
Key Points
-
Clinical severe T-cell immunodeficiencies (in many cases accompanied by deficiencies of other lymphocyte populations) are known to be caused by mutations in almost 40 different genes. These genes encode products vital for processes such as cytokine and T-cell receptor (TCR) signalling, V(D)J recombination and antigen presentation.
-
T-cell immunodeficiencies may not always be absolute, and in many cases an immunodeficient patient may also have symptoms of immune dysregulation, such as autoimmunity. In certain cases, identical mutations can cause severe or partial T-cell immunodeficiency, highlighting the complexity of pathogenesis.
-
For both severe and partial T-cell immunodeficiencies in humans, there are multiple genetic and/or phenotypic counterparts in mice, which are proving invaluable as tools to dissect cellular and biochemical mechanisms of these disorders.
-
Studies of these models have revealed that multiple T-cell tolerance mechanisms are population sensitive. These mechanisms, including thymic negative selection, peripheral suppression by FOXP3+ regulatory T cells and peripheral activation of effector T cells, show reduced efficacy at low T-cell population densities regardless of the properties of individual T cells.
-
Many common variant alleles (in genes such as PTPN22 (protein tyrosine phosphatase, non-receptor type 22) and IL7RA (interleukin-7 receptor α-chain)) have been reproducibly associated with multiple common autoimmune diseases. We propose that a proportion of such alleles (and also rare alleles not detected by genome-wide association studies) contribute to common autoimmune diseases by creating a state of partial T-cell immunodeficiency, which is insufficient in itself to promote autoimmunity but can synergize to promote the activation of self-reactive T cells.
Abstract
Partial T-cell immunodeficiencies constitute a heterogeneous cluster of disorders characterized by an incomplete reduction in T-cell number or activity. The immune deficiency component of these diseases is less severe than that of the severe T-cell immunodeficiencies and therefore some ability to respond to infectious organisms is retained. Unlike severe T-cell immunodeficiencies, however, partial immunodeficiencies are commonly associated with hyper-immune dysregulation, including autoimmunity, inflammatory diseases and elevated IgE production. This causative association is counter-intuitive — immune deficiencies are caused by loss-of-function changes to the T-cell component, whereas the coincident autoimmune symptoms are the consequence of gain-of-function changes. This Review details the genetic basis of partial T -cell immunodeficiencies and draws on recent advances in mouse models to propose mechanisms by which a reduction in T-cell numbers or function may disturb the population-dependent balance between activation and tolerance.
Similar content being viewed by others


Main
Congenital T-cell immunodeficiencies are rare diseases caused by mutations in genes that are essential for T-cell differentiation and which result in a severe deficiency in peripheral T cells. A more complex and poorly understood class of diseases is that which comprises the partial T-cell immunodeficiency diseases, in which effector T cells are present but in reduced numbers or with reduced function. These diseases are genetically heterogeneous with a largely undetermined aetiology, but they include associations with alternative mutations in the same genes that can cause severe T-cell immunodeficiency. Although many diseases in this class are relatively rare, together they are probably more common than the severe T-cell deficiencies. The most striking clinical manifestation of the partial T-cell immunodeficiencies is the frequent association of these diseases with immune dysregulation, such as autoimmune disease and elevated IgE production, both of which are consequences of effector T-cell hyperactivity. The association of effector T-cell hyperactivity symptoms with an effector T-cell hypoactive disease is counter-intuitive. However, recent studies in mouse models have shown that hyperactivation of immune system components can be a direct outcome of a partial decrease in T-cell number or activity.
In this Review, we describe the insights gained from mouse models into immune dysregulation during partial numerical or functional immunodeficiency and propose a model of 'population-dependent' immune tolerance mechanisms to explain the coincident presentation of both partial T-cell immunodeficiency and immune dysregulation. This model is based on the assumption that tolerance mechanisms evolved in the context of a complete peripheral T-cell niche, and that these mechanisms start to break down when they are forced to operate in the suboptimal context of a reduction in the functional T-cell population size. Partial reduction in the number or function of T cells therefore disturbs the tolerogenic balance, generating the unfortunate combination of immunodeficiency and immune dysregulation.
Genetic basis of T-cell immunodeficiency
Nearly 40 different genes have been identified that can cause monogenic severe T-cell immunodeficiencies in humans. These genes can be grouped into five main categories based on the cellular function of the proteins that they encode. Four of these categories include genes that encode proteins involved in functional processes that are essential for the differentiation of T cells (Fig. 1) (see Supplementary information S1 (table)). The first of these four categories comprises those genes of which the protein products are involved in cytokine signalling. The cytokine interleukin-7 (IL-7) is necessary for the differentiation and peripheral maintenance of T cells and therefore mutations in genes encoding molecules for IL-7-induced signalling can cause T-cell immunodeficiencies. The three other functional categories that include proteins essential for T-cell differentiation are antigen presentation, somatic recombination of the T-cell receptor (TCR) and TCR signalling. These three categories represent the requirement of T cells for positive selection signalling through the TCR during thymic development and peripheral activation. Proteins encoded by genes in the antigen presentation category function in the thymic epithelium, whereas proteins encoded by genes in the TCR V(D)J recombination or TCR signalling categories are T-cell intrinsic. As B cells and T cells share common components of the V(D)J recombination machinery, defects in the genes belonging to this category also result in B-cell immunodeficiency, albeit to a varying degree depending on the component that is mutated. Part of this variation may be explained by the additional roles of several components in DNA repair. It is clear why defects in the described functions of the proteins encoded by all of the genes in these four categories would lead to T-cell immunodeficiency, as they are necessary for the function of essential pathways during T-cell differentiation.

Genes with mutations that cause monogenic severe T-cell immunodeficiency in humans can be intrinsic to the thymic epithelium or to T cells. a | Genetic defects that are intrinsic to thymic epithelial cells ultimately affect the antigen-presentation pathway. b | Genetic defects that are intrinsic to T cells include those that affect T-cell receptor (TCR) signalling, cytokine signalling, somatic recombination or basic cellular processes. Other genes that are important to these pathways or processes but have not been linked to severe T-cell deficiency in humans are shown in grey. c | The pie chart shows the proportion of defective genes that affect each of these categories. ADA, adenosine deaminase; ATM, ataxia-telangiectasia mutated; CBL, Casitas B-lineage lymphoma; CIITA, class II transactivator; DCLRE1C, DNA crosslink repair 1C; DNMT3b, DNA cytosine-5 methyltransferase 3β; ER, endoplasmic reticulum; FOXN1, forkhead box N1; GADS, GRB2-related adaptor protein; IL-2Rγ, IL-2 receptor γ-chain; IL-7, interleukin-7; IL-7Rα, IL-7 receptor α-chain; JAK, Janus kinase; LAT, linker for activation of T cells; LIG4, ligase IV; MRE11A, meiotic recombination 11 homologue A; Nibrin, Nijmegen breakage syndrome 1; NP, nucleoside phosphorylase; CRACM1, calcium release-activated calcium modulator 1; RAG, recombination-activating gene; RFX5, regulatory factor X5; RFXANK, RFX-associated ankyrin-containing protein; RFXAP, RFX-associated protein; RMRP, RNA component of mitochondrial RNA-processing endoribonuclease; SLP76, SRC-homology-2-domain-containing leukocyte protein of 76 kDa; SMARCAL1, SWI/SNF-related matrix-associated actin-dependent regulator of chromatin a-like 1; SP110, SP110 nuclear body protein; STAT5B, signal transducer and activator of transcription 5B; TAP, transporter associated with antigen processing; TAPBP, TAP-binding protein; TSAD, T-cell-specific adaptor protein; ZAP70, ζ-chain-associated protein kinase of 70 kDa.
The fifth category of gene defects that can cause moderate to severe T-cell immunodeficiency comprises several apparently unrelated pathways. The proteins produced by these genes are all known to have a role in basic cellular processes that are common to multiple cell types. For example, ADA (adenosine deaminase) and NP (nucleoside phosphorylase; also known as PNP) are both involved in basic cellular metabolism; DNMT3b (DNA cytosine-5 methyltransferase 3β) and SP110 (SP110 nuclear body protein) have broad functions in gene regulation; SMARCAL1 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin subfamily a-like 1) is a chromatin remodelling protein; and RMRP (RNA component of mitochondrial RNA-processing endoribonuclease) is part of the mitochondrial RNA-processing endoribonuclease complex (see Supplementary information S1 (table)). Because each of these genes is expressed by multiple cell types, defects in these genes would not be predicted to give T-cell-specific phenotypes. It is possible that these genes might have currently unknown T-cell-specific functions; however, it is more likely that T cells are uniquely susceptible to mild defects in these basic cellular processes. This possibility is highlighted by the T-cell-specific defects observed in mice with a point mutation in Ncaph2 (non-SMC condensin II complex subunit H2; which encodes kleisin-β)1 and in mice lacking Rpl22 (ribosomal protein L22)2. Kleisin-β is a component of the ubiquitous chromosome condensin II subunit that is involved in cellular proliferation. The point mutation causes no obvious defects in the mice apart from reduced T-cell numbers, a phenotype that is independent of somatic recombination or TCR signalling1. RPL22 is a component of the ubiquitous 60S large ribosomal subunit. Unlike other ribosomal subunits, it is not essential for the function of the large subunit, and loss of the homologue protein in yeast only results in mild growth retardation3,4. In Rpl22−/− mice, the global cellular defect is compensated for in all cell types except in early thymocytes, which die in a p53-dependent manner2. The functional underpinning of such unique susceptibility may lie in the extremely rapid proliferation cycle of early thymocytes, allowing defects that would otherwise be redundant at slow proliferation rates to reach pathological status in thymocytes.
Clinical association with immune dysregulation
Although the most severe forms of T-cell immunodeficiency completely disable the adaptive immune system, a more common clinical manifestation is partial immunodeficiency, in which adaptive immune responses are impaired but retain some function (Table 1). In most of these diseases additional leukocyte subsets are affected, either directly or indirectly, and have an effect on the clinical presentation of these patients (in this Review however we focus exclusively on the T-cell effects). Intuitively, decreased T-cell function owing to partial immunodeficiency would be predicted to lower the incidence of conditions associated with hyperactivity of the adaptive immune system, such as autoimmunity, elevated IgE or inflammatory disease, in these patients. Yet, contrary to these theoretical speculations, up to half of all patients with partial T-cell deficiencies develop autoimmune disease5. This does not include diseases in which primary autoimmunity is likely to be the driving force behind secondary partial immunodeficiency, such as APECED (autoimmune polyendocrinopathy-candidiasis-ectodermal-dystrophy syndrome) and IPEX (immunodysregulation, polyendocrinopathy and enteropathy, X-linked syndrome). APECED and IPEX, together with the more common autoimmune diseases, are caused by primary defects in T-cell tolerance mechanisms. The associated increase in susceptibility to infections in these diseases is likely to be due to secondary effects caused by autoimmunity rather than due to a direct reduction in the capacity of effector T cells to combat infection. The clinical presentation of immune dysregulation in patients with partial T-cell immunodeficiencies can be so striking that the first of these conditions identified was recognized much earlier than the severe T-cell immunodeficiencies.
Monogenic partial immunodeficiencies with a shared genetic basis to severe immunodeficiency. In several cases, mutations in the same genes are known to result in either severe immunodeficiency without further immunological complications or in partial immunodeficiency with autoimmune or allergic manifestations. One example of this is Omenn syndrome, which is characterized by a combination of immunodeficiency and graft-versus-host-disease-like autoimmunity and immune dysregulation with elevated IgE production. Partial loss-of-function variants of the genes RAG1 (recombination-activating gene 1), RAG2, DCLRE1C (DNA crosslink repair 1C; also known as ARTEMIS), IL2RG (IL-2 receptor γ-chain), IL7RA and RMRP are known to cause Omenn syndrome6,7,8,9,10,11. Other related conditions arise from defects in the genes ADA (ADA deficiency), LIG4 (ligase IV deficiency), ZAP70 (ζ-chain-associated protein kinase of 70 kDa; ZAP70 deficiency) and STAT5B (signal transducer and activator of transcription 5B; growth-hormone insensitivity with immunodeficiency). These 10 genes are also linked to severe immunodeficiency without autoimmune complications.
The RAG1, RAG2 and DCLRE1C mutations associated with Omenn syndrome cause impaired V(D)J recombination and the emergence of an oligoclonal T-cell repertoire, which indicates that thymic selection in individuals with these mutations is restricted to those few T cells in which recombinase activity was sufficient to generate a functional TCR. Despite the limited number of functional T cells produced, the absolute peripheral number of T cells is often normalized by niche-filling mechanisms that expand the clones that have a successfully rearranged TCR12. Although in most cases different sets of mutations are responsible for the complete immunodeficiency phenotype, there does seem to be some overlap, as siblings of patients with Omenn syndrome have been observed to have severe T-cell immunodeficiency13, and identical RAG1 or RAG2 mutations have been identified in patients with severe T-cell immunodeficiency or Omenn syndrome14. The reason why identical RAG1 or RAG2 mutations can result in either the severe or partial form of immunodeficiency is currently unknown; it might be due to the stochastic effects of recombinase activity in individual thymocytes on the composition of the oligoclonal TCR repertoire, an alteration of recombinase activity by additional genetic factors or the influence of environmental factors (such as viral infections) on the T-cell clones that manage to expand15,16. Ligase IV is also required for V(D)J recombination and LIG4 mutations can manifest as complete immunodeficiency17; however, the majority of individuals with LIG4 mutations have partial immunodeficiency and include autoimmune components such as autoimmune thrombocytopaenia18,19.
IL2RG and IL7RA have essential roles in IL-7-dependent thymocyte survival, and mutations in these genes have been associated with both Omenn-like syndrome10,11 and severe T-cell deficiency20,21,22,23. In the case of IL2RG, identical alleles have been identified in both diseases11,24. A similar effect may be responsible for the variations in the clinical manifestation of deficiency of STAT5B, a component of the receptor signalling pathway for growth hormones and key haematopoietic-cell cytokines (including IL-7). STAT5B deficiency can manifest as either classical immunodeficiency with growth retardation25 or a complex of conditions including partial immunodeficiency with recurring infections, growth retardation, and the autoimmune conditions of thyroiditis and juvenile arthritis26.
Mutations in RMRP have been observed in two patients with Omenn syndrome9. RMRP encodes the RNA component of the ribonuclease mitochondrial RNA-processing complex, which is required for mitochondrial DNA replication. Although it is not known how defects in RMRP lead to T-cell oligoclonality and partial immunodeficiency, other mutations in RMRP cause cartilage-hair hypoplasia, a disorder that is commonly associated with partial lymphopaenia27 and, in rare cases, with severe T-cell immunodeficiency28,29,30. Complete loss-of-function mutations in RMRP have not been observed.
Likewise, severe mutagenic defects in ADA are associated with severe T-cell immunodeficiency, and patients with partial defects and less severe delayed-onset immunodeficiency have been seen to develop autoimmunity or have elevated levels of IgE31,32,33. An interesting case is that of a family in which one sibling suffered from classical severe T-cell immunodeficiency symptoms, whereas the other sibling showed spontaneous reversion of the ADA mutation in a subset of lymphocytes, and had less severe immunodeficiency and elevated IgE production34. In a similar manner, rare cases of ZAP70 deficiency result in a combination of partial immunodeficiency and production of allergen-specific IgE35.
Monogenic partial immunodeficiencies without genetic association to severe immunodeficiency. Mutations in other genes are also associated with partial T-cell immunodeficiency and immune dysregulation, but these have not been associated with complete T-cell immunodeficiency. These genes include WAS (mutation of which can cause Wiskott–Aldrich syndrome), and IL2RA, (mutation of which causes IL-2Rα deficiency).
Wiskott–Aldrich syndrome is characterized by eczema, thrombocytopaenia and bloody diarrhoea, combined with an increased susceptibility to infection. Around 40–70% of all patients with Wiskott–Aldrich syndrome also develop autoimmunity, most frequently haemolytic anaemia, neutropaenia and arthritis36,37. Shortly after the causative gene, WAS, was identified38, mutations in the same gene were revealed to cause a distinct clinical condition known as X-linked thrombocytopaenia, which is also associated with autoimmunity39,40.
Common allelic variants in IL2RA have been linked to the autoimmune diseases type 1 diabetes and Graves' disease41,42, but not to T-cell immunodeficiency. However, mutations in IL2RA have been identified in two patients, one with partial immunodeficiency (with frequent infections and an inability to reject allogeneic skin grafts) and extensive multi-tissue lymphocytic infiltration43, and another with frequent infections and autoimmunity but no T-cell lymphopaenia44.
Clinical conditions of uncertain genetic basis. Several other conditions combine partial immunodeficiency with immune dysregulation, but have no definitive genetic basis. In particular, three syndromes — hyper-IgE syndrome (the autosomal recessive form, rather than the classical autosomal dominant form that is not associated with autoimmunity), common variable immunodeficiency, and combined immunodeficiency with autoimmunity and spondylometaphyseal dysplasia — have a largely undefined genetic aetiology. These disorders have distinct immunological phenotypes and only a subset of patients exhibit partial T-cell lymphopaenia (which is of relevance to this Review), whereas others have normal T-cell counts. An additional disorder combining immunodeficiency with autoimmunity is 22q11 deletion syndrome (also known as velocardiofacial syndrome and DiGeorge syndrome). The genetic basis of this disorder is a deletion of chromosomal region 22q11, with multiple genes affected. As no mouse models that reproduce the immunological phenotype of these diseases are available, they are not discussed further in this Review. The possibility should be noted, however, that these diseases may result from a failure of immunological tolerance similar to the diseases listed above.
Despite the ambiguities of a common causality in several of the conditions described, it is clear that partial T-cell immunodeficiencies are associated with autoimmune disorders at a more frequent rate than the incidence of either disease alone would predict. Furthermore, the association of multiple genes with both severe immunodeficiency and partial T-cell immunodeficiency with immune dysregulation indicates that the association is not the property of select genes with differential effects, but rather an underlying consequence of partial T-cell immunodeficiency.
Although the clinical combination of partial immunodeficiency and immune dysregulation is relatively common, there are few mouse models available to dissect the association of partial immunodeficiency with autoimmunity. The key reasons for this deficit are likely to be the reliance on gene knockout mouse models and the incomplete understanding of the genetic basis of several diseases.
Mouse models of shared monogenic disorders
Nine of the ten human genes that have been identified both in patients with complete T-cell deficiency and in patients with partial T-cell deficiency have been well studied in mouse models. Mice that lack Rag1 (Ref. 45, 46), Rag2 (Refs 47, 48), Zap70 (Refs 49, 50, 51, 52, 53), Il7ra54 or Dclre1c55,56 show a severe defect in the production of T cells. These knockout mouse models accurately represent the alleles associated with severe disease in humans, but do not reflect the complex clinical presentation associated with alternative alleles. Mice lacking Ada57,58 and Lig4 (Ref. 59) have also been developed, but they have a more severe phenotype than that in human patients and die in utero57,59. This may reflect the retention of hypomorphic rather than null alleles in the human population. Mice deficient in Il2rg or Stat5 develop the same severe T-cell immunodeficiency phenotype as seen in humans and also show signs of immune dysregulation60,61,62,63. Rmrp-knockout mice have not been generated for comparison to the human phenotypes.
Using more sophisticated methods of genetic manipulation, including targeted gene alterations and missense mutations, mice with partial loss-of-function alleles of some of these genes have been generated. So far, 19 mouse strains have been generated that show some combination of partial T-cell immunodeficiency and immune dysregulation (Table 2). Of these strains, three have N-ethyl-N-nitrosourea (ENU)-induced mutations, three have spontaneous mutations, three have designed alleles, one is mediated by Cre-LoxP technology and nine have the genes knocked out.
Partial loss-of-function in somatic recombination genes. Whereas knockout mouse strains of Rag1, Rag2, Dclre1c and Lig4 do not reproduce the dual phenotype observed in some human patients, mouse strains with hypomorphic alleles of Rag1, Rag2 and Lig4 do. A fourth strain, the Tcra−/− mouse strain, may also fit into this category, as loss of TCRα results in a less severe T-cell immunodeficiency than loss of TCRβγ or RAG proteins and is also accompanied by autoimmune symptoms64.
An ENU-induced hypomorphic allele of Lig4 that generates an amino-acid substitution at position 288 (LIG4Y288C) does not affect viability of mice but results in severely reduced numbers of T cells and B cells65. LIG4Y288C mice spontaneously develop antinuclear antibodies at a high incidence (R. Cornall, personal communication), indicating that these mice also develop immune dysregulation, analogous to the autoimmune symptoms of patients with partial ligase IV deficiency. As yet, the mechanisms underlying this immune dysregulation are not clear.
Recently, two mouse models of Omenn syndrome have been developed with hypomorphic RAG alleles. The first used a knock-in allele of Rag2 that results in the RAG2R229Q variant, a mutation that is found in human patients with 'leaky' severe T-cell immunodeficiency and Omenn syndrome and that confers a 150-fold reduction in activity by this recombinase14. RAG2R229Q mice show a severe defect in T-cell development at the double negative 3 (DN3) stage and develop elevated levels of IgE and inflammatory disease66. The second model uses a spontaneous mutant of Rag1 that results in the RAG1R972Q protein variant that has only 12% of wild-type activity67. The corresponding human mutation, RAG1R975Q, has been observed in patients with Omenn syndrome68. Similar to RAG2R229Q mice, the RAG1R972Q mice are lymphopaenic and develop T-cell-dependent increased IgE production and inflammatory disease67.
The mechanism by which hypomorphic Rag alleles precipitate immune dysregulation is unclear, although there have been several interesting experimental results generated from studying these mice. In mice expressing the RAG1R972Q mutant, the activation of CD4+ T cells depends on the lymphopaenic environment, as wild-type CD4+ T cells transferred into RAG1R972Q mice exhibit the same activated phenotype67. In mice expressing the RAG2R229Q mutant, thymic expression of autoimmune regulator (AIRE) is reduced66, as is the case in patients with Omenn syndrome69, leading to the possibility that negative selection of developing thymocytes is dysfunctional70. RAG2R229Q-expressing mice also have a reduced percentage of forkhead box P3 (FOXP3)+ regulatory T cells in the thymus and spleen, and this could precipitate autoimmunity by allowing FOXP3− T cells to proliferate71. Which of these defects are causative of the immune dysregulation is as yet unknown.
Mouse models with cytokine signalling defects. Mouse models of Il7ra, Il2rg or Stat5b deficiency are all available. Although Il7ra- or Il7-knockout mice have severe T-cell immunodeficiency54,72 and Stat5b-knockout mice have T-cell defects without autoimmunity73,74, Il2rg-knockout mice and mice deficient in both STAT5A and STAT5B reproduce both immunodeficiency and immune dysregulation.
Stat5b-knockout mice show growth retardation, minor T-cell lymphopaenia and defects in T-cell function, but they have not been documented to develop autoimmunity73,74. However, mice lacking both Stat5a and Stat5b have a more severe phenotype75. They develop similar symptoms to human patients, with growth-hormone insensitivity, stunted growth and immunodeficiency. The mice also have a severe deficiency in total T-cell numbers and CD4+CD25+ regulatory T-cell numbers, and develop T-cell-dependent autoimmunity affecting the colon, liver and kidney63. As the transfer of wild-type CD4+ T cells prevents disease, the pathogenicity of Stat5a−/−Stat5b−/− T cells is not intrinsic to the effector T cells and is probably either a direct result of lymphopaenia or a defect in regulatory T-cell capacity63.
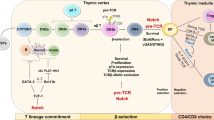
Mouse models with TCR signalling defects. ZAP70 deficiency blocks signalling through the TCR and typically results in severe T-cell immunodeficiency49,50,51. However, a case has been observed in which expression of the homologous kinase SYK (spleen tyrosine kinase) apparently compensated for the loss of ZAP70 and partially restored TCR signalling activity, resulting in the patient developing elevated levels of IgE35. A comprehensive range of mouse models are available for partial TCR signalling blockades, many of which combine partial immunodeficiency with elevated levels of IgE or autoimmunity. Double-knockout models can lead to intermediate effects by simultaneously removing a positive and negative modulator of TCR signalling, such as Lcp2 (lymphocyte cytosolic protein 2; also known as Slp76) and Cbl (Casitas B-lineage lymphoma)76 or Vav1 and Cblb77. Loss of a TCR signalling mediator, such as SH2D2A (SRC-homology-2-domain protein 2A; also known as TSAD) or RASGRP1 (RAS guanyl-releasing protein 1), also partially impairs TCR signalling78,79, as do designed mutations of the TCR adaptor protein LAT (linker for activation of T cells)80,81,82. The block in T-cell differentiation that occurs in mice expressing the LAT mutant (LATY136F) is accompanied by elevated IgE and IgG1 production and multi-organ infiltration of lymphocytes80,81. By contrast, mice expressing LATY7/8/9F (where tyrosines seven, eight and nine were mutated to phenylalanine) have a partial repertoire of γδ T cells with CD4+ αβ T-cell properties that drive elevated production of IgG1 and IgE82. Mice lacking both the calcium sensors Stim1 (stromal interaction molecule 1) and Stim2 have a severe impairment in generating sustained calcium flux83. Despite the resulting defect in NFAT (nuclear factor of activated T cells) translocation, these mice develop a lymphoproliferative syndrome83. Another example of autoimmunity in mice with partial defects in TCR signalling is the Zap70skg mouse strain, which has a spontaneous hypomorphic W163C mutation in ZAP70 and suffers from a partial T-cell differentiation block together with T-cell-dependent autoimmune arthritis84. So, there are multiple mouse models in which partial defects in TCR signalling recapitulate the human syndrome of both immunodeficiency and immune dysregulation.
The proposed mechanisms for the development of immune dysregulation in these TCR-signalling mutant mice include impaired negative selection, and thereby disrupted central tolerance, in the Zap70skg and LATY136F strains84,85, and a defect in FOXP3+ regulatory T-cell commitment or peripheral activation, and therefore defective peripheral tolerance, in the LATY136F strain86 and in strains deficient in both STIM1 and STIM2 (Ref. 83). Peripheral T cells in all of these mouse strains show a profound reduction in activation capacity83,84,85, yet this raises the question of why the decreased immunogenic signalling (the ability of effector T cells to become activated) does not cancel out the tolerogenic signalling defect (Fig. 2). If immunogenic signalling processes (such as stimulatory TCR signalling to activate effector T cells or inhibitory signalling in regulatory T cells) were controlled in the same way as tolerogenic signalling processes (stimulatory TCR signalling in regulatory T cells, or inhibitory TCR signalling resulting in anergy or deletion of effector T cells), any defect in TCR signalling should be neutral in terms of the immunogenic–tolerogenic balance. If the effects are not equal, there are two distinct mechanisms that could explain the phenomenon. The first is that tolerogenic and immunogenic TCR signalling have differential signalling requirements (such as their reliance on signalling subpathways), and the specific alleles that cause autoimmunity are separation-of-function alleles that impair tolerogenic signalling to a greater extent than immunogenic signalling, for example by slightly reducing effector T-cell activation but greatly impeding negative selection. The second is that the alleles are simple reduction-of-function alleles, but the balance between tolerance and immunity is based on complex interactions rather than on proportional interactions. In this scenario, even equal reductions in tolerogenic and immunogenic signalling can result in autoimmunity, if, for example, stimulatory TCR signalling was equally reduced in effector and regulatory T cells, but regulatory T cells lost function more rapidly than effector T cells.

The TCR signalling pathway leads to both immunogenic and tolerogenic outcomes. a | TCR signalling is required for thymocyte positive selection, effector T-cell lineage commitment and effector T-cell proliferation or activation. However, TCR signalling is also crucial for tolerogenic processes, such as the negative selection and anergy induction of autoreactive T cells and the selection and activation of regulatory T cells. The balance between these two processes results in immunological tolerance while still allowing immunogenic responses when necessary. b | Separation-of-function defects in TCR signalling may induce simultaneous immunodeficiency and autoimmunity by reducing immunogenic signalling pathways but having a greater impact on tolerogenic signalling pathways. c | Partial loss-of-function defects in TCR signalling may also induce simultaneous immunodeficiency and autoimmunity. In this case, signalling to immunogenic and tolerogenic pathways is equally reduced, but the outcome to each branch is nonlinear and tolerogenic mechanisms fail under a loss of signalling that still allows immunogenic mechanisms to operate at a reduced level. FOXP3, forkhead box P3; LAT, linker for activation of T cells; SLP76, SRC-homology-2-domain-containing leukocyte protein of 76 kDa; TReg cell, regulatory T cell; ZAP70, ζ-chain-associated protein kinase of 70 kDa.
In the case of the LATY136F mouse strain, the lack of FOXP3+ regulatory T cells is due to a separation-of-function change, probably because the Y136F mutation in LAT eliminates its ability to recruit phospholipase Cγ1 (PLCγ1)81. This causes a selective defect in the NFAT and Ca2+ signalling pathways but leaves the extracellular-signal-regulated kinase (ERK) signalling pathway intact81. The tolerogenic process of FOXP3 induction seems to be more reliant on the NFAT and Ca2+ signalling pathways than the immunogenic processes, as the observed defect in FOXP3+ regulatory T cells is greater in LATY136F mice than in ZAP70 hypomorphic mice, whereas both the LATY136F and ZAP70 hypomorphic mice have a similar reduction in positive selection87. It also seems that the separation-of-function change can act in an intrinsic manner to unbalance immunogenic and tolerogenic signalling in effector T cells, as LATY136F effector T cells still show immune dysregulation in the presence of wild-type FOXP3+ regulatory T cells and the absence of MHC class II interaction88. This mouse strain shows that, in principle, separation-of-function TCR signalling alleles can result in both immunodeficiency and immune dysregulation.
Two other mouse strains, Lcp2twp and Zap70mrd/mrt, demonstrate that pure reduction-of-function alleles are also able to promote autoimmunity. In the Lcp2twp strain, an ENU-induced mutation in a splice donor site reduces the amount of full-length mRNA encoding SLP76 (SH2-domain-containing leukocyte protein of 76 kDa) that can be produced (C. Goodnow and L. Miosge, personal communication). An intermediate level of T-cell immunodeficiency occurs and the mice develop enhanced IgE levels without any coding change in the TCR signalling machinery. In the Zap70mrd/mrt strain, two hypomorphic ENU-induced mutations are combined. The Zap70mrd mutation encodes an I367F amino-acid substitution and causes a minor reduction in ZAP70 activity, whereas the Zap70mrt mutation encodes a W504R amino-acid substitution that leaves only marginal ZAP70 activity. Neither parental strain develops hyper-production of IgE or autoantibodies (negating specific allele effects), yet the compound heterozygous mice have a level of immunodeficiency that is intermediate between that of the two homozygous strains and spontaneously produce autoantibodies87. These results indicate that it is the degree of immunodeficiency that is responsible for spontaneous autoimmunity.
Mouse models of unique monogenic disorders
As detailed in Table 1, two monogenic partial T-cell immunodeficiencies have been identified in which the causative gene has not been associated with a classical severe T-cell immunodeficiency phenotype — IL-2Rα deficiency and Wiskott–Aldrich syndrome. Knockout mice have been generated for both genes, but only Was−/− mice mimic the immunological aspects of the human condition.
Was-knockout mice develop both partial immunodeficiency and autoimmunity, including severe inflammation of the colon89,90. The loss of WAS protein (WASP) leads to severe defects in T-cell signalling due to the function of WASP in actin organization during the formation of the immunological synapse90,91,92. The reduction in TCR signalling capacity is linked to defects in the production93, homeostasis94 and function93,95,96 of FOXP3+ regulatory T cells, and this may contribute to the observed autoimmunity in patients with Wiskott–Aldrich syndrome. It is also probable that loss of WASP causes a B-cell-intrinsic defect, as Was−/− mice have reduced responses to T-cell-independent antigens and B-cell lines from patients with Wiskott–Aldrich syndrome show reduced chemotaxis towards CXC-chemokine ligand 13 (CXCL13). As the most frequently observed autoimmune diseases in WAS are autoantibody mediated, these B-cell defects may contribute to disease; however, autoantibody production may instead be secondary to the T-cell phenotype. Mice that lack WASP-interacting partner (Wip) develop a similar phenotype, with loss of T-cell proliferation and the generation of immune dysregulation97,98. For defects in both Was and Wip, a model for how the regulatory T-cell defect overcomes the defect in effector T cells has not yet been proposed.
IL-2Rα deficiency has only been observed in two patients, one with numerical T-cell-deficiency and extensive lymphocytic infiltrates43 and one with normal T-cell counts but frequent infections and IPEX-like autoimmunity44. Mice that lack Il2ra, Il2rb and Il2 have been generated, but although they all exhibit immunopathology, the mice are not immunodeficient99,100,101. The primary effect of loss of IL-2 responsiveness is a reduction in the thymic production and peripheral maintenance of FOXP3+ T cells, resulting in a loss of suppression of low-affinity, self-reactive T-cell clones102,103. Although IL-2 is a potent immune stimulator, Il2-knockout mice do not develop immunodeficiency. As yet, there are insufficient patients with IL2RA deficiency to determine whether loss of IL-2R signalling in humans generates primary immunodeficiency due to differences in the relative role of IL-2 in humans and mice, or whether the immunodeficiency observed in these patients is secondary, akin to IPEX.
Mechanistic models
Mouse models that result in intermediate TCR signalling or somatic recombination capacity indicate that the degree of T-cell immunodeficiency sets the conditions for uncontrolled T-cell activation and immune dysregulation. Mild or very severe immunodeficiency does not trigger immune dysregulation, whereas intermediate levels do. If both immunogenic and tolerogenic activities were equally impaired owing to immunodeficiency, this outcome would be paradoxical (Fig. 3a). However, these opposing activities do not need to respond equally, as tolerogenic mechanisms evolved in the context of homeostatic T-cell numbers with no evolutionary pressure for them to operate correctly at sub-homeostatic T-cell levels. The occurrence of hyperimmune activity only during an intermediate range of immunodeficiency can then be explained by distinct patterns of the response to various degrees of immunodeficiency. If the tolerogenic and immunogenic mechanisms respond with different kinetics, the balance that occurs during homeostasis can be lost only during partial immunodeficiency, resulting in the observed opposing clinical outcomes (Fig. 3b).

Competing immunogenic and tolerogenic processes alter the net ability of T cells to be activated (y axis). a | When both processes are proportionally modified by a reduction in T-cell receptor (TCR) signalling ability (x axis), an intuitive net effect occurs. b | When immunogenic and tolerogenic processes respond to TCR-signalling variants with unique kinetics, counter-intuitive net effects can occur.
Multiple and not mutually contradictory disconnections may be responsible for these effects. Immune dysregulation in Lcp2twp mice depends on the poor function of their FOXP3+ regulatory T cells, as immune dysregulation is suppressed on transfer of wild-type but not Foxp3−/− T cells (C. Goodnow and L. Miosge, personal communication), whereas immune dysregulation in Zap70mrd/mrt mice is intrinsic to effector T cells, suggesting that the immune defect is due to impaired negative selection87. There are several mechanisms by which these processes may require homeostatic conditions for attaining sufficient tolerogenic function. For TCR signalling mutants, both FOXP3+ regulatory T-cell commitment and negative selection have altered signalling requirements compared with effector T-cell activation86,104,105,106. This could allow a partial defect in TCR signalling capacity that causes a more potent impairment of both negative selection and induction of FOXP3 expression than will occur for positive selection and peripheral activation. These differential signalling requirements potentially give loss-of-function TCR signalling mutations a disproportionate effect on tolerogenic processes over immunogenic processes (Fig. 2).
Other complex interactions do not need to cause changes in signalling capacity and may be directly affected by the size of the T-cell population. For these population-dependent mechanisms, any mutation that reduces T-cell differentiation will impair the effectiveness of tolerogenic processes, whether the mutation affects TCR signalling, cytokine signalling or V(D)J recombination. For example, the effectiveness of regulatory T cells in carrying out their suppressive function may be disproportionately reduced by restrictions in the T-cell repertoire. FOXP3+ T cells require antigen for activation of their suppressor function107,108, so a broad repertoire of TCRs expressed by the FOXP3+ T-cell population is probably required for effective suppression of potential autoimmune effector T cells (Fig. 4a). Restriction of both the effector and regulatory T-cell populations to an oligoclonal repertoire would reduce the overlap between regulatory and effector T-cell specificities, facilitating the escape of autoreactive oligoclonal effector T cells (Fig. 4b). This principle has been demonstrated by the reconstitution of a lymphopaenic recipient mouse with different numbers of CD4+CD25+ regulatory T cells. Whereas the transfer of either low or high numbers of precursors resulted in the same absolute number of CD4+CD25+ regulatory T cells after lymphopaenic expansion, mice that received a low number of T-cell precursors were subsequently unable to control introduced naive CD4+ T cells and developed autoimmunity, allergy and elevated levels of serum IgE109,110. So, although the regulatory T-cell populations that arise in the recipient mice are genetically and numerically identical, when they originate from a diverse TCR repertoire pool they are protective, but when they arise from a limited TCR repertoire pool immune dysregulation can ensue.

Regulatory T-cell repertoire diversity, thymic negative selection and peripheral T-cell activation have the potential to act in population-dependent manners. a | Under homeostatic conditions, regulatory T cells control autoreactive T cells, through the expression of complementary T-cell receptor (TCR) repertoires specific for antigen sets. b | Under conditions of partial thymic immunodeficiency, a reduced number of successful thymocytes mature and enter the periphery. This results in oligoclonal expansion and hence 'holes' in the TCR repertoire for both regulatory T cells and autoreactive T cells, which by chance can create gaps in antigen-specific tolerance as indicated by asterisks (*), whereby the regulatory T-cell repertoire is limited but the autoreactive T-cell repertoire is present. c | Thymic crosstalk between single-positive (SP) thymocytes and medullary thymic epithelial cells (mTECs) via lymphotoxin, supports a well-structured thymic microarchitecture and efficient elimination of autoreactive T cells. d | Restriction of positive selection results in decreased production of SP thymocytes, reduced lymphotoxin levels and a disordered thymic microarchitecture. This may result in an increased number of autoreactive T cells escaping negative selection in the thymus. e | Under conditions of homeostasis, the limiting levels of interleukin-7 (IL-7) and co-stimulation signals from dendritic cells (through CD80 and CD86) increase the activation threshold for naive T cells, preventing naive autoreactive T cells from becoming activated. f | Under conditions of peripheral lymphopaenia, the reduced numbers of T cells allows each T cell increased access to the constant level of IL-7. Under lymphopaenic conditions, dendritic cells increase surface levels of CD80 and CD86. The combined effect of these two factors lowers the activation threshold for naive T cells. This may allow naive autoreactive T cells that would otherwise remain quiescent to become activated and pathogenic.
Another process that may depend on T-cell population size is the efficiency of negative selection in the thymus. In the analogous process of B-cell negative selection, the population size modulates the efficiency of apoptosis in response to antigen stimulation111. A similar interaction may occur during T-cell negative selection. Microarchitecture in the thymic medulla depends on the crosstalk between mature thymocytes and the medullary epithelium through the lymphotoxin pathway112,113. Lymphotoxin is also required for the expression of chemokines to guide thymocyte migration, and the ability of epithelial cells to respond to lymphotoxin increases the efficiency of negative selection114 (Fig. 4c). Early studies suggested that lymphotoxin promotes AIRE expression115, however recent analyses have suggested that this effect is secondary to the role of lymphotoxin in the development of thymic epithelial cells112 and that the key inducer for AIRE expression is RANKL (receptor activator of nuclear factor-κB ligand)116. RANKL is expressed by lymphoid-tissue inducer cells rather than thymocytes, making it a population-insensitive pathway116. Under conditions in which the number of mature thymocytes is restricted (such as during partial T-cell immunodeficiency), lower amounts of lymphotoxin are available to epithelial cells, reducing the effectiveness of thymocyte–epithelial-cell communication, and the thymic microarchitecture becomes disordered113. This is likely to make efficiency of negative selection a population-sensitive process, as the quantity of T cells, above and beyond the properties of the individual cells, alters the efficiency of negative selection (Fig. 4d). Previous studies have shown that even slight changes to the thymic expression of peripheral antigens can reduce the effectiveness of negative selection117, which demonstrates that even subtle defects in negative selection can have pathogenic consequences.
Both of the mechanisms above are sensitive to numerical T-cell defects in the thymus, regardless of whether the size of the T-cell pool is normalized in the periphery. Other mechanisms are likely to be sensitive to the size of the peripheral T-cell pool. The activation of effector T cells, for example, has been shown to occur with greater ease and towards lower affinity antigens under conditions of lymphopaenia118,119. Several different factors may contribute to this reduced threshold for activation. One is an increased availability of serum IL-7, with fewer T cells competing for a constant resource120. Another factor is the expression of co-stimulatory factors by dendritic cells, which respond to T-cell lymphopaenia by increasing their expression of CD80 and CD86, among other factors, and this lowers the activation threshold and potentially allows autoreactive T cells to proliferate121. Peripheral lymphopaenia not only increases the capacity of T cells to expand in response to low-affinity ligands (such as positively selecting self ligands), but it also reduces the efficiency of peripheral deletion mechanisms in response to self antigens122. All of these mechanisms, and probably additional ones, act in a population-dependent manner, in that the efficacy is reduced simply by virtue of the reduced population size, rather than by changes at the cellular level.
Concluding remarks
A survey of the published clinical literature of the partial T-cell immunodeficiencies highlights the surprising frequency with which these conditions are accompanied by immune dysregulation in the form of elevated production of IgE, inflammatory disease or autoimmunity. For at least a subset of these conditions, it is likely that the immune dysregulation component is a direct consequence of the partial nature of the immunodeficiency.
The growing number of 'designer' alleles and point mutations has allowed the generation of multiple mouse strains with immunodeficiency in the intermediate range. These mice develop a range of immune dysregulation symptoms due to defects in negative selection and regulatory T-cell function under conditions of partial immunodeficiency. The use of a titration of Zap70 hypomorphic alleles indicates that immune dysregulation can be a spontaneous condition that is dependent on the level of immunodeficiency rather than on the consequences of particular alleles. The degree to which heterozygous polymorphisms in different genes can synergistically produce a similar effect remains an open question. Taken together, the mouse models indicate that a single common mechanism — reduced efficiency of tolerance mechanisms at suboptimal functional T-cell population sizes — can be used to explain the development of immune dysregulation as a consequence of partial T-cell immunodeficiency. This opens up the study of which tolerance mechanisms depend on the size of the functional population, and to what degree they are sensitive to changes in functional T-cell population density. An understanding of the population sensors involved may allow manipulation of the effectiveness of these tolerance mechanisms, and hence susceptibility to immune dysregulation.
The translation of observations made in the mouse model to human disease presents a major challenge for the future. However, if replicated, these findings open up several interesting possibilities. First, they suggest a mechanism for this close clinical association of apparently opposite conditions. Second, they suggest that non-familial cases of partial immunodeficiency and immune dysregulation may result from the inheritance of particular combinations of hypomorphic alleles that act synergistically to lower T-cell numbers or function to the point at which immune dysregulation occurs spontaneously. In this regard, the increase in large-scale sequencing will soon allow us to sequence all or most of the genes involved in T-cell development and signalling in humans. Third, these findings suggest potential new avenues for investigation of common autoimmune and allergic diseases. Can minor defects in the T-cell population act in a synergistic manner with disease-specific polymorphisms to increase susceptibility to disease? The contribution of peripheral lymphopaenia to autoimmune diabetes that occurs spontaneously in the BB rat model indicates that it is at least plausible123. The association of polymorphisms in PTPN22 (protein tyrosine phosphatase, non-receptor type 22)124,125 and SH2D2A126, which are known to lower TCR signalling capacity, with common autoimmune diseases extends the potential for this contribution to human patients. Whether these alleles function by inducing a mild degree of T-cell deficiency awaits direct testing. The direct measurement of minor degrees of T-cell deficiency in autoimmunity is made difficult, as thymic tissue is generally unavailable for direct detection of defects in T-cell differentiation, and peripheral inflammation and compensation mechanisms can mask the initiating defect. A sensitive and reliable surrogate measure to test for mild immunodeficiency in common autoimmune diseases could be the measurement of TCR repertoire diversity in the peripheral blood.
An even more striking example of translation to the polygenic scenario in humans is the association of IL7RA polymorphisms with multiple sclerosis and type 1 diabetes127,128. As described above, severe loss-of-function alleles of IL7RA cause severe T-cell deficiency20,21, whereas moderate loss-of-function alleles can cause Omenn syndrome10. The IL7RA allele associated with common autoimmunity alters the splicing efficiency of the mRNA and results in a twofold upregulation of IL7RA transcripts lacking exon 6 (Ref. 127). This exon encodes the transmembrane domain, so the IL7RA polymorphism results in an increase in a soluble form of IL-7Rα, which probably reduces the availability of serum IL-7. As this allele is insufficient to induce autoimmunity alone, but acts as a susceptibility allele for several common autoimmune diseases, it provides a direct connection between the rare monogenic conditions discussed in this Review and more prevalent polygenic autoimmune diseases.
Note added in proof
In a recent publication by Vignali and colleagues, the effect of intermediate levels of TCR signalling capacity was tested by generating mice with different numbers of immunoreceptor tyrosine-based activation motifs (ITAMs) in the TCR–CD3 complex, ranging from the normal complement of 10 to the null signal level of 0 (Ref. 140). Conforming to the models described in this Review, they found that minor loss of signalling capacity (9–7 ITAMs) was compensated for, extreme loss of signalling (0–1 ITAMs) resulted in profound immunodeficiency, and an intermediate level of signalling capacity (6–2 ITAMs) resulted in the dual presentation of partial immunodeficiency and autoimmunity140.
References
Gosling, K. M. et al. A mutation in a chromosome condensin II subunit, kleisinβ, specifically disrupts T cell development. Proc. Natl Acad. Sci. USA 104, 12445–12450 (2007). This study describes the sensitivity of thymocytes to global impairments in basic cellular processes, with a partial block at the double-negative stage of thymocyte development in mice with a point mutation in the ubiquitious chromosome component kleisin-β.
Anderson, S. J. et al. Ablation of ribosomal protein L22 selectively impairs αβT cell development by activation of a p53-dependent checkpoint. Immunity 26, 759–772 (2007).
Deutschbauer, A. M. et al. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics 169, 1915–1925 (2005).
Giaever, G. et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 (2002).
Fischer, A. et al. Naturally occurring primary deficiencies of the immune system. Annu. Rev. Immunol. 15, 93–124 (1997).
Villa, A. et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell 93, 885–896 (1998). A seminal paper characterizing the mechanism of Omenn syndrome, revealed as an incomplete reduction of V(D)J recombination.
Ege, M. et al. Omenn syndrome due to ARTEMIS mutations. Blood 105, 4179–4186 (2005).
Gennery, A. R. et al. Omenn's syndrome occurring in patients without mutations in recombination activating genes. Clin. Immunol. 116, 246–256 (2005).
Roifman, C. M., Gu, Y. & Cohen, A. Mutations in the RNA component of RNase mitochondrial RNA processing might cause Omenn syndrome. J. Allergy Clin. Immunol. 117, 897–903 (2006).
Giliani, S. et al. Omenn syndrome in an infant with IL7RA gene mutation. J. Pediatr. 148, 272–274 (2006).
Shibata, F. et al. Skin infiltration of CD56bright CD16− natural killer cells in a case of X-SCID with Omenn syndrome-like manifestations. Eur. J. Haematol. 79, 81–85 (2007).
Santagata, S. et al. N-terminal RAG1 frameshift mutations in Omenn's syndrome: internal methionine usage leads to partial V(D)J recombination activity and reveals a fundamental role in vivo for the N-terminal domains. Proc. Natl Acad. Sci. USA 97, 14572–14577 (2000).
de Saint-Basile, G. et al. Restricted heterogeneity of T lymphocytes in combined immunodeficiency with hypereosinophilia (Omenn's syndrome). J. Clin. Invest. 87, 1352–1359 (1991).
Corneo, B. et al. Identical mutations in RAG1 or RAG2 genes leading to defective V(D)J recombinase activity can cause either T-B-severe combined immune deficiency or Omenn syndrome. Blood 97, 2772–2776 (2001). The remarkable observation that identical alleles of RAG1 or RAG2 can give rise to the profoundly different conditions of SCID (severe combined immunodeficient) or Omenn syndrome, highlighting the contribution of unknown genetic and/or environmental influences.
Ehl, S. et al. A variant of SCID with specific immune responses and predominance of γδ T cells. J. Clin. Invest. 115, 3140–3148 (2005).
de Villartay, J. P. et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J. Clin. Invest. 115, 3291–3299 (2005).
van der Burg, M. et al. A new type of radiosensitive T−B−NK+ severe combined immunodeficiency caused by a LIG4 mutation. J. Clin. Invest. 116, 137–145 (2006).
Enders, A. et al. A severe form of human combined immunodeficiency due to mutations in DNA ligase IV. J. Immunol. 176, 5060–5068 (2006).
O'Driscoll, M. et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell 8, 1175–1185 (2001).
Puel, A., Ziegler, S. F., Buckley, R. H. & Leonard, W. J. Defective IL7R expression in T−B+NK+ severe combined immunodeficiency. Nature Genet. 20, 394–397 (1998).
Roifman, C. M., Zhang, J., Chitayat, D. & Sharfe, N. A partial deficiency of interleukin-7Rα is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Blood 96, 2803–2807 (2000).
Puck, J. M. et al. The interleukin-2 receptor γ chain maps to Xq13.1 and is mutated in X-linked severe combined immunodeficiency, SCIDX1. Hum. Mol. Genet. 2, 1099–1104 (1993).
Noguchi, M. et al. Interleukin-2 receptor γ chain: a functional component of the interleukin-7 receptor. Science 262, 1877–1880 (1993).
Jones, A. M. et al. B-cell-negative severe combined immunodeficiency associated with a common γ chain mutation. Hum. Genet. 99, 677–680 (1997).
Kofoed, E. M. et al. Growth hormone insensitivity associated with a STAT5b mutation. N. Engl. J. Med. 349, 1139–1147 (2003).
Hwa, V. et al. Growth hormone insensitivity and severe short stature in siblings: a novel mutation at the exon 13-intron 13 junction of the STAT5b gene. Horm. Res. 68, 218–224 (2007).
Makitie, O., Kaitila, I. & Savilahti, E. Susceptibility to infections and in vitro immune functions in cartilage-hair hypoplasia. Eur. J. Pediatr. 157, 816–820 (1998).
Barth, R. F., Vergara, G. G., Khurana, S. K., Lowman, J. T. & Beckwith, J. B. Rapidly fatal familial histiocytosis associated with eosinophilia and primary immunological deficiency. Lancet 2, 503–506 (1972).
Berthet, F. et al. Bone marrow transplantation in cartilage-hair hypoplasia: correction of the immunodeficiency but not of the chondrodysplasia. Eur. J. Pediatr. 155, 286–290 (1996).
Kuijpers, T. W. et al. Short-limbed dwarfism with bowing, combined immune deficiency, and late onset aplastic anaemia caused by novel mutations in the RMPR gene. J. Med. Genet. 40, 761–766 (2003).
Hirschhorn, R. & Candott, F. in Primary Immunodeficiency diseases, 2nd Edn (eds H. D. Ochs, C. I. E. Smith & J. Puck) 169–196 (Oxford Univ. Press, 2007).
Shovlin, C. L. et al. Adult presentation of adenosine deaminase deficiency. Lancet 341, 1471 (1993).
Hirschhorn, R., Yang, D. R., Israni, A., Huie, M. L. & Ownby, D. R. Somatic mosaicism for a newly identified splice-site mutation in a patient with adenosine deaminase-deficient immunodeficiency and spontaneous clinical recovery. Am. J. Hum. Genet. 55, 59–68 (1994).
Hirschhorn, R. et al. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nature Genet. 13, 290–295 (1996).
Toyabe, S., Watanabe, A., Harada, W., Karasawa, T. & Uchiyama, M. Specific immunoglobulin E responses in ZAP70-deficient patients are mediated by Syk-dependent T-cell receptor signalling. Immunology 103, 164–171 (2001).
Sullivan, K. E., Mullen, C. A., Blaese, R. M. & Winkelstein, J. A. A multiinstitutional survey of the Wiskott–Aldrich syndrome. J. Pediatr. 125, 876–885 (1994).
Schurman, S. H. & Candotti, F. Autoimmunity in Wiskott–Aldrich syndrome. Curr. Opin. Rheumatol. 15, 446–453 (2003).
Derry, J. M., Ochs, H. D. & Francke, U. Isolation of a novel gene mutated in Wiskott–Aldrich syndrome. Cell 78, 635–644 (1994). This study described the first causative mutation of a clinical immunodeficiency with immune dysregulation.
Imai, K. et al. Clinical course of patients with WASP gene mutations. Blood 103, 456–464 (2004).
Villa, A. et al. X-linked thrombocytopenia and Wiskott–Aldrich syndrome are allelic diseases with mutations in the WASP gene. Nature Genet. 9, 414–417 (1995).
Vella, A. et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 76, 773–779 (2005).
Brand, O. J. et al. Association of the interleukin-2 receptor α (IL-2Rα)/CD25 gene region with Graves' disease using a multilocus test and tag SNPs. Clin. Endocrinol. 66, 508–512 (2007).
Sharfe, N., Dadi, H. K., Shahar, M. & Roifman, C. M. Human immune disorder arising from mutation of the α chain of the interleukin-2 receptor. Proc. Natl Acad. Sci. USA 94, 3168–3171 (1997).
Caudy, A. A., Reddy, S. T., Chatila, T., Atkinson, J. P. & Verbsky, J. W. CD25 deficiency causes an immune dysregulation, polyendocrinopathy, enteropathy, X-linked-like syndrome, and defective IL-10 expression from CD4 lymphocytes. J. Allergy Clin. Immunol. 119, 482–487 (2007).
Mombaerts, P. et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell 68, 869–877 (1992).
Spanopoulou, E. et al. Functional immunoglobulin transgenes guide ordered B-cell differentiation in Rag-1-deficient mice. Genes Dev. 8, 1030–1042 (1994).
Hao, Z. & Rajewsky, K. Homeostasis of peripheral B cells in the absence of B cell influx from the bone marrow. J. Exp. Med. 194, 1151–1164 (2001).
Shinkai, Y. et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68, 855–867 (1992).
Arpaia, E., Shahar, M., Dadi, H., Cohen, A. & Roifman, C. M. Defective T cell receptor signaling and CD8+ thymic selection in humans lacking ZAP-70 kinase. Cell 76, 947–958 (1994).
Elder, M. E. et al. Human severe combined immunodeficiency due to a defect in ZAP-70, a T cell tyrosine kinase. Science 264, 1596–1599 (1994).
Chan, A. C. et al. ZAP-70 deficiency in an autosomal recessive form of severe combined immunodeficiency. Science 264, 1599–1601 (1994).
Wiest, D. L. et al. A spontaneously arising mutation in the DLAARN motif of murine ZAP-70 abrogates kinase activity and arrests thymocyte development. Immunity 6, 663–671 (1997).
Negishi, I. et al. Essential role for ZAP-70 in both positive and negative selection of thymocytes. Nature 376, 435–438 (1995).
Peschon, J. J. et al. Early lymphocyte expansion is severely impaired in interleukin 7 receptor-deficient mice. J. Exp. Med. 180, 1955–1960 (1994).
Li, L. et al. Targeted disruption of the Artemis murine counterpart results in SCID and defective V(D)J recombination that is partially corrected with bone marrow transplantation. J. Immunol. 174, 2420–2428 (2005).
Rooney, S. et al. Leaky Scid phenotype associated with defective V(D)J coding end processing in Artemis-deficient mice. Mol. Cell 10, 1379–1390 (2002).
Wakamiya, M. et al. Disruption of the adenosine deaminase gene causes hepatocellular impairment and perinatal lethality in mice. Proc. Natl Acad. Sci. USA 92, 3673–3677 (1995).
Migchielsen, A. A. et al. Adenosine-deaminase-deficient mice die perinatally and exhibit liver-cell degeneration, atelectasis and small intestinal cell death. Nature Genet. 10, 279–287 (1995).
Frank, K. M. et al. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 396, 173–177 (1998).
Cao, X. et al. Defective lymphoid development in mice lacking expression of the common cytokine receptor γ chain. Immunity 2, 223–238 (1995).
DiSanto, J. P., Muller, W., Guy-Grand, D., Fischer, A. & Rajewsky, K. Lymphoid development in mice with a targeted deletion of the interleukin 2 receptor γ chain. Proc. Natl Acad. Sci. USA 92, 377–381 (1995).
Ohbo, K. et al. Modulation of hematopoiesis in mice with a truncated mutant of the interleukin-2 receptor γ chain. Blood 87, 956–967 (1996).
Snow, J. W. et al. Loss of tolerance and autoimmunity affecting multiple organs in STAT5A/5B-deficient mice. J. Immunol. 171, 5042–5050 (2003).
Wen, L. et al. Immunoglobulin synthesis and generalized autoimmunity in mice congenitally deficient in αβ+T cells. Nature 369, 654–658 (1994).
Nijnik, A. et al. DNA repair is limiting for haematopoietic stem cells during ageing. Nature 447, 686–690 (2007).
Marrella, V. et al. A hypomorphic R229Q Rag2 mouse mutant recapitulates human Omenn syndrome. J. Clin. Invest. 117, 1260–1269 (2007).
Khiong, K. et al. Homeostatically proliferating CD4 T cells are involved in the pathogenesis of an Omenn syndrome murine model. J. Clin. Invest. 117, 1270–1281 (2007). In references 66 and 67, mouse models of Omenn syndrome were generated through limited activity of RAG1 and RAG2. Both strains developed partial T-cell immunodeficiency and spontaneous immune dysregulation.
Villa, A. et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood 97, 81–88 (2001).
Cavadini, P. et al. AIRE deficiency in thymus of 2 patients with Omenn syndrome. J. Clin. Invest. 115, 728–732 (2005).
Liston, A., Lesage, S., Wilson, J., Peltonen, L. & Goodnow, C. C. Aire regulates negative selection of organ-specific T cells. Nature Immunol. 4, 350–354 (2003).
Kim, J. M., Rasmussen, J. P. & Rudensky, A. Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nature Immunol. 8, 191–197 (2007).
von Freeden-Jeffry, U. et al. Lymphopenia in interleukin (IL)-7 gene-deleted mice identifies IL-7 as a nonredundant cytokine. J. Exp. Med. 181, 1519–1526 (1995).
Yao, Z. et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc. Natl Acad. Sci. USA 103, 1000–1005 (2006).
Imada, K. et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J. Exp. Med. 188, 2067–2074 (1998).
Teglund, S. et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 93, 841–850 (1998).
Chiang, Y. J. et al. Inactivation of c-Cbl reverses neonatal lethality and T cell developmental arrest of SLP-76-deficient mice. J. Exp. Med. 200, 25–34 (2004).
Krawczyk, C. et al. Cbl-b is a negative regulator of receptor clustering and raft aggregation in T cells. Immunity 13, 463–473 (2000).
Layer, K. et al. Autoimmunity as the consequence of a spontaneous mutation in Rasgrp1. Immunity 19, 243–255 (2003).
Drappa, J. et al. Impaired T cell death and lupus-like autoimmunity in T cell-specific adapter protein-deficient mice. J. Exp. Med. 198, 809–821 (2003).
Aguado, E. et al. Induction of T helper type 2 immunity by a point mutation in the LAT adaptor. Science 296, 2036–2040 (2002).
Sommers, C. L. et al. A LAT mutation that inhibits T cell development yet induces lymphoproliferation. Science 296, 2040–2043 (2002). References 80 and 81 independently describe the LATY136F knock-in mouse, the first demonstration of a recurrent theme that mutations in positive regulators of TCR signalling can cause immune dysregulation.
Nuñez-Cruz, S. et al. LAT regulates γδ T cell homeostasis and differentiation. Nature Immunol. 4, 999–1008 (2003).
Oh-Hora, M. et al. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nature Immunol. 9, 432–443 (2008). This study shows that deletion of STIM1 and STIM2 in T cells dramatically reduced their capacity for Ca2+ flux and NFAT signalling. Despite the defect in effector T cells, the mice developed lymphoproliferation, which could be corrected by the transfer of wild-type regulatory T cells.
Sakaguchi, N. et al. Altered thymic T-cell selection due to a mutation of the ZAP-70 gene causes autoimmune arthritis in mice. Nature 426, 454–460 (2003). In this study, the skg mouse was characterized, showing a hypomorphic W163C mutation in Zap70 and the spontaneous development of autoimmune arthritis.
Sommers, C. L. et al. Mutation of the phospholipase C-γ1-binding site of LAT affects both positive and negative thymocyte selection. J. Exp. Med. 201, 1125–1134 (2005).
Koonpaew, S., Shen, S., Flowers, L. & Zhang, W. LAT-mediated signaling in CD4+CD25+ regulatory T cell development. J. Exp. Med. 203, 119–129 (2006).
Siggs, O. M. et al. Opposing function of the T cell receptor kinase ZAP-70 in immunity and tolerance differentially titrate in response to nucleotide substitutions. Immunity 27, 912–926 (2007). This study used ENU-induced Zap70 mutants to show that simple partial loss-of-function of ZAP70 can result in immune dysregulation when ZAP70 activity decreases to within a critical range.
Wang, Y. et al. Th2 lymphoproliferative disorder of LatY136F mutant mice unfolds independently of TCR-MHC engagement and is insensitive to the action of Foxp3+ regulatory T cells. J. Immunol. 180, 1565–1575 (2008).
Snapper, S. B. et al. Wiskott–Aldrich syndrome protein-deficient mice reveal a role for WASP in T but not B cell activation. Immunity 9, 81–91 (1998).
Zhang, J. et al. Antigen receptor-induced activation and cytoskeletal rearrangement are impaired in Wiskott–Aldrich syndrome protein-deficient lymphocytes. J. Exp. Med. 190, 1329–1342 (1999).
Silvin, C., Belisle, B. & Abo, A. A role for Wiskott–Aldrich syndrome protein in T-cell receptor-mediated transcriptional activation independent of actin polymerization. J. Biol. Chem. 276, 21450–21457 (2001).
Sims, T. N. et al. Opposing effects of PKCθ and WASp on symmetry breaking and relocation of the immunological synapse. Cell 129, 773–785 (2007).
Maillard, M. H. et al. The Wiskott–Aldrich syndrome protein is required for the function of CD4+CD25+Foxp3+ regulatory T cells. J. Exp. Med. 204, 381–391 (2007).
Humblet-Baron, S. et al. Wiskott–Aldrich syndrome protein is required for regulatory T cell homeostasis. J. Clin. Invest. 117, 407–418 (2007).
Marangoni, F. et al. WASP regulates suppressor activity of human and murine CD4+CD25+FOXP3+ natural regulatory T cells. J. Exp. Med. 204, 369–380 (2007).
Adriani, M. et al. Impaired in vitro regulatory T cell function associated with Wiskott–Aldrich syndrome. Clin. Immunol. 124, 41–48 (2007).
Anton, I. M. et al. WIP deficiency reveals a differential role for WIP and the actin cytoskeleton in T and B cell activation. Immunity 16, 193–204 (2002).
Curcio, C. et al. WIP null mice display a progressive immunological disorder that resembles Wiskott–Aldrich syndrome. J. Pathol. 211, 67–75 (2007).
Sadlack, B. et al. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell 75, 253–261 (1993).
Suzuki, H. et al. Deregulated T cell activation and autoimmunity in mice lacking interleukin-2 receptor β. Science 268, 1472–1476 (1995).
Willerford, D. M. et al. Interleukin-2 receptor α chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3, 521–530 (1995).
Fontenot, J. D., Rasmussen, J. P., Gavin, M. A. & Rudensky, A. Y. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nature Immunol. 6, 1142–1151 (2005).
Liston, A., Siggs, O. M. & Goodnow, C. C. Tracing the action of IL-2 in tolerance to islet-specific antigen. Immunol. Cell Biol. 85, 338–342 (2007).
Neilson, J. R., Winslow, M. M., Hur, E. M. & Crabtree, G. R. Calcineurin B1 is essential for positive but not negative selection during thymocyte development. Immunity 20, 255–266 (2004).
Gong, Q. et al. Disruption of T cell signaling networks and development by Grb2 haploid insufficiency. Nature Immunol. 2, 29–36 (2001).
McCarty, N. et al. Signaling by the kinase MINK is essential in the negative selection of autoreactive thymocytes. Nature Immunol. 6, 65–72 (2005).
Thornton, A. M. & Shevach, E. M. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J. Immunol. 164, 183–190 (2000).
Takahashi, T. et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 10, 1969–1980 (1998).
Milner, J. D., Ward, J. M., Keane-Myers, A. & Paul, W. E. Lymphopenic mice reconstituted with limited repertoire T cells develop severe, multiorgan, Th2-associated inflammatory disease. Proc. Natl Acad. Sci. USA 104, 576–581 (2007). This study looked at the efficiency of a limited repertoire of regulatory T cells to prevent autoimmunity. They found that a numerically-equal population of regulatory T cells expanded from a limited number of precursor cells has a reduced capacity for systemic T-cell regulation.
Milner, J., Ward, J., Keane-Myers, A., Min., B. & Paul, W. E. Repertoire-dependent immunopathology. J. Autoimmun. 29, 257–261 (2007).
Lesley, R. et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity 20, 441–453 (2004). This study showed that the deletion of autoreactive B cells varies in efficiency with population size, and that the molecular mechanism was due to competition for the B-cell survival factor BAFF (B-cell activating factor).
Venanzi, E. S., Gray, D. H., Benoist, C. & Mathis, D. Lymphotoxin pathway and Aire influences on thymic medullary epithelial cells are unconnected. J. Immunol. 179, 5693–5700 (2007).
Gray, D. H. et al. Controlling the thymic microenvironment. Curr. Opin. Immunol. 17, 137–143 (2005).
Zhu, M., Chin, R. K., Tumanov, A. V., Liu, X. & Fu, Y. X. Lymphotoxin receptor is required for the migration and selection of autoreactive T cells in thymic medulla. J. Immunol. 179, 8069–8075 (2007). This paper showed that lymphotoxin-dependent crosstalk between thymocytes and thymic epithelial cells is required for efficient deletion of autoreactive T cells. The effect was not mediated by defects in the AIRE-dependent expression pathway, but rather due to alterations in the thymic microarchitecture.
Chin, R. K. et al. Lymphotoxin pathway directs thymic Aire expression. Nature Immunol. 4, 1121–1127 (2003).
Rossi, S. W. et al. RANK signals from CD4+3− inducer cells regulate development of Aire-expressing epithelial cells in the thymic medulla. J. Exp. Med. 204, 1267–1272 (2007).
Liston, A. et al. Gene dosage limiting role of Aire in thymic expression, clonal deletion and organ-specific autoimmunity. J. Exp. Med. 200, 1015–1026 (2004).
Kieper, W. C. & Jameson, S. C. Homeostatic expansion and phenotypic conversion of naive T cells in response to self peptide/MHC ligands. Proc. Natl Acad. Sci. USA 96, 13306–13311 (1999).
Goldrath, A. W. & Bevan, M. J. Low-affinity ligands for the TCR drive proliferation of mature CD8+ T cells in lymphopenic hosts. Immunity 11, 183–190 (1999).
Schluns, K. S., Kieper, W. C., Jameson, S. C. & Lefrancois, L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nature Immunol. 1, 426–432 (2000).
Fazekas de St. Groth, B. DCs and peripheral T cell tolerance. Semin. Immunol. 13, 311–322 (2001).
Shklovskaya, E. & Fazekas de St. Groth, B. Severely impaired clonal deletion of CD4+ T cells in low-dose irradiated mice: role of T cell antigen receptor and IL-7 receptor signals. J. Immunol. 177, 8320–8330 (2006). This study found that when TCR-transgenic cells are transferred into lymphopaenic mice, they show a reduced efficiency in clonal deletion and increased efficiency of clonal expansion. Importantly, the TCR transgene itself does not support spontaneous lymphopaenia-induced clonal expansion.
Ramanathan, S. & Poussier, P. BB rat lyp mutation and type 1 diabetes. Immunol. Rev. 184, 161–171 (2001).
Vang, T. et al. Autoimmune-associated lymphoid tyrosine phosphatase is a gain-of-function variant. Nature Genet. 37, 1317–1319 (2005).
Bottini, N. et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nature Genet. 36, 337–338 (2004). References 124 and 125 describe the initial association of the R620W gain-of-function variant of PTPN22 with type 1 diabetes, implying that reductions in T-cell activation can contribute to autoimmune disease.
Smerdel, A. et al. Genetic association between juvenile rheumatoid arthritis and polymorphism in the SH2D2A gene. Genes Immun. 5, 310–312 (2004).
Gregory, S. G. et al. Interleukin 7 receptor α chain (IL7R) shows allelic and functional association with multiple sclerosis. Nature Genet. 39, 1083–1091 (2007). In this gene-association study, polymorphisms in the IL7RA gene are found to be associated with risk of developing multiple sclerosis. The susceptibility allele of IL7RA has altered splicing efficiency and increased production of a mRNA transcript encoding soluble IL-7R α.
Todd, J. A. et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nature Genet. 39, 857–864 (2007).
Santagata, S., Villa, A., Sobacchi, C., Cortes, P. & Vezzoni, P. The genetic and biochemical basis of Omenn syndrome. Immunol. Rev. 178, 64–74 (2000).
Aleman, K., Noordzij, J. G., de Groot, R., van Dongen, J. J. & Hartwig, N. G. Reviewing Omenn syndrome. Eur. J. Pediatr. 160, 718–725 (2001).
Dupuis-Girod, S. et al. Autoimmunity in Wiskott–Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics 111, e622–e627 (2003).
Kolluri, R. et al. Identification of WASP mutations in patients with Wiskott–Aldrich syndrome and isolated thrombocytopenia reveals allelic heterogeneity at the WAS locus. Hum. Mol. Genet. 4, 1119–1126 (1995).
Schopfer, K. et al. Systemic lupus erythematosus in Staphylococcus aureus hyperimmunoglobulinaemia E syndrome. Br. Med. J. (Clin. Res. Ed) 287, 524–526 (1983).
Renner, E. D. et al. Autosomal recessive hyperimmunoglobulin E syndrome: a distinct disease entity. J. Pediatr. 144, 93–99 (2004).
Cunningham-Rundles, C. & Bodian, C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin. Immunol. 92, 34–48 (1999).
Sullivan, K. E. et al. Juvenile rheumatoid arthritis-like polyarthritis in chromosome 22q11.2 deletion syndrome (DiGeorge anomalad/velocardiofacial syndrome/conotruncal anomaly face syndrome). Arthritis Rheum. 40, 430–436 (1997).
Jawad, A. F., McDonald-Mcginn, D. M., Zackai, E. & Sullivan, K. E. Immunologic features of chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). J. Pediatr. 139, 715–723 (2001).
Chinen, J., Rosenblatt, H. M., Smith, E. O., Shearer, W. T. & Noroski, L. M. Long-term assessment of T-cell populations in DiGeorge syndrome. J. Allergy Clin. Immunol. 111, 573–579 (2003).
Roifman, C. M. & Melamed, I. A novel syndrome of combined immunodeficiency, autoimmunity and spondylometaphyseal dysplasia. Clin. Genet. 63, 522–529 (2003).
Holst, J. et al. Scalable signaling mediated by T cell antigen receptor–CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nature Immunol. 9, 658–666 (2008).
Acknowledgements
We thank C. Goodnow and K. Randal for stimulating discussions, and R. Cornall, L. Miosge and C. Goodnow for sharing unpublished data. This work is supported by the CJ Martin and RG Menzies Foundations (A.L.) and the Deutsche Forschungsgemeinschaft (A.E.).
Author information
Authors and Affiliations
Corresponding author
Supplementary information
Supplementary information
Human genetic variants responsible for severe T-cell immunodeficiency (PDF 313 kb)
Related links
Glossary
- Immunodeficiency
-
A state in which the capacity of the immune system to respond to infectious disease is compromised or completely absent.
- Positive selection
-
One step in the process of T-cell differentiation in the thymus. T cells that express T-cell receptors with moderate to high affinity for self antigens receive a survival signal and continue to develop towards becoming double positive (CD4+CD8+) T cells. Positive selection occurs through antigens presented by resident stromal cells and dendritic cells in the thymic cortex and is followed by negative selection.
- V(D)J recombination
-
Somatic rearrangement of variable (V), diversity (D) and joining (J) regions of the genes that encode antigen receptors, leading to repertoire diversity of both T-cell and B-cell receptors.
- APECED
-
(Autoimmune polyendocrinopathy-candidiasis-ectodermal-dystrophy syndrome). A rare human autoimmune disorder that is inherited in an autosomal recessive manner and is characterized by various endocrine deficiencies, chronic mucocutaneous candidiasis and ectodermal dystrophies. It is caused by several different mutations in the gene that encodes autoimmune regulator (AIRE).
- IPEX
-
(Immunodysregulation, polyendocrinopathy and enteropathy, X-linked syndrome). A disease caused by mutations in FOXP3 (forkhead box P3) and characterized by refractory enteritis and, in some patients, autoimmune endocrinopathies, autoimmune diabetes and thyroiditis.
- Graft-versus-host disease
-
Tissue damage in a recipient of allogeneic tissue (usually a bone-marrow transplant) that results from the activity of donor cytotoxic T cells recognizing the tissues of the recipient as foreign. This disease varies markedly in extent, but it can be life threatening in severe cases. Damage to the liver, skin and gut mucosa are common clinical manifestations.
- Autoimmune thrombocytopaenia
-
A reduced number of circulating platelets, owing to increased immune-mediated clearance from the blood, predominantly in the spleen.
- Graves' disease
-
A type of autoimmune disease in which autoantibodies produced by the immune system overstimulate the thyroid gland, causing hyperthyroidism.
- N-ethyl-N-nitrosourea
-
(ENU). An ethylating chemical that produces a high rate of genome-wide point mutations at random in the germline.
- Cre-LoxP technology
-
A site-specific recombination system. Two short DNA sequences (LoxP sites) are engineered to flank the target DNA. Expression of Cre recombinase leads to excision of the intervening sequence. Depending on the type of promoter, Cre can be expressed at specific times during development or in specific sets of cells.
- Antinuclear antibodies
-
Autoantibodies generated against antigens within the nucleus, including DNA. They are indicative of systemic lupus erythematosus, but are also common in patients with rheumatoid arthritis, Sjogren's syndrome and other autoimmune diseases, and are rarely found in healthy individuals.
- Negative selection
-
The deletion of self-reactive thymocytes in the thymus. Thymocytes expressing T-cell receptors that strongly recognize self peptide bound to self MHC molecules undergo apoptosis in response to the signalling generated by high-affinity binding.
- FOXP3+ regulatory T cells
-
A rare population of CD4+ T cells that naturally express high levels of CD25 (the interleukin-2 receptor α-chain) and the transcription factor forkhead box P3 (FOXP3), and that have suppressive regulatory activity towards effector T cells and other immune cells. Absence or dysfunction of these cells is associated with severe autoimmunity.
- γδ T cells
-
T cells that express a T-cell receptor consisting of a γ-chain and a δ-chain.
- Immunogenic signalling
-
Signalling that results in immunity by enhancing the immune response. Immunogenic T-cell receptor signalling can be defined as signalling that increases the activation of effector T cells or reduces the activity of regulatory T cells.
- Tolerogenic signalling
-
Signalling that results in the suppression of immunity or the induction of immune tolerance. Tolerogenic T-cell receptor signalling can be defined as signalling that reduces the activation of effector T cells or increases the activity of regulatory T cells.
- Immunological synapse
-
A region that can form between two cells of the immune system in close contact. The immunological synapse originally referred to the interaction between a T cell and an antigen-presenting cell. It involves adhesion molecules, as well as antigen receptors and cytokine receptors.
Rights and permissions
About this article
Cite this article
Liston, A., Enders, A. & Siggs, O. Unravelling the association of partial T-cell immunodeficiency and immune dysregulation. Nat Rev Immunol 8, 545–558 (2008). https://doi.org/10.1038/nri2336
Issue Date:
DOI: https://doi.org/10.1038/nri2336
This article is cited by
-
Age-Related Changes to the Immune System Exacerbate the Inflammatory Response to Pandemic H1N1 Infection
Bulletin of Mathematical Biology (2022)
-
Disrupted Ca2+ homeostasis and immunodeficiency in patients with functional IP3 receptor subtype 3 defects
Cellular & Molecular Immunology (2022)
-
Rheumatological manifestations in inborn errors of immunity
Pediatric Research (2020)
-
An injectable bone marrow–like scaffold enhances T cell immunity after hematopoietic stem cell transplantation
Nature Biotechnology (2019)
-
B cell autoimmunity at the extremes
Nature Immunology (2017)
