granzyme-containing granules. Second, T cells can induce tumour necrosis factor receptor (TNFR) family-dependent apoptosis of infected cells by expressing CD95 ligand (also known as FAS ligand) or TNF-related apoptosis-inducing ligand (TRAIL). Third, T cells can produce pro-inflammatory mediators (and regulatory mediators (see below)) in response to encounter with virus-infected cells. Much of our current knowledge of these processes and their role in virus clearance has come from studies on the host adaptive immune T cell response to infection by a limited number of respiratory viruses, most notably influenza virus.
Several studies suggest a crucial role for the cytolytic functions of CD8+ effector T cells in influenza virus infection. These functions trigger the direct lysis of virus-infected cells and require T cell receptor-mediated recognition of processed viral antigens on the infected target cell48,49,50. With certain notable exceptions51,52,53,the release of particular pro-inflammatory mediators (such as IFNγ) by CD8+ T cells has only a modest impact on virus clearance and recovery.
There is also recent evidence from the influenza virus model that virus-infected respiratory tract cells (specifically infected alveolar epithelial cells) may be eliminated during the host response through the action of activated lytic macrophages that are capable of triggering the apoptosis of virus-infected cells through a TRAIL-dependent mechanism54. Accordingly, activated inflammatory mononuclear cells (rather than effector CD8+ T cells) that express TRAIL and are recruited into the virus-infected lungs could engage alveolar epithelial cells, which upregulate TRAIL receptor early in response to infection, and trigger alveolar epithelial cell apoptosis. The contribution of this mechanism to the control of virus replication and to the development of alveolar damage in the infected respiratory tract awaits further evaluation.
Effector CD4+ T cells have been demonstrated to exhibit cytotoxic activity in vitro, but the contribution of CD4+ T cell cytotoxicity to virus clearance in vivo in the lungs is modest55,56. This cytotoxicity is restricted to viral antigen-bearing cells that express MHC class II molecules; such cells include CD45+ inflammatory mononuclear phagocytic cells and a few CD45− lung parenchymal cell types that constitutively (or inducibly) express MHC class II molecules (such as type II alveolar epithelial cells)57 (Table 1). Results from experimental infection studies suggest that the primary role of antiviral CD4+ T cells is to support the activation and differentiation of B cells, which leads to antibody production58,59.
Modulating effector T cell responses in situ. The classical view of antiviral effector T cell generation posits that the proliferative expansion necessary for the differentiation of activated naive antiviral T cells into effector T cells occurs primarily, if not exclusively, in the draining lymph nodes. The first hint that this earlier view may be incomplete in the case of respiratory virus infection came from studies demonstrating that activated effector T cells can undergo extensive proliferation in the respiratory tract60,61. More recent studies indicate a requirement for effector T cells in the infected lungs to interact with CD45+ inflammatory mononuclear phagocytes, which produce cytokines (such as IL-15, which they trans-present) and/or express co-stimulatory molecules (such as CD70) to support the proliferation and sustain the viability of the effector T cells62,63,64.
In addition, a recent study demonstrated that virus-specific CD8+ effector T cells can produce the regulatory cytokine IL-10 in response to antigenic stimulation but do so only following their migration into the virus-infected lungs65. It is noteworthy that a burst of T cell-derived IL-10 production was shown to occur at the time of the initial influx of effector T cells into the lungs. This burst coincides with the peak of production of pro-inflammatory mediators such as IFNγ by the CD8+ effector T cells and with the onset of virus clearance65. IL-10-producing effector CD8+ T cells have been shown to simultaneously produce the pro-inflammatory cytokine IFNγ, and the production of both of these cytokines by CD8+ T cells decreases with virus clearance65. It remains to be determined whether this transition from an effector T cell capable of producing pro-inflammatory cytokines in the draining lymph nodes to an effector T cell that can produce both pro-inflammatory and regulatory cytokines after entering the virus-infected lungs represents a change in the activation state or in the differentiation state of the activated T cell. However, an examination of IL-10 production by CD8+ effector T cells in vivo in infected lungs66 revealed that the production of this regulatory cytokine required exposure of the T cells in the infected lungs to IL-2 derived from antiviral CD4+ effector T cells and IL-27 produced by inflammatory mononuclear cells and neutrophils. These and other observations67 favour the view that the exposure of CD8+ effector T cells to pro-inflammatory (IL-27) and survival (IL-2) stimuli in the infected lungs enhances T cell activation, thereby facilitating Il10 gene expression.
An early in vitro analysis of the regulation of T cell effector activity suggested that there was an expressional hierarchy in T cell effector activities that was dependent on the strength of the activating stimulus68. According to this analysis, CD8+ T cell cytotoxic activity had the least stringent requirement for co-stimulatory and cytokine signals, followed by T cell proliferation, with pro-inflammatory cytokine production requiring the strongest co-stimulatory signals. Very recently, evidence has emerged from an in vivo analysis of T cell responses to respiratory virus infection to support this concept49. In this study, effector CD8+ T cell activation by influenza virus-derived antigens that were presented by infected CD45− respiratory epithelial cells resulted in T cell-mediated killing of the infected epithelial cells without the release of pro-inflammatory cytokines. By contrast, effector CD8+ T cell activation by viral antigens that were presented by interstitial CD45+ inflammatory cells (primarily LY6ChiCD11c+ inflammatory mononuclear phagocytes) induced cell-mediated cytotoxicity and pro-inflammatory cytokine production by the effector CD8+ T cells. The mechanism (or mechanisms) that account for this difference in the T cell response to APC or target cell recognition remains to be fully elucidated.
However, one crucial difference between the recognition of CD45+ APCs and that of CD45− target cells by effector T cells is the expression of co-stimulatory ligands by APCs but not by target cells49,69. Indeed, blockade of the co-stimulatory molecules CD80 and CD86 in vitro or in vivo suppresses the production of pro-inflammatory cytokines by effector CD8+ T cells without affecting their cytotoxic activity in vitro or virus clearance in vivo49. Thus, the engagement of co-stimulatory receptors on effector CD8+ T cells by APCs would provide the additional signal strength necessary for the production of pro-inflammatory cytokines by the T cells. Several reports have evaluated the contribution of specific co-stimulatory receptor–ligand interactions in regulating the host adaptive immune response and the outcome of respiratory virus infection70,71,72,73. However, in most instances no specific distinction was made between the contribution of these co-stimulatory receptor–ligand interactions during the induction phase of the response in the draining lymph nodes and during the effector phase in the infected lungs.
The above results may have important implications for therapeutic intervention in respiratory tract infections with viruses (such as influenza virus) in which productive infection (that is production of infectious virions) is restricted to CD45− respiratory epithelial cells. More generally, these observations add to the emerging picture that, after entering the infected lungs, effector T cells are altered or moulded by the inflammatory milieu encountered in situ. Such alterations could include changes in activation state (such as increased avidity of the T cell receptor for its target peptide–MHC complex on APCs), in effector activity (such as the types and/or amounts of mediators produced) and in viability, and possibly the differentiation of certain effector T cells into memory T cells49,63,69,71,74.
Acute control of pulmonary inflammation
Multiple cell types express immunosuppressive molecules75 to maintain pulmonary homeostasis. This suppressive state is disrupted by the action of innate and adaptive immune cells responding to infection. Their responses facilitate virus clearance but also can produce excessive pulmonary inflammation and tissue damage (reviewed in Ref. 76) (Fig. 2).

Molecules derived from both innate and adaptive immune cells contribute to the regulation of excessive pulmonary inflammation during acute respiratory virus infection. CD4+ T helper 1 (TH1) cells and type 1 CD8+ cytotoxic T cells (TC1 cells) express the transcription factors T-bet and BLIMP1 (B lymphocyte-induced maturation protein 1) and produce high levels of the regulatory cytokine interleukin-10 (IL-10), in addition to effector cytokines and cytolytic molecules, during respiratory virus infection. Regulatory T (TReg) cells in the lungs also express the 'effector' transcription factors T-bet and BLIMP1, produce various immunoregulatory cytokines (including IL-10 and transforming growth factor-β1 (TGFβ1)) and express the inhibitory receptor cytotoxic T-lymphocyte antigen 4 (CTLA4). Lung epithelial cells express CD200 and are required for the control of exuberant activation of classically activated macrophages. By contrast, alternatively activated macrophages contribute to the control of excessive pulmonary inflammation. Plasmacytoid dendritic cells (pDCs) also contribute to the suppression of excessive T cell-mediated inflammation through an unidentified mechanism. iNOS, inducible nitric oxide synthase; PPARγ, peroxisome proliferator-activated receptor-γ; TNF, tumour necrosis factor; TRAIL, TNF-related apoptosis-inducing ligand.
Forkhead box P3 (FOXP3)+ regulatory T (TReg) cells have an important role in maintaining immune homeostasis by suppressing inflammation in naive hosts. However, it has recently been demonstrated that activated TReg cells can acquire unique phenotypes that enhance their ability to control exaggerated inflammation77,78. Following respiratory virus infection, TReg cells in the lungs have been shown to express 'effector' TReg cell markers. For instance, the expression of the TH1 cell-associated transcription factor T-bet is upregulated in lung TReg cells during influenza virus infection65 and is associated with enhanced TReg cell-mediated suppression of excessive TH1-type inflammatory responses79. Similarly, BLIMP1 (B lymphocyte-induced maturation protein 1; also known as PRDM1), which is required for the terminal differentiation of effector T cells, is necessary for the enhanced production of IL-10 by TReg cells during influenza virus infection65,67. The depletion of TReg cells leads to excessive pulmonary inflammation and injury during respiratory syncytial virus (RSV) infection80,81,82. Furthermore, the adoptive transfer of TReg cells into recombination-activating gene (RAG)-deficient mice (which lack B and T cells) controls the otherwise lethal inflammation mediated by innate immune cells during influenza virus infection83. These data suggest that TReg cells may be crucial for controlling acute host inflammation and for restoring lung homeostasis following respiratory virus infection, although the mechanisms underlying these functions are poorly understood.
In addition to effector TReg cells, conventional effector CD4+ and CD8+ T cells have recently been shown to exert a 'regulatory' function through the production of high levels of IL-10 during influenza virus, RSV or simian virus 5 infection65,84,85. Blocking T cell-derived IL-10 results in excessive pulmonary inflammation characterized by increased accumulation of CD45+ inflammatory cells and enhanced production of pro-inflammatory mediators during influenza virus or RSV infection65,85,86. As mentioned above, the production of IL-10 by effector T cells is primarily restricted to the site of infection rather than the site of T cell priming, suggesting that the local environment of the infected lungs can instruct the function of effector T cells65,84.
Various innate immune cell types may also act to suppress excessive inflammation arising from antiviral immune responses (Table 1). pDCs have a crucial role in restricting excessive T cell-mediated inflammation during experimental RSV infection through an unknown mechanism87. In addition, neutrophils can facilitate viral clearance and dampen inflammation88, and lung epithelial cells can inhibit the over-exuberant inflammatory activities of macrophages through the expression of the inhibitory molecule CD200 during influenza virus infection11. Furthermore, classically activated phagocytes (such as inflammatory DCs and macrophages) can be significant contributors to disease pathogenesis during influenza virus infection24,25,54, whereas alternatively activated macrophages have a regulatory function in RSV infection, dampening virus-induced inflammation driven by innate immune cells89. Of note, activation of the nuclear receptor peroxisome proliferator-activated receptor-γ (PPARγ) — a transcription factor that controls the differentiation of monocyte precursors into alternatively activated macrophages90 — suppresses lethal inflammation during influenza virus infection25. Therefore, the differentiation state of the responding immune cell may have a crucial role in the resolution of inflammation and the outcome of virus infection (Table 1).
The immunosuppressive cytokine TGFβ1 is constitutively expressed by many lung cell types, most notably epithelial cells, which express the integrins required to convert latent TGFβ1 into the active form and can therefore maintain immune homeostasis in the normal lung75. Administration of TGFβ1 to mice during influenza virus or RSV infection impairs virus clearance but suppresses the associated lung inflammation91, whereas inhibition of TGFβ1 during influenza virus infection results in lethal tissue injury92. Interestingly, whereas the neuraminidase protein from certain influenza virus strains can process latent TGFβ1 into its active form and thereby potentially dampen pulmonary inflammation during infection, neuraminidase from the highly pathogenic H5N1 influenza virus cannot process TGFβ1 (Ref. 92). Therefore the ability (or inability) of a virus to activate or inhibit host immunoregulatory mechanisms (such as TGFβ1 activation) may contribute to viral pathogenesis. So, there appear to be multiple layers of immunoregulation during acute respiratory virus infection at both the cellular level (such as TReg cells and other cells) and the molecular level (such as IL-10, CD200 and TGFβ1) (Fig. 2).
Bacterial superinfection following virus clearance
Epidemiological evidence has clearly suggested that respiratory virus infection, in particular type A influenza virus infection, increases the incidence and severity of secondary bacterial infections in the respiratory tract93. Histological analyses of infected lung tissue indicates that bacterial superinfection resulting in secondary bacterial pneumonia was the predominant cause of death during the 1918 type A influenza virus pandemic and the more recent H1N1 pandemic94,95. In a mouse model of bacterial superinfection, type A influenza virus infection increases the susceptibility of mice to various bacterial infections, including respiratory challenge with Streptococcus pneumoniae, Staphylococcus aureus or Haemophilus influenzae and systemic Listeria monocytogenes infection96,97. Increased susceptibility to bacterial superinfection typically starts as early as day 3 and continues until at least 2 weeks post type A influenza virus infection. The prevailing opinion to explain this susceptibility to superinfection has, for many years, been that type A influenza virus infection damages epithelial cells and degrades the barrier function of the epithelium, thereby facilitating bacterial colonization of the respiratory tract93. However, evidence has emerged over the past decade suggesting that type A influenza virus infection also alters the function of innate and adaptive immune cells, resulting in a failure to effectively control bacterial replication (Fig. 3).

Multiple distinct mechanisms have been postulated to account for the increased susceptibility to bacterial superinfection and bacterial pneumonia following infection with respiratory viruses such as type A influenza viruses. Influenza virus infection induces the production of type I interferons (IFNs), which inhibit the recruitment of circulating neutrophils and macrophages to the lung following bacterial challenge. Type I IFNs also inhibit the differentiation of antibacterial T helper 17 (TH17) cells from naive T cells (TH0 cells) or other TH cell types (such as TH1 and TH2 cells)110 and thereby potentiate host susceptibility to secondary bacterial infection. IFNγ production by influenza virus-specific effector T cells decreases the expression of macrophage receptor with collagenous structure (MARCO) by alveolar macrophages and inhibits the ingestion of bacteria by these cells. Moreover, interleukin-10 (IL-10) production by influenza virus-specific effector T cells may inhibit the ability of innate immune cells, in particular macrophages, to kill bacteria. Finally, the direct interaction and/or infection of innate immune cells — such as macrophages, neutrophils and natural killer (NK) cells — with influenza virus suppresses the ability of these cells to take up and kill bacteria.
Among the various immune cell types, lung-resident alveolar macrophages are the first line of defence against bacteria in the respiratory tract, as they are capable of both limiting bacterial outgrowth and orchestrating the recruitment and activation of other immune cells to restrict bacterial growth. Type A influenza virus infection reduces the numbers of alveolar macrophages by promoting their apoptosis and also suppresses the function of these cells through direct mechanisms (such as infection of the macrophages) and indirect mechanisms (via host factors induced by the infection)98,99. Alveolar macrophages from lungs infected with type A influenza virus show increased expression of the CD200 receptor (CD200R), ligation of which suppresses alveolar macrophage activation in response to secondary bacterial challenge100. Furthermore, alveolar macrophages from virus-infected lungs exhibit decreased expression of the scavenger receptor MARCO (macrophage receptor with collagenous structure) and a diminished ability to ingest bacteria99. They also produce only low levels of the cytokines and chemokines required for orchestrating the responses of other cell types against bacterial infection, such as neutrophil recruitment and activation98. The diminished chemokine production by alveolar macrophages from virus-infected lungs is associated with decreased translocation of nuclear factor-κB (NF-κB) to the nucleus of alveolar macrophages as a result of as yet unidentified mechanisms98. The desensitization of alveolar macrophages can last for several months after influenza virus infection. Therefore, given the important role of alveolar macrophages in the initial control of bacterial infection, these data suggest that individuals who have recovered from severe influenza virus infection may have compromised immunity to subsequent bacterial infections for extended time periods.
In addition to suppressing alveolar macrophages, influenza virus infection inhibits the recruitment of circulating monocytes and macrophages during responses to bacterial challenge through type I IFN-dependent mechanisms101. Neutrophils have a crucial role in clearing pulmonary bacterial infection, but those isolated from influenza virus-infected lungs exhibit impaired responses to bacterial challenge102. Indeed, influenza virus infection of (or binding to) neutrophils in vitro results in a direct inhibition of neutrophil chemotaxis and superoxide production103. In vivo, influenza virus can directly infect neutrophils in the lung and, thus, may directly suppress the function of neutrophils during secondary bacterial challenge104. Furthermore, type I IFN production induced during primary influenza virus infection can directly inhibit neutrophil infiltration into the lung and/or increase the apoptosis of neutrophils105.
Natural killer (NK) cells, via their release of antibacterial factors, can also serve as important effector cells during certain bacterial infections106. However, NK cells isolated from influenza virus-infected lungs show decreased production of the antibacterial effector cytokine TNF106, although the mechanisms underlying this defect in NK cell function during influenza virus infection remain elusive. NK cells are reported to be susceptible to infection by influenza virus, and infected NK cells have various defects in effector activities such as cytotoxicity and cytokine and chemokine production107.
Influenza virus infection induces highly polarized TH1-type immune responses characterized by the production of IFNγ. Of note, neutralization of IFNγ during influenza virus infection restores MARCO expression by macrophages, increases bacterial ingestion and killing by macrophages and enhances host resistance to secondary bacterial infection99. Highly polarized CD4+ TH1 cells and type 1 CD8+ cytotoxic T cells are also a major source of the anti-inflammatory cytokine IL-10 during influenza virus infection65,108. IL-10 has been implicated in the susceptibility to secondary bacterial infections following influenza virus infection. In one study, the blockade of IL-10 enhanced the clearance of a secondary infection with S. pneumoniae109. However, the role of IL-10 during secondary bacterial infection remains controversial, as two recent reports using IL-10-deficient mice found a minimal role for IL-10 in regulating host susceptibility to secondary bacterial challenge99,105. TH17 cell responses are also crucial for host resistance to challenges with certain bacteria, including S. pneumoniae and S. aureus110, and influenza virus-induced production of type I IFNs has been shown to suppress the subsequent development of TH17 cells in response to bacterial superinfection110.
In summary, emerging evidence suggests that one or more of the regulatory mechanisms (for example, the expression of CD200R, IL-10 and/or TGFβ1) that are used by innate and adaptive immune cells to suppress excess inflammation during acute respiratory virus infections may facilitate bacterial colonization by diminishing the recruitment and function of phagocytes and the subsequent destruction of bacterial pathogens (Fig. 3). If immune mechanisms aimed at controlling virus replication and/or excessive inflammation in the virus-infected lungs predispose the individual to bacterial superinfection, then therapeutic strategies to prevent bacterial superinfection in the lungs must balance the need to prevent virus replication and excessive inflammation during virus clearance with the need to optimize the antibacterial functions of innate immune phagocytes.
Lower respiratory tract virus infections can, if severe, also alter lung function for an extended period after virus clearance and potentially predispose to, or at least exacerbate, chronic pulmonary inflammatory diseases such as asthma and chronic obstructive pulmonary disease (Box 1).
Conclusions
The recent advances in the identification and characterization of distinct subsets of DCs within the respiratory tract, along with the elucidation of their functions as APCs before and during virus infection, offer the promise of developing newer and more effective vaccination strategies against viral and other respiratory tract pathogens. The emerging evidence that the spectrum of effector activity exhibited by antiviral CD8+ T cells may differ depending on the level of co-stimulation that they receive raises the possibility that in the case of respiratory viruses, for which replication is restricted to respiratory epithelium, excessive inflammation associated with severe infection can be controlled by blocking co-stimulatory signals, without altering immune-mediated virus clearance.
More generally, recent evidence strongly suggests that the activation state and function of effector T cells in virus-infected lungs are sculpted by the local milieu generated by the resident and infiltrating inflammatory cells and the factors that they produce in response to virus infection. This additional moulding of the immune response at the site of infection will have an impact on the process of virus clearance and an equally important effect on the regulation of acute inflammation, and possibly immunological memory.
Resolution of respiratory virus infection requires not only the elimination of the virus but also the repair and regeneration of normal lung structures and, most importantly, the restoration of normal pulmonary function. Although at present the resolution response is only poorly understood and appreciated, the cells and molecules of the innate and adaptive immune system that respond to virus infection in the lower respiratory tract will almost certainly be shown to contribute. Whether and how these cells and molecules regulate processes such as epithelial cell to mesenchymal cell transitions, as well as the transformation of fibroblasts and fibrocytes into myofibroblasts, will become an area of intense research, as dysregulation of these processes in response to lung inflammation is associated with progressive pulmonary injury and lung fibrosis.
References
Chen, Y. et al. A novel subset of putative stem/progenitor CD34+Oct-4+ cells is the major target for SARS coronavirus in human lung. J. Exp. Med. 204, 2529–2536 (2007).
Mallick, B., Ghosh, Z. & Chakrabarti, J. MicroRNome analysis unravels the molecular basis of SARS infection in bronchoalveolar stem cells. PLoS ONE 4, e7837 (2009).
Shinya, K. et al. Avian flu: influenza virus receptors in the human airway. Nature 440, 435–436 (2006).
Yamada, S. et al. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature 444, 378–382 (2006).
Khatri, M., O'Brien, T. D., Goyal, S. M. & Sharma, J. M. Isolation and characterization of chicken lung mesenchymal stromal cells and their susceptibility to avian influenza virus. Dev. Comp. Immunol. 34, 474–479 (2010).
McGill, J., Heusel, J. W. & Legge, K. L. Innate immune control and regulation of influenza virus infections. J. Leukoc. Biol. 86, 803–812 (2009).
Pang, I. K. & Iwasaki, A. Inflammasomes as mediators of immunity against influenza virus. Trends Immunol. 32, 34–41 (2011).
Takeuchi, O. & Akira, S. Innate immunity to virus infection. Immunol. Rev. 227, 75–86 (2009).
Holt, P. G., Strickland, D. H., Wikstrom, M. E. & Jahnsen, F. L. Regulation of immunological homeostasis in the respiratory tract. Nature Rev. Immunol. 8, 142–152 (2008).
Waffarn, E. E. & Baumgarth, N. Protective B cell responses to flu — no fluke! J. Immunol. 186, 3823–3829 (2011).
Snelgrove, R. J. et al. A critical function for CD200 in lung immune homeostasis and the severity of influenza infection. Nature Immunol. 9, 1074–1083 (2008). This paper shows that CD200R is crucial for lung macrophage immune homeostasis in the resting state and limits the amplitude and duration of the inflammatory response during pulmonary influenza virus infection.
Kirby, A. C., Coles, M. C. & Kaye, P. M. Alveolar macrophages transport pathogens to lung draining lymph nodes. J. Immunol. 183, 1983–1989 (2009).
Plantinga, M., Hammad, H. & Lambrecht, B. N. Origin and functional specializations of DC subsets in the lung. Eur. J. Immunol. 40, 2112–2118 (2010).
Kim, T. S. & Braciale, T. J. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS ONE 4, e4204 (2009). This paper demonstrates that different subsets of migratory respiratory DCs differ in their efficiency at presenting virus-derived antigens to naive CD4+ and CD8+ T cells in the draining nodes in response to respiratory virus infection.
del Rio, M. L., Rodriguez-Barbosa, J. I., Kremmer, E. & Forster, R. CD103− and CD103+ bronchial lymph node dendritic cells are specialized in presenting and cross-presenting innocuous antigen to CD4+ and CD8+ T cells. J. Immunol. 178, 6861–6866 (2007).
Sung, S. S. et al. A major lung CD103 (αE)-β7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J. Immunol. 176, 2161–2172 (2006). This paper provides the first evidence that CD103+ and CD11bhi respiratory DCs are the major DC populations in the lung.
GeurtsvanKessel, C. H. et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b− but not plasmacytoid dendritic cells. J. Exp. Med. 205, 1621–1634 (2008). This paper demonstrates that langerin+CD11b− migratory respiratory DCs are required for efficient CD8+ T cell activation and for virus clearance in vivo.
Bursch, L. S. et al. Identification of a novel population of Langerin+ dendritic cells. J. Exp. Med. 204, 3147–3156 (2007).
Helft, J., Ginhoux, F., Bogunovic, M. & Merad, M. Origin and functional heterogeneity of non-lymphoid tissue dendritic cells in mice. Immunol. Rev. 234, 55–75 (2010).
Edelson, B. T. et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8α+ conventional dendritic cells. J. Exp. Med. 207, 823–836 (2010).
Ginhoux, F. et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 206, 3115–3130 (2009).
Beaty, S. R., Rose, C. E. Jr & Sung, S. S. Diverse and potent chemokine production by lung CD11bhigh dendritic cells in homeostasis and in allergic lung inflammation. J. Immunol. 178, 1882–1895 (2007).
Jakubzick, C. et al. Blood monocyte subsets differentially give rise to CD103+ and CD103− pulmonary dendritic cell populations. J. Immunol. 180, 3019–3027 (2008).
Lin, K. L., Suzuki, Y., Nakano, H., Ramsburg, E. & Gunn, M. D. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 180, 2562–2572 (2008).
Aldridge, J. R. Jr et al. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc. Natl Acad. Sci. USA 106, 5306–5311 (2009).
Jenkins, S. J. et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 332, 1284–1288 (2011).
Hammad, H. & Lambrecht, B. N. Dendritic cells and epithelial cells: linking innate and adaptive immunity in asthma. Nature Rev. Immunol. 8, 193–204 (2008).
Kitamura, H. et al. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8-mediated activation of TGF-β. J. Clin. Invest. 121, 2863–2875 (2011).
Hammad, H. et al. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nature Med. 15, 410–416 (2009).
Marsolais, D. et al. Local not systemic modulation of dendritic cell S1P receptors in lung blunts virus-specific immune responses to influenza. Mol. Pharmacol. 74, 896–903 (2008).
Jakubzick, C., Tacke, F., Llodra, J., van Rooijen, N. & Randolph, G. J. Modulation of dendritic cell trafficking to and from the airways. J. Immunol. 176, 3578–3584 (2006).
Lawrence, C. W. & Braciale, T. J. Activation, differentiation, and migration of naive virus-specific CD8+ T cells during pulmonary influenza virus infection. J. Immunol. 173, 1209–1218 (2004).
Yoon, H., Legge, K. L., Sung, S. S. & Braciale, T. J. Sequential activation of CD8+ T cells in the draining lymph nodes in response to pulmonary virus infection. J. Immunol. 179, 391–399 (2007).
Khanna, K. M. et al. In situ imaging reveals different responses by naive and memory CD8 T cells to late antigen presentation by lymph node DC after influenza virus infection. Eur. J. Immunol. 38, 3304–3315 (2008).
Kim, T. S., Hufford, M. M., Sun, J., Fu, Y. X. & Braciale, T. J. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J. Exp. Med. 207, 1161–1172 (2010).
Belz, G. T. et al. Distinct migrating and nonmigrating dendritic cell populations are involved in MHC class I-restricted antigen presentation after lung infection with virus. Proc. Natl Acad. Sci. USA 101, 8670–8675 (2004).
Lukens, M. V., Kruijsen, D., Coenjaerts, F. E., Kimpen, J. L. & van Bleek, G. M. Respiratory syncytial virus-induced activation and migration of respiratory dendritic cells and subsequent antigen presentation in the lung-draining lymph node. J. Virol. 83, 7235–7243 (2009).
Beauchamp, N. M., Busick, R. Y. & Alexander-Miller, M. A. Functional divergence among CD103+ dendritic cell subpopulations following pulmonary poxvirus infection. J. Virol. 84, 10191–10199 (2010).
Tournier, J. N. & Mohamadzadeh, M. Key roles of dendritic cells in lung infection and improving anthrax vaccines. Trends Mol. Med. 16, 303–312 (2010).
Ballesteros-Tato, A., Leon, B., Lund, F. E. & Randall, T. D. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8+ T cell responses to influenza. Nature Immunol. 11, 216–224 (2010). This paper shows that CD11bhi migratory respiratory DCs dominate antigen presentation at the peak of infection.
Moltedo, B., Li, W., Yount, J. S. & Moran, T. M. Unique type I interferon responses determine the functional fate of migratory lung dendritic cells during influenza virus infection. PLoS Pathog. 7, e1002345 (2011).
Desch, A. N. et al. CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell-associated antigen. J. Exp. Med. 208, 1789–1797 (2011).
Ho, A. W. et al. Lung CD103+ dendritic cells efficiently transport influenza virus to the lymph node and load viral antigen onto MHC class I for presentation to CD8 T cells. J. Immunol. 187, 6011–6021 (2011).
Nakano, H. et al. Blood-derived inflammatory dendritic cells in lymph nodes stimulate acute T helper type 1 immune responses. Nature Immunol. 10, 394–402 (2009).
Zammit, D. J., Turner, D. L., Klonowski, K. D., Lefrancois, L. & Cauley, L. S. Residual antigen presentation after influenza virus infection affects CD8 T cell activation and migration. Immunity 24, 439–449 (2006).
Takamura, S. et al. The route of priming influences the ability of respiratory virus-specific memory CD8+ T cells to be activated by residual antigen. J. Exp. Med. 207, 1153–1160 (2010).
Jelley-Gibbs, D. M. et al. Unexpected prolonged presentation of influenza antigens promotes CD4 T cell memory generation. J. Exp. Med. 202, 697–706 (2005).
Brincks, E. L., Katewa, A., Kucaba, T. A., Griffith, T. S. & Legge, K. L. CD8 T cells utilize TRAIL to control influenza virus infection. J. Immunol. 181, 4918–4925 (2008).
Hufford, M. M., Kim, T. S., Sun, J. & Braciale, T. J. Antiviral CD8+ T cell effector activities in situ are regulated by target cell type. J. Exp. Med. 208, 167–180 (2011). This study demonstrates that the expression of cytotoxic T lymphocyte effector activity at the site of virus infection is dictated by the type of target cell encountered.
Topham, D. J., Tripp, R. A. & Doherty, P. C. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J. Immunol. 159, 5197–5200 (1997).
Graham, M. B. et al. Response to influenza infection in mice with a targeted disruption in the interferon γ gene. J. Exp. Med. 178, 1725–1732 (1993).
Cook, D. N. et al. Requirement of MIP-1 α for an inflammatory response to viral infection. Science 269, 1583–1585 (1995).
Peper, R. L. & Van Campen, H. Tumor necrosis factor as a mediator of inflammation in influenza A viral pneumonia. Microb. Pathog. 19, 175–183 (1995).
Herold, S. et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J. Exp. Med. 205, 3065–3077 (2008). This paper identifies epithelial cell apoptosis induced by TRAIL-expressing macrophages as a major mechanism by which macrophages that are recruited to the lung induce alveolar leakage and enhance mortality associated with influenza virus pneumonia.
Brown, D. M., Dilzer, A. M., Meents, D. L. & Swain, S. L. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J. Immunol. 177, 2888–2898 (2006).
Graham, M. B., Braciale, V. L. & Braciale, T. J. Influenza virus-specific CD4+ T helper type 2 T lymphocytes do not promote recovery from experimental virus infection. J. Exp. Med. 180, 1273–1282 (1994).
Debbabi, H. et al. Primary type II alveolar epithelial cells present microbial antigens to antigen-specific CD4+ T cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 289, L274–L279 (2005).
Topham, D. J. & Doherty, P. C. Clearance of an influenza A virus by CD4+ T cells is inefficient in the absence of B cells. J. Virol. 72, 882–885 (1998).
Topham, D. J., Tripp, R. A., Hamilton-Easton, A. M., Sarawar, S. R. & Doherty, P. C. Quantitative analysis of the influenza virus-specific CD4+ T cell memory in the absence of B cells and Ig. J. Immunol. 157, 2947–2952 (1996).
Lawrence, C. W., Ream, R. M. & Braciale, T. J. Frequency, specificity, and sites of expansion of CD8+ T cells during primary pulmonary influenza virus infection. J. Immunol. 174, 5332–5340 (2005).
McGill, J. & Legge, K. L. Cutting edge: contribution of lung-resident T cell proliferation to the overall magnitude of the antigen-specific CD8 T cell response in the lungs following murine influenza virus infection. J. Immunol. 183, 4177–4181 (2009).
Hendriks, J., Xiao, Y. & Borst, J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J. Exp. Med. 198, 1369–1380 (2003).
McGill, J., Van Rooijen, N. & Legge, K. L. IL-15 trans-presentation by pulmonary dendritic cells promotes effector CD8 T cell survival during influenza virus infection. J. Exp. Med. 207, 521–534 (2010).
van Gisbergen, K. P. et al. The costimulatory molecule CD27 maintains clonally diverse CD8+ T cell responses of low antigen affinity to protect against viral variants. Immunity 35, 97–108 (2011).
Sun, J., Madan, R., Karp, C. L. & Braciale, T. J. Effector T cells control lung inflammation during acute influenza virus infection by producing IL-10. Nature Med. 15, 277–284 (2009). This study demonstrates that effector T cells (in particular, effector CD8+ T cells) can regulate inflammation at the site of infection by producing IL-10 in response to influenza virus infection.
Sun, J., Dodd, H., Moser, E. K., Sharma, R. & Braciale, T. J. CD4+ T cell help and innate-derived IL-27 induce Blimp-1-dependent IL-10 production by antiviral CTLs. Nature Immunol. 12, 327–334 (2011).
Cretney, E. et al. The transcription factors Blimp-1 and IRF4 jointly control the differentiation and function of effector regulatory T cells. Nature Immunol. 12, 304–311 (2011).
Valitutti, S., Muller, S., Dessing, M. & Lanzavecchia, A. Different responses are elicited in cytotoxic T lymphocytes by different levels of T cell receptor occupancy. J. Exp. Med. 183, 1917–1921 (1996).
Dolfi, D. V. et al. Dendritic cells and CD28 costimulation are required to sustain virus-specific CD8+ T cell responses during the effector phase in vivo. J. Immunol. 186, 4599–4608 (2011).
Humphreys, I. R. et al. A critical role for ICOS co-stimulation in immune containment of pulmonary influenza virus infection. Eur. J. Immunol. 36, 2928–2938 (2006).
Humphreys, I. R. et al. A critical role for OX40 in T cell-mediated immunopathology during lung viral infection. J. Exp. Med. 198, 1237–1242 (2003).
Lin, G. H. et al. Endogenous 4–1BB ligand plays a critical role in protection from influenza-induced disease. J. Immunol. 182, 934–947 (2009).
Snell, L. M. et al. CD8 T cell-intrinsic GITR is required for T cell clonal expansion and mouse survival following severe influenza infection. J. Immunol. 185, 7223–7234 (2010).
McGill, J., Van Rooijen, N. & Legge, K. L. Protective influenza-specific CD8 T cell responses require interactions with dendritic cells in the lungs. J. Exp. Med. 205, 1635–1646 (2008).
Snelgrove, R. J., Godlee, A. & Hussell, T. Airway immune homeostasis and implications for influenza-induced inflammation. Trends Immunol. 32, 328–334 (2011).
La Gruta, N. L., Kedzierska, K., Stambas, J. & Doherty, P. C. A question of self-preservation: immunopathology in influenza virus infection. Immunol. Cell Biol. 85, 85–92 (2007).
Campbell, D. J. & Koch, M. A. Phenotypical and functional specialization of FOXP3+ regulatory T cells. Nature Rev. Immunol. 11, 119–130 (2011).
Ohkura, N. & Sakaguchi, S. Maturation of effector regulatory T cells. Nature Immunol. 12, 283–284 (2011).
Koch, M. A. et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nature Immunol. 10, 595–602 (2009).
Ruckwardt, T. J., Bonaparte, K. L., Nason, M. C. & Graham, B. S. Regulatory T cells promote early influx of CD8+ T cells in the lungs of respiratory syncytial virus-infected mice and diminish immunodominance disparities. J. Virol. 83, 3019–3028 (2009).
Fulton, R. B., Meyerholz, D. K. & Varga, S. M. Foxp3+ CD4 regulatory T cells limit pulmonary immunopathology by modulating the CD8 T cell response during respiratory syncytial virus infection. J. Immunol. 185, 2382–2392 (2010).
Lee, D. C. et al. CD25+ natural regulatory T cells are critical in limiting innate and adaptive immunity and resolving disease following respiratory syncytial virus infection. J. Virol. 84, 8790–8798 (2010).
Antunes, I. & Kassiotis, G. Suppression of innate immune pathology by regulatory T cells during influenza A virus infection of immunodeficient mice. J. Virol. 84, 12564–12575 (2010).
Palmer, E. M., Holbrook, B. C., Arimilli, S., Parks, G. D. & Alexander-Miller, M. A. IFNγ-producing, virus-specific CD8+ effector cells acquire the ability to produce IL-10 as a result of entry into the infected lung environment. Virology 404, 225–230 (2010).
Sun, J. et al. Autocrine regulation of pulmonary inflammation by effector T-cell derived IL-10 during infection with respiratory syncytial virus. PLoS Pathog. 7, e1002173 (2011).
Weiss, K. A., Christiaansen, A. F., Fulton, R. B., Meyerholz, D. K. & Varga, S. M. Multiple CD4+ T cell subsets produce immunomodulatory IL-10 during respiratory syncytial virus infection. J. Immunol. 187, 3145–3154 (2011).
Smit, J. J., Rudd, B. D. & Lukacs, N. W. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J. Exp. Med. 203, 1153–1159 (2006).
Tate, M. D. et al. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J. Immunol. 183, 7441–7450 (2009).
Shirey, K. A. et al. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4Rα-, TLR4-, and IFN-β-dependent. Mucosal Immunol. 3, 291–300 (2010). This study demonstrates that alternatively activated macrophages may mediate the resolution of RSV-induced lung injury.
Odegaard, J. I. et al. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447, 1116–1120 (2007).
Williams, A. E. et al. TGF-β prevents eosinophilic lung disease but impairs pathogen clearance. Microbes Infect. 7, 365–374 (2005).
Carlson, C. M. et al. Transforming growth factor-β: activation by neuraminidase and role in highly pathogenic H5N1 influenza pathogenesis. PLoS Pathog. 6, e1001136 (2010).
McCullers, J. A., Speck, K. M., Williams, B. F., Liang, H. & Mirro, J. Jr. Increased influenza vaccination of healthcare workers at a pediatric cancer hospital: results of a comprehensive influenza vaccination campaign. Infect. Control Hosp. Epidemiol. 27, 77–79 (2006).
Morens, D. M., Taubenberger, J. K. & Fauci, A. S. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198, 962–970 (2008).
Bautista, E. et al. Clinical aspects of pandemic 2009 influenza A (H1N1) virus infection. N. Engl. J. Med. 362, 1708–1719 (2010).
Ballinger, M. N. & Standiford, T. J. Postinfluenza bacterial pneumonia: host defenses gone awry. J. Interferon Cytokine Res. 30, 643–652 (2010).
Jamieson, A. M., Yu, S., Annicelli, C. H. & Medzhitov, R. Influenza virus-induced glucocorticoids compromise innate host defense against a secondary bacterial infection. Cell Host Microbe 7, 103–114 (2010).
Didierlaurent, A. et al. Sustained desensitization to bacterial Toll-like receptor ligands after resolution of respiratory influenza infection. J. Exp. Med. 205, 323–329 (2008).
Sun, K. & Metzger, D. W. Inhibition of pulmonary antibacterial defense by interferon-γ during recovery from influenza infection. Nature Med. 14, 558–564 (2008). This study demonstrates that IFNγ can promote secondary bacterial infection following influenza virus infection by downregulating the expression of the class A scavenger receptor MARCO on alveolar macrophages.
Goulding, J. et al. Lowering the threshold of lung innate immune cell activation alters susceptibility to secondary bacterial superinfection. J. Infect. Dis. 204, 1086–1094 (2011).
Nakamura, R. et al. Interleukin-15 is critical in the pathogenesis of influenza A virus-induced acute lung injury. J. Virol. 84, 5574–5582 (2010).
McNamee, L. A. & Harmsen, A. G. Both influenza-induced neutrophil dysfunction and neutrophil-independent mechanisms contribute to increased susceptibility to a secondary Streptococcus pneumoniae infection. Infect. Immun. 74, 6707–6721 (2006).
Cooper, J. A. Jr, Carcelen, R. & Culbreth, R. Effects of influenza A nucleoprotein on polymorphonuclear neutrophil function. J. Infect. Dis. 173, 279–284 (1996).
Manicassamy, B. et al. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl Acad. Sci. USA 107, 11531–11536 (2010).
Shahangian, A. et al. Type I IFNs mediate development of postinfluenza bacterial pneumonia in mice. J. Clin. Invest. 119, 1910–1920 (2009). This study demonstrates that type I IFNs can promote secondary bacterial infection following influenza virus infection by inhibiting the production of the chemokines that recruit neutrophils to the lung.
Small, C. L. et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J. Immunol. 184, 2048–2056 (2010).
Guo, H. et al. The functional impairment of natural killer cells during influenza virus infection. Immunol. Cell Biol. 87, 579–589 (2009).
McKinstry, K. K. et al. IL-10 deficiency unleashes an influenza-specific Th17 response and enhances survival against high-dose challenge. J. Immunol. 182, 7353–7363 (2009).
van der Sluijs, K. F. et al. IL-10 is an important mediator of the enhanced susceptibility to pneumococcal pneumonia after influenza infection. J. Immunol. 172, 7603–7609 (2004).
Kudva, A. et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 186, 1666–1674 (2011). This study demonstrates that influenza virus infection inhibits the development of antibacterial T H 17 cell responses through the induction of type I IFNs.
Zheng, T. et al. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J. Clin. Invest. 106, 1081–1093 (2000).
Barnes, P. J. The cytokine network in asthma and chronic obstructive pulmonary disease. J. Clin. Invest. 118, 3546–3556 (2008).
Lukacs, N. W. et al. Differential immune responses and pulmonary pathophysiology are induced by two different strains of respiratory syncytial virus. Am. J. Pathol. 169, 977–986 (2006).
Grayson, M. H. et al. Induction of high-affinity IgE receptor on lung dendritic cells during viral infection leads to mucous cell metaplasia. J. Exp. Med. 204, 2759–2769 (2007).
Kim, E. Y. et al. Persistent activation of an innate immune response translates respiratory viral infection into chronic lung disease. Nature Med. 14, 633–640 (2008).
Wills-Karp, M. & Finkelman, F. D. Innate lymphoid cells wield a double-edged sword. Nature Immunol. 12, 1025–1027 (2011).
Chang, Y. J. et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nature Immunol. 12, 631–638 (2011).
Saenz, S. A., Noti, M. & Artis, D. Innate immune cell populations function as initiators and effectors in Th2 cytokine responses. Trends Immunol. 31, 407–413 (2010).
Monticelli, L. A. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nature Immunol. 12, 1045–1054 (2011).
GeurtsvanKessel, C. H. et al. Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus-infected mice. J. Exp. Med. 206, 2339–2349 (2009).
Halle, S. et al. Induced bronchus-associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J. Exp. Med. 206, 2593–2601 (2009).
Gereke, M., Jung, S., Buer, J. & Bruder, D. Alveolar type II epithelial cells present antigen to CD4+ T cells and induce Foxp3+ regulatory T cells. Am. J. Respir. Crit. Care Med. 179, 344–355 (2009).
Strickland, D. H., Kees, U. R. & Holt, P. G. Suppression of T-cell activation by pulmonary alveolar macrophages: dissociation of effects on TcR, IL-2R expression, and proliferation. Eur. Respir. J. 7, 2124–2130 (1994).
Daughety, T. W., Marrack, P., Kappler, J. W. & Chiller, J. M. The capacity of murine alveolar macrophages to stimulate antigen-dependent T-lymphocyte activation and proliferation. Cell. Immunol. 79, 374–382 (1983).
GeurtsvanKessel, C. H. et al. Both conventional and interferon killer dendritic cells have antigen-presenting capacity during influenza virus infection. PLoS ONE 4, e7187 (2009).
Yoo, J. K., Galligan, C. L., Virtanen, C. & Fish, E. N. Identification of a novel antigen-presenting cell population modulating antiinfluenza type 2 immunity. J. Exp. Med. 207, 1435–1451 (2010).
Acknowledgements
We thank the members of the Braciale laboratory for their editorial assistance and insightful discussions, and colleagues in the field who provided recent publications and personal communications. As a result of space limitations we apologize for being unable to cite many primary references relevant to the topic of this Review. This study was supported by grants to T.J.B. from the US National Institutes of Health (RO1 AI-15608, RO1 AI-37293, RO1 HL-33391 and U-19 AI-83024). T.S.K. and J.S. are recipients of Senior Research Training Fellowships from the American Lung Association.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information S1 (table)
Viruses that induce infections in humans that are primarily localized to the respiratory tract (PDF 82 kb)
Supplementary information S2 (box)
Non-immune barriers and innate immune defence to virus infection in the lung (PDF 105 kb)
Related links
Glossary
- Type I and type II alveolar epithelial cells
-
Type I alveolar epithelial cells (also known as squamous alveolar cells and type I pneumocytes) form the structure of the alveolar wall and are responsible for gas exchange in the alveoli. Type II alveolar epithelial cells (also known as great alveolar cells) continually secrete pulmonary surfactant to lower the surface tension of pulmonary fluids, thereby increasing gas exchange.
- Acute respiratory distress syndrome
-
(ARDS). A severe inflammatory disease of the lung that is usually triggered by another pulmonary pathology. The uncontrolled inflammation leads to impaired gas exchange, alveolar flooding and/or collapse, and systemic inflammatory response syndrome.
- Plasmacytoid DCs
-
(pDCs). A dendritic cell (DC) subtype defined by the expression of CD11c and B220 and the lack of CD8 and CD11b. pDCs are specialized to produce large amounts of type I interferons in response to viral infection, and they therefore have an important role in the immune response to viruses.
- Integrin
-
A member of a group of proteins that regulate the attachment of cells to one another (cell–cell adhesion) and to the surrounding network of proteins and other molecules (cell–matrix adhesion). Integrins also transmit chemical signals that regulate cell growth and the activity of certain genes.
- Sphingosine-1-phosphate
-
(S1P). A sphingolipid that is involved in signalling. In the immune system, S1P induces the egress of lymphocytes from lymphoid organs by binding to S1P receptors on the cells.
- Cross-presentation
-
A mechanism that can initiate a CD8+ T cell response to an antigen that is not present within antigen-presenting cells (APCs). This exogenous antigen must be taken up by APCs and then re-routed to the MHC class I pathway of antigen presentation.
- Efferocytosis
-
The phagocytic clearance of apoptotic cells before they undergo secondary necrosis. The process usually triggers an anti-inflammatory response.
- Inflammatory DCs
-
Dendritic cells (DCs) that are not normally present in the steady state but develop as a result of inflammation or microbial stimuli. For example, one such subset is tumour necrosis factor- and inducible nitric oxide synthase-producing DCs (TIP DCs). Inflammatory monocytes can also give rise to inflammatory DCs.
- Effector TReg cell
-
A regulatory T (TReg) cell that expresses particular transcription factors, such as T-bet, and follows a differentiation pathway to acquire a phenotype or function for efficiently controlling a specific subset of effector T cells, such as TH1 cells.
- Alternatively activated macrophages
-
(Also known as M2 macrophages). Macrophages that are stimulated by IL-4 or IL-13 and that express arginase 1, the mannose receptor CD206 and IL-4 receptor. There may be pathogen-associated molecular patterns expressed by helminths that can also drive the alternative activation of macrophages.
- Innate lymphoid cells
-
(ILCs; also known as natural helper cells and nuocytes). Cells that combine innate and adaptive immune functions and are part of the first line of defence against mucosal infections. ILCs are distinguished from adaptive lymphocytes by their independence from recombination-activating genes and the resulting absence of specific antigen receptors. They produce large quantities of type 2 cytokines such as IL-5 and IL-13.
- Fibrocytes
-
Mature fibroblasts that no longer produce fibres or intercellular substance in connective tissue. Fibroblasts are large, flat cells that form the collagenous and elastic fibres and intercellular substance of loose connective tissue.
Rights and permissions
About this article
Cite this article
Braciale, T., Sun, J. & Kim, T. Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol 12, 295–305 (2012). https://doi.org/10.1038/nri3166
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri3166
This article is cited by
-
A phase 1/2 clinical trial of invariant natural killer T cell therapy in moderate-severe acute respiratory distress syndrome
Nature Communications (2024)
-
Immunomodulatory Role of Microelements in COVID-19 Outcome: a Relationship with Nutritional Status
Biological Trace Element Research (2023)
-
Circulation of Respiratory Viruses in the City: Towards an Agent-Based Ecosystem model
Bulletin of Mathematical Biology (2023)
-
Metatranscriptome of human lung microbial communities in a cohort of mechanically ventilated COVID-19 Omicron patients
Signal Transduction and Targeted Therapy (2023)
-
Management of asthma patients during the COVID-19 pandemic: pathophysiological considerations to address the challenges
Beni-Suef University Journal of Basic and Applied Sciences (2022)
