
Key Points
-
The dominant rationale to generate new (and to assess old) anticancer chemotherapeutic agents is to determine their cell-autonomous effects — that is, their capacity to reduce the growth (cytostasis) and to induce the death (cytotoxicity) of tumour cells in vitro and in vivo (usually in immunodeficient mice that have been xenotransplanted with human tumours). Experimental data obtained from immuncompetent mice and clinical data obtained from patients indicate that several chemotherapeutic agents have unexpected effects on the immune system. At least in some instances, these 'side effects' can contribute to the therapeutic effects of anticancer drugs.
-
A non-exhaustive list of examples of drugs that combine anticancer and immunostimulatory effects includes: imatinib mesylate, cyclophosphamide, anthracyclines and 5-fluorouracil.
-
Imatinib mesylate, a protein tyrosine kinase inhibitor, can induce caspase-independent death of tumour cells, can enhance natural killer (NK)-cell activities and can induce the expansion of a specific NK-cell subset that also bears dendritic-cell markers, known as interferon-producing killer dendritic cells, which in turn have tumoricidal effects.
-
Cyclophosphamide, a DNA-alkylating agent, induces non-apoptotic cell death of tumour cells and T cells. In addition, it can deplete regulatory T cells, thereby overriding their antitumour immune responses.
-
Anthracyclines can induce an immunogenic variant of apoptosis in tumour cells, thereby eliciting an antitumour immune response that is mediated by dendritic cells and cytotoxic T cells.
-
5-Fluorouracil and other p53-activating cytotoxic drugs promote increased expression of tumour-associated antigens and co-stimulatory molecules on tumour cells.
-
These agents illustrate the therapeutic feasibility of an 'immunogenic chemotherapy'; that is, a programme of chemotherapy that aims at stimulating the antitumour immune response as a warranted side effect of the therapy. Moreover, it might be possible to combine agents that induce direct cancer-cell-specific and immunostimulatory effects for an optimal therapeutic outcome.
-
Theoretically, the induction of immunogenic cancer-cell death or other immunogenic effects should be one of the aims of anticancer chemotherapy so that the immune system can contribute through a 'bystander effect' to eradicate chemotherapy-resistant cancer cells and cancer stem cells.
Abstract
Accumulating evidence indicates that the innate and adaptive immune systems make a crucial contribution to the antitumour effects of conventional chemotherapy-based and radiotherapy-based cancer treatments. Moreover, the molecular and cellular bases of the immunogenicity of cell death that is induced by cytotoxic agents are being progressively unravelled, challenging the guidelines that currently govern the development of anticancer drugs. Here, we review the immunological aspects of conventional cancer treatments and propose that future successes in the fight against cancer will rely on the development and clinical application of combined chemo- and immunotherapies.
Similar content being viewed by others
Main
Primary tumours are currently treated by a combination of therapies, in most cases including surgery, local radiotherapy and chemotherapy. Even when the tumour has apparently been defeated, micrometastases of dormant tumour cells (or cancer stem cells) frequently lead to tumour relapse and therapeutic failure. To win the fight against cancer, it is necessary not only to develop strategies to kill all cancer (stem) cells efficiently, by using the correct combination and schedule of chemotherapeutic agents, but also to attempt to stimulate an immune response so that the immune system can keep residual tumour cells in check.
Cancer is widely considered to be a cell-autonomous genetic disease that results from alterations in oncogenes, tumour-suppressor genes and genome-stability genes. However, the tumour-cell microenvironment, the stroma and immunity also have a major role in cancer. Indeed, for the development of full-blown neoplasia, cancer cells have to overcome intrinsic (cell autonomous) and extrinsic (immune mediated) barriers to oncogenesis1. Only when tumour cells overcome immune control can they progress and kill the host. Accordingly, the increased incidence of some solid tumours in immunosuppressed patients, reports of spontaneous tumour regression, and the positive prognostic impact of tumour-specific cytotoxic T lymphocytes (CTLs) or antibodies support the idea that the immune system has an effect on tumour progression in humans1.
For historical reasons, drug discovery programmes for cancer therapy have neglected the possibility that immune reactions might contribute to the efficacy of treatment. For example, since 1976, the National Cancer Institute, USA, has used a drug screening and validation strategy in which human tumour cells are xenotransplanted into immunodeficient mice2 (Timeline). Although cancer chemotherapy and radiotherapy is often viewed as a strategy that mainly affects tumour cells, accumulating evidence indicates that cytotoxic drugs also affect the immune system to contribute to tumour regression.

In this Review, we summarize current knowledge on the contribution of the immune system to conventional cancer therapies. The immune system is elicited in two ways by conventional therapies. Some therapeutic programmes can elicit specific cellular responses that render tumour-cell death immunogenic. Other drugs may have side effects that stimulate the immune system, through transient lymphodepletion, by the subversion of immunosuppressive mechanisms or through direct or indirect stimulatory effects on immune effectors. Moreover, vaccination against cancer-specific antigens can sensitize the tumour to subsequent chemotherapeutic treatment. The challenge is to hijack the host immune system so that it can control any residual disease, or as stated by Prendergast and Jaffee, to stop “segregating cancer immunology from cancer genetics and cell biology”3. We anticipate that the physician of the future will need to integrate parameters that pertain to the host and the tumour in order to provide optimal management of the disease.
Theoretical bases for antagonistic effects
Many of the therapeutic procedures that are used in oncology today can curtail the immune response against tumour cells. For example, although it is well established from animal studies that the lymph nodes provide the optimal environment for T-cell priming, surgical oncologists frequently re-sect the tumour-draining lymph nodes, with the goal of performing histological staging of the tumour and removing local metastases4. More importantly, many chemotherapeutic drugs have notable immunosuppressive side effects, either directly, by inhibiting or killing effector cells, or indirectly, by provoking anergy or immune paralysis (Table 1).
Immunosuppressive side effects of chemotherapeutics. Several of the cancer chemotherapeutics that are used today are also used as immunosuppressants for the treatment of severe systemic autoimmune diseases. This applies to cyclophosphamide5 and methotrexate6 (Box 1), which impair the proliferative and/or effector functions of peripheral T cells. Protein tyrosine kinase inhibitors may also affect the T-cell arm of adaptive immunity. At high doses, imatinib mesylate (Gleevec; Novartis), which specifically blocks signalling through the receptors KIT, c-ABL (cellular Abelson leukaemia-virus protein) and its oncogenic fusion derivative BCR (B-cell receptor)–ABL, suppresses T-cell proliferation and activation, presumably through inhibition of the protein tyrosine kinase LCK7. Studies that were carried out in mouse models have revealed that imatinib mesylate selectively curtails the expansion of memory CTLs, but does not affect primary T- and B-cell responses8. Treatment of patients with imatinib mesylate may also suppress the graft-versus-leukaemia effect from allogeneic transplantations and increase susceptibility to viral and bacterial infections9.
Owing to their capacity to trigger lymphocyte apoptosis, glucocorticoids are also important components of the chemotherapeutic cocktails that are used in treating several lymphoproliferative diseases. High doses of glucocorticoids are prescribed to patients with cancer to attenuate chemotherapy-associated nausea and vomiting. Glucocorticoids suppress the production of pro-inflammatory cytokines (such as interferon-α (IFNα), IFNβ, interleukin-1α (IL-1α) and IL-1β) and chemokines (such as CXC-chemokine ligand 8 (CXCL8), CC-chemokine ligand 7 (CCL7), CCL8, CCL11, CCL13, CCL17, CCL19 and CCL20) by blood mononuclear cells from healthy donors10. Although glucocorticoids induce the expression of the pattern-recognition receptors Toll-like receptor 2 (TLR2) and TLR4, they severely impair the differentiation of and antigen presentation by dendritic cells (DCs) in vitro and in vivo11. Moreover, glucocorticoids are known to repress the expression of genes that are associated with the adaptive immune response (including those that encode components of the MHC class II machinery, the T-cell receptor (TCR) genes TCRA, TCRB, TCRD and TCRG, signal transducer and activator of transcription 1 (STAT1) and CD40), alter T-cell development and function, suppress the development of TH1 cells (T helper 1 cells) and bias responses towards the TH2-cell type, thereby precluding the elicitation of effector and memory antitumour immunity. Several members of the transforming growth factor-β (TGFβ) family that suppress T-cell and natural killer (NK)-cell effector functions are upregulated by glucocorticoids. Glucocorticoids inhibit the cell-surface expression of the main natural cytotoxicity receptors (NKp30 and NKp44) and reduce IL-2- and IL-15-triggered NK-cell proliferation while compromising the NK-cell cytotoxicity that is mediated by the NKp46, NKG2D (natural-killer group 2, member D) or 2B4 receptors12.
Cell-death modalities: apoptosis and tolerance. Chemotherapy-induced tumour-cell death occurs frequently (but not exclusively) through apoptosis, a cell-death modality that is reputedly non-immunogenic or tolerogenic (Box 2). Based on the assumption that apoptosis, a morphologically defined phenomenon (with nuclear condensation and fragmentation as prominent hallmarks), is biochemically homogeneous, it is assumed that chemotherapy would elicit non-immunogenic cell death. Accordingly, cancer cells that are treated with mitomycin C, which induces classical apoptosis, fail to elicit an immune response when injected subcutaneously into histocompatible hosts in the absence of adjuvant. Similarly, 20 apoptosis-inducing compounds that operate through distinct modes of action failed to induce immunogenic cancer-cell death. This applies to drugs that kill cancer cells through mitochondrial, lysosomal or endoplasmic reticulum stress (ER stress), as well as protein tyrosine kinase inhibitors, proteasome inhibitors or DNA-damaging agents (such as alkylating agents or topoisomerase inhibition)13. One explanation of the non-immunogenic nature of apoptosis resides in the way in which cell corpses are handled by the immune system. For example, apoptosis is accompanied by the exposure of phosphatidylserine in the outer leaflet of the plasma membrane. Phosphatidylserine exposure functions as an 'eat-me' signal, which triggers phagocytosis by macrophages, and stimulates the production of anti-inflammatory cytokines. Likewise, the standard apoptotic programme is linked to the absence of immunostimulatory signals, as discussed in a later section.
Immunosuppression by massive tumour-antigen release. The sudden and systemic release of numerous dying tumour cells resulting from chemotherapy might have deleterious consequences on subsequent tumour-specific immune responses. Regionally advanced melanomas of the limbs can be treated by isolated limb perfusion (ILP) with tumour-necrosis factor (TNF) and the drug melphalan (Alkeran; Celgene) (Box 1). This results in local responses that are up to 80% complete, but does not prolong the survival of the patients14. So, despite the marked cell death that is induced by vasodilatation (vasoplegia), the induction of pro-inflammatory mediators, the expression of the adhesion molecules on endothelial cells, and the recruitment of lymphocytes, granulocytes and macrophages into infused tumour beds15, these patients fail to mount a protective immune response. Instead, pro-inflammatory cytokines (such as IL-1β, TNF and IL-6) that are produced by either the tumour or host immune cells may promote tumour progression through a molecular signalling pathway that involves nuclear factor-κB (NF-κB)16.
In addition, although this is theoretical, the massive release of tumour antigens during ILP may cause a sort of high-dose antigen-mediated tolerance. Indeed, the delivery of large amounts of antigen could be deleterious for mounting reactive effector T cells. It has been shown that there is a direct correlation between the amount of antigens that are expressed in the periphery and the degree of T-cell proliferation and number of tolerogenic antigen-specific CD8+ T cells in the draining lymph nodes17. Studies to evaluate melanoma-specific CD8+ T-cell responses after ILP are underway and may confirm these hypotheses.
Therefore, owing to their chemical nature (for example, as alkylating agents and glucocorticoids), their mode of action (for example, in which tumour-cell death is not preceded by cellular stress, and vasoplegia) or their handling (for example, their dosing and route of administration), several components of the therapeutic arsenal can be detrimental to the immune system.
Agonistic interplay
Accumulating evidence has indicated that radiotherapy and some cytotoxic compounds promote specific anticancer immune responses that contribute to the therapeutic effects of conventional therapy.
DNA damage: a fast track to immunity? DNA-damaging agents, such as ionizing irradiation or topoisomerase inhibitors, stimulate a complex response that involves the activation of tumour-suppressor proteins, such as the protein kinases ATM (ataxiatelangiectasia mutated) and CHK1 (checkpoint kinase 1), as well as the transcription factor p53. This DNA-damage response induces the expression of NKG2D ligands on tumour cells in an ATM-dependent and CHK1-dependent (but p53-independent) manner18. NKG2D is an activating receptor that is involved in tumour immunosurveillance by NK cells, NKT cells, γδ T cells and resting (in mice) and/or activated (in humans) CD8+ T cells.
Although p53 is not required for the expression of NKG2D ligands in cells undergoing DNA damage, a recent study has highlighted an important cooperation between p53-induced tumour-cell senescence and the innate immune system19. It was found that restoration of p53 function in established liver cancers led to tumour regression, but only when the mice had functional NK cells, neutrophils and macrophages. Reactivation of p53 in hepatocellular cancers induced the expression of pro-inflammatory cytokines (such as IL-15 and macrophage colony-stimulating factor (M-CSF)), adhesion molecules (such as intercellular adhesion molecule 1 (ICAM1) and vascular cell-adhesion molecule 1 (VCAM1)) and chemokines (such as CCL2 and CXCL1), which could contribute to the p53-induced recruitment of neutrophils, macrophages and NK cells into tumour beds19. This example illustrates how molecules that are activated during the DNA-damage response (ATM, CHK1 and p53) can alert the innate immune system to mediate an antitumour response.
Distant effects of radiotherapy. Besides its direct cytotoxic properties towards tumour cells, irradiation mediates many effects on cells and tissues, some of which may stimulate an immune response. Low doses of ionizing irradiation upregulate the expression of MHC class I molecules, tumour-associated antigens (carcinoembryonic antigen and mucin 1)20 and CD95 (also known as FAS) by tumour cells, as well as adhesion molecules by endothelial cells21, thereby boosting CTL activity22 and T-cell trafficking towards irradiated tumour sites23. Local irradiation of a single tumour site can reduce the size of non-irradiated metastases that are located at a distant site, a phenomenon known as the 'abscopal effect', when mediated by the immune system. In an elegant study, Reits et al.24 showed that irradiation enhances tumour antigenicity in three ways by modulating the repertoire of tumour-derived peptides that are presented to the immune system. First, irradiation enhances the degradation of existing proteins, thereby increasing the intracellular pool of peptides for MHC class I presentation. Second, activation of mammalian target of rapamycin (mTOR) in irradiated tumour cells stimulates protein translation and increases peptide production. Third, irradiation stimulates the synthesis of new proteins and therefore antigenic peptides, which can be presented for recognition by different T-cell repertoires24. Moreover, potent synergistic effects against established tumours between passively transferred CTLs24 or TLR9 ligands25 and ionizing radiation have been reported24.
High doses of alkylating agents for lymphoablation. The therapeutic induction of lymphopaenia has raised considerable interest in the context of adoptive transfer therapies and vaccination against melanomas26. Transient lymphopaenia is thought to enhance the efficiency of these therapies by activating homeostatic mechanisms that stimulate the tumour-reactive effector T cells and by counteracting tumour-induced suppression. Common methods to induce lymphopaenia include low-dose total body irradiation for reversible myelosuppression, or treatment with cyclophosphamide alone or in combination with fludarabine, which promotes long-term lymphopaenia in humans27. In addition to eradicating the cells that may suppress antitumour responses, such as regulatory T cells28, lymphoid reconstitution could overcome cancer-induced defects in T-cell signalling, endow host antigen-presenting cells with co-stimulatory functions, and increase the production and availability of cytokines, such as IL-7, IL-15 and IL-21, resulting in enhanced CD8+ T-cell activity28. Lymphodepletion also enhances T-cell homing into tumour beds and intratumoral proliferation of effector cells27,29. Finally, total body irradiation could cause mucosal barrier injury, resulting in microbial translocation and systemic release of the TLR4 ligand lipopolysaccharide (LPS), which has been shown to increase DC and T-cell activation and to promote tumour regression30. Animal studies have shown that lymphoablation enhances the effectiveness of adoptively transferred tumour-specific CD8+ T cells31. This strategy has been successful in a clinical trial that involved 35 patients with advanced metastatic melanomas that were refractory to conventional treatments29.
Lymphodepletion can also be combined with vaccination strategies that promote the differentiation of central memory T cells that recognize self tumour antigens32,33. Moreover, lethal myeloablation followed by autologous haematopoietic stem cell (HSC) rescue further enhances the efficacy of adoptive transfer therapies for tumours33. Profound lymphodepletion results in the expansion of both adoptively transferred T cells and host naive T cells.
So, chemotherapy-induced or irradiation-induced lymphodepletion, combined with additional manipulations, such as adoptive cell transfer, vaccination or HSC rescue, may be useful approaches for generating antitumour immune responses.
Low-dose cyclophosphamide for suppression of regulatory T cells. Although high doses of cyclophosphamide have direct tumoricidal effects and cause immunosuppression by lymphoablation (see above), low doses of the same compound mediate immunostimulation. Low doses of cyclophosphamide can potentiate delayed-type hypersensitivity (DTH) responses34 by acting on a cyclophosphamide-sensitive regulatory T-cell subset28. Low doses of cyclophosphamide decrease the number and inhibitory function of CD4+CD25+ regulatory T cells (TReg cells) by downregulating the expression of key functional markers of TReg cells, forkhead box P3 (FOXP3) and glucocorticoid-induced TNF-receptor-related protein (GITR)35. The effects of cyclophosphamide on TReg cells and cyclophosphamide-stimulated IFNα production36 might account for the augmented antibody responses and the persistence of memory T cells. All these effects contribute to the eradication of immunogenic tumours in synergy with specific immunotherapies37,38. However, the ablation of regulatory cells is likely to be of varying importance, depending on the tumour type, stage and location39.
In small clinical studies, the combination of low doses of intravenous cyclophosphamide with vaccines has been shown to augment DTH responses40,41, decrease the proportion of CD4+2H4+ (CD45+) suppressor T cells42 and prolong the survival of patients with metastatic cancer40. A daily dose of oral cyclophosphamide, also referred to as a metronomic programme, given for 1 month to patients with end-stage cancer could suppress TReg-cell inhibitory functions, restore the proliferative capacity of effector T cells and restore the cytotoxicity of NK cells43,44. However, the optimized schedule of cyclophosphamide yielding immunostimulatory effects in patients with cancer awaits further randomized investigation.
Anthracyclines as immunostimulators? Immune modulation by the anthracycline doxorubicin has been studied for decades45,46. Recently, it has been shown that doxorubicin enhances the antitumour potency of the GM-CSF-transfected tumour-cell vaccine when given before or after immunization47. The antileukaemic effects of a combination of IL-12 and various cytotoxic agents (cisplatin, cyclophosphamide, paclitaxel or doxorubicin) were evaluated in immunocompetent mice with syngeneic L1210 leukaemia cells, and it was demonstrated that significantly prolonged survival of the mice is only achieved with a combination of IL-12 and doxorubicin48. The therapeutic effect of IL-12 plus doxorubicin was lost when the mice were irradiated or injected with silica, indicating that radiosensitive immune cells (possibly T cells) and macrophages are required for the therapeutic effect. As a possible mechanism to account for the immune-mediated anticancer effects of anthracylines, it has recently been discovered that anthracyclines — unlike many other cytotoxic agents — can elicit immunogenic cell death (see below)13,49.
Taxanes: TLR4 ligands and beyond. The taxanes paclitaxel (Taxol; Bristol–Myers Squibb) and docetaxel (Taxotere; Sanofi–Aventis) both bind to the β-subunit of tubulin and affect microtubule polymerization, leading to cell-cycle arrest at the G2/M stage and subsequent cell death. Paclitaxel binds to mouse TLR4 (but not to human TLR4) and so can mimic bacterial LPS by activating mouse macrophages and DCs through a pathway that requires TLR4, MD2 (a key component of the LPS receptor complex) and myeloid differentiation primary-response gene 88 (MyD88)50.
In mice that are transgenic for human epidermal growth-factor receptor 2 (HER2; also known as NEU and ERBB2), which is overexpressed in some breast cancers, and that are therefore tolerant to HER2, treatment with paclitaxel increases the efficacy of tumour vaccines that express HER2 and GM-CSF51. Similarly, mice that have a mammary adenocarcinoma transfected with a candidate tumour antigen respond more efficiently to intratumour DC inoculations when they receive paclitaxel52. The optimal scheduling of docetaxel treatment combined with a GM-CSF-producing tumour vaccine was assessed in the 3LL tumour model53. When mice that have established 3LL tumours were pre-treated with docetaxel, followed by vaccination with irradiated GM-CSF-transfected 3LL tumour cells, significant tumour regression and prolonged survival were observed in comparison with chemotherapy alone. If docetaxel was administered after tumour-cell vaccination, treatment with taxane abrogated the antitumour effects of the vaccine, presumably by negatively affecting the dividing effector T cells. Mice that survived the treatment developed memory CTLs specific for a 3LL epitope (MUT-1) and resisted rechallenge with live 3LL tumour cells. In humans, there is circumstantial evidence that taxanes can stimulate the anticancer immune response. T-cell proliferation and NK-cell cytolysis assays have revealed that a cohort of patients with breast cancer (stage II/III) treated with taxanes showed enhanced T-cell and NK-cell functions compared with patients treated without taxanes54. Therefore, experimental data obtained in mice and humans contravene traditional thinking that taxanes suppress immune-cell functions.
Multiple immunostimulatory effects of gemcitabine. Gemcitabine (Gemzar; Celgene) is a synthetic pyrimidine nucleoside analogue that is efficient in the treatment of pancreatic, breast and lung cancers (Box 1). Its phosphorylated metabolites inhibit ribonucleotide reductase and DNA polymerase-α, which results in the depletion of the deoxynucleotide pool, thereby stopping DNA synthesis. Gemcitabine inhibits B-cell proliferation and antibody production in response to tumour antigens55, a phenomenon that may skew antitumour immunity towards beneficial T-cell responses56. Moreover, gemcitabine reduces the frequency of CD11b+GR1+ myeloid suppressor cells57. Gemcitabine-induced apoptosis of established tumours may enhance the DC-dependent cross-presentation of tumour antigens to T cells58. Consistent with these data, gemcitabine can function in synergy with CD40 stimulation of T cells to cure established mouse tumours59. The immunostimulatory effects of gemcitabine have been confirmed in patients. In patients with pancreatic60, non-small-cell lung61 or colon62 cancer, gemcitabine combined with recombinant cytokines or vaccines could enhance the frequency of tumour-specific CTL precursors and result in objective response rates.
5-Fluorouracil. 5-Fluorouracil is a fluoropyrimidine that is commonly used against breast cancer and gastrointestinal malignancies (Box 1). In vitro, 5-fluorouracil induces the expression of heat-shock proteins (HSPs) in tumour cells and facilitates antigen uptake by DCs and subsequent cross-presentation of tumour antigens63. Human colon carcinoma cell lines that were treated with 5-fluorouracil acquired CD95 and ICAM1 expression and became more sensitive to lysis by CTLs. In a mouse model, intratumoral inoculation of DCs after systemic chemotherapy that was based on 5-fluorouracil induced the T-cell-dependent eradication of the DC-treated site and, importantly, also of tumours located at a distant site, resulting in the long-term survival of the mice. Such effects can only be obtained by treatment with a combination of DCs and 5-fluorouracil, not with each treatment alone64. In another model, 5-fluorouracil was also able to augment the immunizing efficacy of a peptide-based vaccine directed against thymidilate synthase, its own molecular target65. These mouse data have prompted the design of a sizeable randomized clinical trial in patients with metastatic colon cancer (X. Cao, personal communication).
5-Aza-2′-deoxycitidine. The DNA methyltransferase inhibitor 5-aza-2′-deoxycitidine (DAC) is used in the treatment of leukaemias and myelodysplastic syndrome66. The antineoplastic effect of DAC is thought to involve reversing the hypermethylation of cancer-associated promoters, which inhibits gene transcription. Treatment with DAC restores the expression of MHC class I molecules and cancer testis antigens on tumour cells, rendering the tumour cells susceptible to CTL attack67. Moreover, hypermethylation of the tumour-suppressor gene that encodes death-associated protein kinase (DAPK) can be suppressed by treatment of leukaemic cells with DAC, thereby restoring the IFNγ-mediated apoptotic-cell-death pathway66. Cytokines that induce IFNγ secretion could therefore potentially be in synergy with DAC. Indeed, combining IL-12 with DAC was synergistic in a T-cell-dependent manner against L1210 leukaemia and B16F10 melanoma in mice68.
Antivascular flavonoids. Vascular-disrupting agents are distinct from anti-angiogenic agents in that they target pre-existing tumour vessels rather than neoangiogenesis. Flavone acetic-acid derivatives are known to have antivascular properties69. However, the flavonoid 5,6-dimethylxanthenone-4-acetic acid (DMXAA) has been shown to have immunological side effects and to elicit type-I-IFN-dependent NK-cell responses, as well as CD4+ T-cell-dependent antitumour responses69. In models of large thoracic tumours, DMXAA was able to promote the infiltration of CD11b+ cells and CD8+ T cells into tumour beds that release immunostimulatory cytokines and chemokines (CXCL10, RANTES, IL-6, IFNγ, TNF, CCL3, CCL2, CXCL9 and inducible nitric-oxide synthase). As the antineoplastic effects of DMXAA depend on perforin and CD8+ T cells, DMXAA could induce the activation of tumour-associated macrophages that contribute to the recruitment of other antitumour effectors70. Recently, DMXAA has been shown to be a novel and specific activator of the TANK-binding kinase 1 (TBK1)–IFN-regulatory factor 3 (IRF3) signalling pathway. Therefore, treatment of primary mouse macrophages with DMXAA has resulted in robust IRF3 activation and a marked increase in the production of the mRNA encoding IFNβ (Ref.71).
Immunostimulatory side effects of imatinib mesylate. Imatinib mesylate is a clinically approved drug for the treatment of gastrointestinal stromal tumours (GISTs), which are associated with activating mutations in KIT or PDGFRA (platelet-derived growth factor receptor-α). This treatment is based on the rationale that imatinib mesylate directly targets the pathognomonic mutations in these receptor protein-tyrosine kinases. However, a subset of patients with GISTs who have no KIT or PDGFRA mutation still respond to imatinib mesylate72. These antitumour effects were found to be related to NK-cell activation that is promoted by the side effects of imatinib mesylate on immune cells43,72. Indeed, imatinib mesylate mediated NK-cell-dependent antitumour effects in vivo in mice with cancer cells that did not respond to imatinib mesylate in vitro73. Imatinib mesylate blocks KIT signalling in host DCs, thereby triggering DC-mediated, NK-cell activation and NK-cell-dependent antitumour effects. In patients with GISTs, NK-cell IFNγ secretion that is induced by imatinib mesylate constitutes a positive prognostic parameter, suggesting that NK cells also contribute to the therapeutic effect of imatinib mesylate in humans72. So, depending on the local concentrations and the duration of exposure to imatinib mesylate, one might expect either inhibitory functions of imatinib mesylate on the differentiation of DCs and memory T cells or stimulatory effects on NK cells.
In mice, the combination of imatinib mesylate plus IL-2 induces the expansion of a unique population of tumour-infiltrating effector cells. These effector cells have been termed IFN-producing killer DCs (IKDCs)74 because they share phenotypic and functional properties with myeloid DCs and NK cells and can produce IFNγ. Unlike B220− NK cells, these CD11c+B220+NK1.1+ cells can lyse various target cells regardless of the expression of MHC class I molecules or NKG2D ligands. IKDCs also upregulate MHC class II expression on contact with transformed cells and produce large amounts of IFNγ, which is in contrast to B220− NK cells74. Adoptive transfer into B16F10 tumour-bearing immunodeficient (Rag−/− Il2rg−/−) mice has revealed that IKDCs but not B220− NK cells significantly delay tumour progression. Together, these data indicate that imatinib mesylate may exert part of its antitumour effects through the expansion and stimulation of IKDCs.
Mechanisms of T-cell-mediated immunity
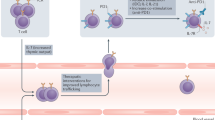
The immuno-adjuvant effects of cytotoxic compounds discussed in this article may primarily rely on the capacity of antigen-presenting cells to engulf dying tumour cells and then to process and present tumour antigens to naive and/or central memory T cells. Therefore, and as previously reviewed75, the signals that are delivered by stressed or dying tumour or stromal cells under the influence of cytotoxic compounds can be expected to regulate antigen uptake (eat-me signals and antigen transfer), as well as antigen processing and presentation, thereby influencing DC maturation, co-stimulation, polarization and trafficking (Fig. 1; Table 2). In addition, stressed tumour cells may upregulate stimulatory ligands for NK cells (for example, NKGD2 ligands after DNA damage) or death receptors (for example, CD95 and TRAILR), thereby increasing their susceptibility to lysis by endogenous immune effectors (Fig. 2; Table 2).

Following tumour insult by cytotoxic agents, tumour cells rapidly translocate intracellular calreticulin (CRT) to the cell surface (within 1 hour in vitro), which acts as a mandatory eat-me signal for dendritic cells (DCs) and induces immunogenic cell death. By 12 hours, molecular chaperones, such as heat-shock protein 90 (HSP90), can appear on the tumour-cell surface, contributing to tumour-cell–DC adhesion and the first steps of DC maturation. Next, by 18 hours, the release by dying tumour cells of the chromatin-binding protein high-mobility group box 1 protein (HMGB1) is required for optimal Toll-like receptor 4 (TLR4)-dependent processing of the phagocytic cargo by DCs. Finally, maturation signals (possibly type I interferons (IFNs) or interleukin-1 (IL-1)) that are delivered by stressed tumour cells complete the maturation programme that is elicited by the earlier events on DCs.

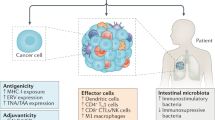
Tumour cells may be lysed by activated cytotoxic cells, such as natural killer (NK) cells, NKT cells, interferon-producing killer dendritic cells (IKDCs), γδ T cells and cytotoxic T lymphocytes (CTLs). However, this direct killing activity may be indirectly facilitated by the intervention of various effector mechanisms that sensitize tumour cells to express stress or danger signals that promote their recognition by particular effector cells. The DNA-damage response promotes the expression of ligands for NKG2D (NK group 2, member D), while restoration of functional p53 might induce the recruitment and activation of innate immune cells through the induction of CC-chemokine ligand 2 (CCL2) and interleukin-15 (IL-15). Inhibitors of histone deacetylases (HDACs) and inhibitors of the phosphatase PP1 contribute to the expression of NKG2D ligands and of cell-surface calreticulin, respectively, eliciting innate immune responses and phagocytosis, respectively. The release of high-mobility group box 1 protein (HMGB1) by tumour cells can be promoted by anthracyclines, oxaliplatin and irradiation, as well as by soluble TRAIL (tumour-necrosis factor-related apoptosis-inducing ligand) (and potentially activated NK cells and IKDCs that express TRAIL), and is mandatory for dendritic cell (DC)-mediated cross-presentation of apoptotic tumours to T cells. Proteasome inhibitors, such as bortezomib, induce cell-surface expression of heat-shock protein 90 (HSP90) by tumour cells, facilitating antigen uptake and maturation of DCs. Many chemotherapeutic agents can upregulate the expression of MHC class I molecules, tumour antigens and CD95 on tumour cells, leading to enhanced susceptibility to CTLs. ATM, ataxia-telangiectasia mutated; CRT, calreticulin; DAC, 5-aza-2′-deoxycitidine.
Chaperones. A common response to cell stress, including that induced by chemotherapy, is the transcriptional activation of a series of molecular chaperones that belong to the class of inducible HSPs. Such HSPs protect against cell death by re-folding damaged proteins and by directing damaged proteins to proteasome-mediated degradation. In addition, they inhibit apoptosis at the pre- and post-mitochondrial levels. HSPs can also stimulate the immune system at several levels.
First, HSP70 and HSP90 (as well as gp96 and calreticulin) can act on the scavenger receptor CD91 on the surface of DCs, thereby transmitting a maturation signal76. As well as binding damaged proteins, HSPs may bind peptides, including tumour-specific antigens, and may direct them to MHC class I and II pathways for presentation to T cells. Purified peptide–HSP complexes can induce protective immunity against the tumour from which they derive77. Peptide–HSP complexes may be particularly efficient as a source of antigen for cross-priming, as has been shown in vivo in suitable mouse models78. Moreover, encouraging results from HSP-based vaccine trials have been reported79.
Second, in addition to their function as vehicles for peptide antigens, chaperones may have an important role in DC activation as eat-me signals. For example, HSP90 appears on the surface of human myeloma cells after treatment with the proteasome inhibitor bortezomib (Velcade; Millennium Pharmaceuticals)80 and then serves as a contact-dependent activation signal for autologous DCs. Of note, this HSP90 exposure depends on the cytotoxic agent bortezomib and does not occur after γ-irradiation or treatment with steroids, indicating that there are stimulus-dependent differences in the type of stress response induced.
Third, another chaperone, calreticulin, can also be exposed on the surface of dying cells and act as an eat-me signal for macrophages, presumably by interacting with CD91 on the engulfing cell81. In response to some cell-death inducers, and in particular anthracyclines and ionizing irradiation, calreticulin can translocate from the lumen of the ER to the surface of cells at an early pre-apoptotic stage13,82. This early exposure of calreticulin and its aggregation in discrete foci on the cell surface (ecto-calreticulin) is not a general feature of apoptosis (Fig. 1). When tumour cells are treated for a few hours with anthracyclines and then injected subcutaneously into mice, they are highly efficient in inducing a specific DC and antitumour T-cell response49. Ecto-calreticulin is crucial for the recognition and engulfment of dying tumour cells by DCs. Thus, anthracyclines and γ-irradiation that induce ecto-calreticulin cause immunogenic cell death, whereas other pro-apoptotic agents (such as mitomycin C and etoposide) neither induce ecto-calreticulin nor immunogenic cell death. Depletion of calreticulin expression by transfection with specific small interfering RNAs (siRNAs) abolishes the immunogenicity of cell death that is elicited by anthracyclines, whereas an exogenous supply of calreticulin (as a recombinant protein) or the use of pharmacological agents that favour calreticulin translocation (such as PP1 phosphatase inhibitors) can enhance the immunogenicity of cell death13,80. Such PP1 inhibitors (for example, tautomycin and caliculin A) can increase the therapeutic efficacy of mitomycin C and etoposide in vivo by eliciting a specific immune response83.
Therefore, calreticulin exposure by tumour cells may allow prediction of the therapeutic outcome, and studies are ongoing to apply this knowledge to human disease82. The pharmacological reestablishment of calreticulin exposure may ameliorate the efficacy of chemotherapy, and the development of non-toxic PP1-inhibitory molecules that can induce calreticulin exposure is in progress.
HMGB1 and TLR4. Therapies that are based on anthracycline, irradiation and oxaliplatin (Eloxatin; Sanofi–Aventis) are more efficient at inhibiting the growth of established syngeneic tumours in immunocompetent mice than in athymic (nude) littermates, indicating that an intact immune system enhances the therapeutic efficacy of conventional anticancer treatments84,85. This result has prompted the search for a link between innate and adaptive immune responses and has led to the systematic screening of the role of TLRs in the efficacy of chemotherapy.
Surprisingly, the only TLR deficiency that compromises the efficacy of chemotherapy or radiotherapy in vivo is TLR4 and its downstream effector MyD88 (Refs 84, 85). Indeed, dying tumour cells can be cross-presented by DCs to naive T cells in vitro and in vivo, and they promote the differentiation of tumour-specific CTLs only when host DCs harbour a functional TLR4–MyD88 pathway. TLR4-deficient DCs are unable to present antigen from dying cells (taken up by phagocytosis) although they are normal in their ability to present antigen from soluble proteins (taken up by pinocytosis)84,85, which suggests a specific defect in antigen presentation after phagocytosis. TLR4 has been reported to inhibit the lysosome-dependent degradation of phagosomes in macrophages86. Accordingly, the kinetics of fusion between phagosomes and lysosomes is slower in wild-type DCs than in TLR4-deficient DCs loaded with dying tumour cells, which suggests that the TLR4 defect causes rapid lysosomal degradation of phagocytic material. Antigen presentation by TLR4-deficient DCs can be restored by inhibiting the activity of lysosomes, either with chloroquine (a lysosomotropic alkaline) or bafilomycin A1 (a specific inhibitor of the vacuolar ATPase responsible for lysosomal acidification). In addition, chloroquine can reverse the adverse effects of TLR4 deficiency on chemotherapeutic responses in vivo84,85.
Further studies have revealed that the ligand of TLR4 that is produced by the dying tumour cells is high-mobility group box 1 protein (HMGB1), a nuclear protein that is released from dying cells during late-stage apoptosis84,85 (Figs 1, 2). Depletion of HMGB1 from dying tumour cells with siRNAs or neutralization of HMGB1 with specific antibodies abolishes the TLR4-dependent, DC-mediated presentation of antigens from dying tumour cells in vitro and in vivo84. So, HMGB1 release is required for the immunogenicity of cell death through its effect on TLR4. However, neither HMGB1 nor calreticulin (nor a combination of both) can promote complete DC maturation, indicating that the search for immunostimulatory molecules produced by dying cells must continue.
Importantly, a polymorphism in TLR4 (rs4,986,790), which leads to a single-nucleotide exchange (A896G) and a single amino-acid substitution (Asp299Gly) in the extracellular domain of TLR4, was found to reduce the binding of HMGB1 to human TLR4. Accordingly, DCs that are derived from patients who carry the TLR4 Asp299Gly allele were far less efficient in cross-presenting antigens from dying melanoma cells to CTLs than were DCs from patients who carry the normal TLR4 allele84,85. This DC defect could be overcome by adding chloroquine. In a retrospective study, we analysed the time to metastasis in a cohort of 280 patients who had been treated for breast cancer with local lymph-node invasion, following a standard protocol of local surgery, local radiotherapy and systemic anthracycline. Patients who had the TLR4 Asp299Gly allele developed metastasis more quickly than patients who had the normal TLR4 allele, establishing TLR4 Asp299Gly as an independent predictive factor of early disease progression84. These results support the concept that a selective immune defect (in the DC-mediated presentation of antigen from dying cells) can compromise the response to anticancer chemotherapy85.
Clinical trials of combination therapies
Numerous clinical studies have combined conventional anticancer treatments with immunotherapies (Table 3). Pending confirmation through randomized, controlled clinical trials, some of these studies suggest that chemotherapy and immunotherapy can create a synergy.
Two clinical studies in which DC vaccines were administered before salvage chemotherapy have indicated that synergistic antitumour effects between vaccination and cytotoxic compounds can be achieved in late-stage cancers. In one study, 29 patients with end-stage small-cell lung cancer that was resistant to first-line platin-based therapy were enrolled in a vaccination protocol that involved autologous DCs infected with adenoviral vectors encoding p53 (Ref. 87). The tumour progressed in 23 patients, who then received salvage chemotherapy with paclitaxel or carboplatin. The response rate to these second-line chemotherapies was 61.5% and 38%, respectively, survival at 1 year post-vaccination, a result that was not expected from historical controls. Another, retrospective study examined the impact of vaccination with peptide-loaded or lysate-loaded DCs on the efficacy of conventional therapy for glioblastoma. Chemotherapy acted in synergy with previous therapeutic vaccination to extend patient survival. The authors suggested that the immunological targeting of tumour antigen tyrosinase-related protein 2 (TRP2) expressed by glioblastoma cells increased the sensitivity to inducers of cell death87.
Similar intriguing synergistic effects were reported when combining DNA vaccines and chemotherapy in prostate cancers and other end-stage tumours88,89,90. Patients who succeeded in mounting vaccine-specific immune responses responded to salvage chemotherapy, whereas those who failed did not. The aforementioned clinical studies and those summarized in Table 3 illustrate that the combination of chemotherapy and immunotherapy may provide a valid therapeutic option for the treatment of neoplastic disease. However, defining the appropriate doses and schedule for the optimal synergy between the two strategies, as well as the key indications and tumour types53, represents our current challenge.
Concluding remarks and future outlook
There is accumulating evidence that conventional therapy for cancer may profit from the participation of the immune system. This immune contribution is elicited in two ways by conventional therapies. On the one hand, some therapeutic programmes can elicit specific cellular responses — beyond the stereotypical apoptotic pathway — that render tumour-cell death immunogenic. These immunogenic modifications include: pre-apoptotic calreticulin translocation in response to anthracyclines; induction of expression of MHC molecules, tumour-specific antigens or death receptors in response to epigenetic modifiers; HSP90 expression in response to bortezomib; and post-apoptotic HMGB1 secretion in response to anthracyclines and oxaliplatin. Other chemotherapeutic agents that induce immunogenic tumour-cell death may elicit yet more mechanisms. On the other hand, some drugs may have side effects (beyond their effect on the tumour itself) that stimulate the immune system, through transient lymphodepletion, the subversion of immunosuppressive mechanisms and the direct or indirect stimulatory effects of immune effectors.
In addition, many experimental therapies that have been tested in mice and a few clinical trials suggest that vaccination against cancer-specific antigens can sensitize the tumour against subsequent chemotherapeutic treatment. Although the mechanisms that underlie such a synergistic effect have not been elucidated, we speculate that the vaccination-induced increase in the frequency of primed T cells, although by itself irrelevant for tumour progression, may constitute a major advantage as soon as the tumour is insulted by cytotoxic drugs.
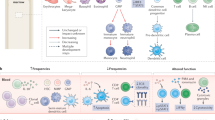
Based on these premises, we anticipate that, in the future, physicians will need to integrate four parameters to design an optimal therapeutic programme (Fig. 3) — the first three pertaining to the host and the last one to the tumour.

For improved clinical management of cancer, four parameters should be integrated in cancer diagnosis and treatment. First, cancer immuno-epidemiology indicates that polymorphisms in the genes that dictate immune responses, such as Toll-like receptor 4 (TLR4) and interleukin-1β (IL1b), should influence the design of the therapy. Second, tumour-induced tolerance relies on suppressor cells or their mediators, such as transforming growth factor-β (TGFβ) and IL-10, which could be antagonized by dedicated therapies that have the aim of restoring antitumour immune responses. Careful immunomonitoring of patients with cancer should reveal which among these immunosuppressive pathways needs to be counterbalanced. Third, the deleterious effects of conventional therapies (including the use of steroids, lymph-node resections and escalating doses of chemotherapeutic agents with immunosuppressive and myelosuppressive side effects) should be avoided. Fourth, by examining the intrinsic characteristics of tumour cells using 'omics' approaches, one should be able to identify the immunosuppressive characteristics of the tumour, its antigenic profile and its capacity to succumb to immunogenic cell death. FcγR, Fc receptor for IgG; IDO, indoleamine 2,3-dioxygenase; ILT4, immunoglobulin-like transcript 4; iNOS, inducible nitric-oxide synthase; KIR, killer-cell immunoglobulin-like receptor; NKG2D, natural-killer group 2, member D; PDL1, programmed cell death ligand 1; STAT3, signal transducer and activator of transcription 3.
First, the emerging field of cancer immuno-epidemiology91 will determine which polymorphisms and mutations dictate therapeutic outcome in patients with cancer. As it stands, it seems that polymorphisms in the genes that encode TLR4, IL-10 and IL-18 may affect the therapeutic response in breast cancer84, lymphoma92 and ovarian cancer93, respectively. Similarly, polymorphisms in the Fc receptor for IgG affect the response to therapeutic monoclonal antibodies94.
Second, mechanisms of tumour-induced tolerance, including suppressor cells (such as TReg cells, T regulatory 1 cells, myeloid suppressor cells and tolerogenic DCs) or their effector mechanisms (such as TGFβ, arginase and IL-10) have to be overcome using specific therapies that may include depleting antibodies, cytokine antagonists or small molecules with immunostimulatory properties.
Third, deleterious effects that are provoked by chemotherapy or radiotherapy on the host's defence system have to be prevented or avoided. For example, the prescription of glucocorticoids during chemotherapy, as well as the ablation of tumour-draining lymph nodes, may be counterproductive because they suppress or avoid the immune responses. Similarly, a neoadjuvant therapy of radiochemotherapy may be immunologically more relevant than adjuvant therapies, and chemotherapies may have to be scheduled so that they liberate a maximum of tumour antigen while avoiding unwarranted immunosuppression.
Fourth, the intrinsic characteristics of tumour cells will have to be determined using 'omics' methods, not only to find the tumour's Achilles' heel for an optimal cytotoxic drug combination (or ideally a 'targeted' therapy) but also to determine the immunological characteristics of the tumour. Precise knowledge on the capacity of the tumour to translocate calreticulin, to expose HSP90, and to express antigen and ligands for recognition by cytotoxic effector cells may yield invaluable information for the optimal design of immunochemotherapies.
References
Zitvogel, L., Tesniere, A. & Kroemer, G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nature Rev. Immunol. 6, 715–727 (2006).
Kelland, L. R. Of mice and men: values and liabilities of the athymic nude mouse model in anticancer drug development. Eur. J. Cancer 40, 827–836 (2004).
Prendergast, G. C. & Jaffee, E. M. Cancer immunologists and cancer biologists: why we didn't talk then but need to now. Cancer Res. 67, 3500–3504 (2007).
Marzo, A. L. et al. Tumor antigens are constitutively presented in the draining lymph nodes. J. Immunol. 162, 5838–5845 (1999).
Weiner, H. L. & Cohen, J. A. Treatment of multiple sclerosis with cyclophosphamide: critical review of clinical and immunologic effects. Mult. Scler. 8, 142–154 (2002).
Weinblatt, M. E. et al. Efficacy of low-dose methotrexate in rheumatoid arthritis. N. Engl. J. Med. 312, 818–822 (1985).
Seggewiss, R. et al. Imatinib inhibits T-cell receptor-mediated T-cell proliferation and activation in a dose-dependent manner. Blood 105, 2473–2479 (2005).
Mumprecht, S., Matter, M., Pavelic, V. & Ochsenbein, A. F. Imatinib mesylate selectively impairs expansion of memory cytotoxic T cells without affecting the control of primary viral infections. Blood 108, 3406–3413 (2006). The first observation of the deleterious role of imatinib mesylate on memory T-cell responses in mice.
Mattiuzzi, G. N. et al. Development of Varicella–Zoster virus infection in patients with chronic myelogenous leukemia treated with imatinib mesylate. Clin. Cancer Res. 9, 976–980 (2003).
Galon, J. et al. Gene profiling reveals unknown enhancing and suppressive actions of glucocorticoids on immune cells. FASEB J. 16, 61–71 (2002).
Rozkova, D., Horvath, R., Bartunkova, J. & Spisek, R. Glucocorticoids severely impair differentiation and antigen presenting function of dendritic cells despite upregulation of Toll-like receptors. Clin. Immunol. 120, 260–271 (2006).
Chiossone, L. et al. Molecular analysis of the methylprednisolone-mediated inhibition of NK-cell function: evidence for different susceptibility of IL-2- versus IL-15-activated NK cells. Blood 109, 3767–3775 (2007).
Obeid, M. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nature Med. 13, 54–61 (2007). The first demonstration of the role of cell-surface calreticulin in the uptake of dying tumour cells by DCs and, therefore, its involvement in antitumour immune responses.
Grunhagen, D. J. et al. One hundred consecutive isolated limb perfusions with TNF-α and melphalan in melanoma patients with multiple in-transit metastases. Ann. Surg. 240, 939–947; discussion 947–948 (2004).
Thom, A. K. et al. Cytokine levels and systemic toxicity in patients undergoing isolated limb perfusion with high-dose tumor necrosis factor, interferon γ, and melphalan. J. Clin. Oncol. 13, 264–273 (1995).
Lin, W. W. & Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J. Clin. Invest. 117, 1175–1183 (2007).
Morgan, D. J., Kreuwel, H. T. & Sherman, L. A. Antigen concentration and precursor frequency determine the rate of CD8+ T cell tolerance to peripherally expressed antigens. J. Immunol. 163, 723–727 (1999).
Gasser, S., Orsulic, S., Brown, E. J. & Raulet, D. H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 436, 1186–1190 (2005). The important link between the intrinsic and the extrinsic tumour-suppressor mechanisms is highlighted by showing that the DNA-damage response induces expression of NKG2D ligands in an ATM- or ATR-dependent manner.
Xue, W. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007). This paper shows the pivotal role of p53 in established and ongoing tumorigenesis. Restoration of p53 function in established tumours leads to tumour regression by promoting tumour-cell senescence and an inflammatory cascade, leading to the recruitment of neutrophils, macrophages and NK cells.
Hareyama, M. et al. Effect of radiation on the expression of carcinoembryonic antigen on the membranes of human gastric adenocarcinoma cells — immunological study using monoclonal antibodies. Nippon Igaku Hoshasen Gakkai Zasshi 48, 1572–1574 (1988).
Gaugler, M. H. et al. Late and persistent up-regulation of intercellular adhesion molecule-1 (ICAM-1) expression by ionizing radiation in human endothelial cells in vitro. Int. J. Radiat. Biol. 72, 201–209 (1997).
Garnett, C. T. et al. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 64, 7985–7994 (2004).
Lugade, A. A. et al. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 174, 7516–7523 (2005).
Reits, E. A. et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 203, 1259–1271 (2006). This paper describes the immunological side effects of irradiation in vivo , showing that distant tumours can regress as a result of local irradiation and the synergistic antitumour effects between adoptive cell therapy and irradiation. Also, a comprehensive study of antigen processing and the presentation machinery following irradiation of tumour cells is provided.
Milas, L. et al. CpG oligodeoxynucleotide enhances tumor response to radiation. Cancer Res. 64, 5074–5077 (2004).
Muranski, P. et al. Increased intensity lymphodepletion and adoptive immunotherapy — how far can we go? Nature Clin. Pract. Oncol. 3, 668–681 (2006).
Dudley, M. E. et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298, 850–854 (2002). A seminal clinical demonstration that lymphodepletion followed by adoptive T-cell therapy can have a positive impact on advanced melanomas.
North, R. J. Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J. Exp. Med. 155, 1063–1074 (1982). This study shows that cyclophosphamide can inhibit tumour-induced suppressor T cells, thereby working in synergy with adoptive cell transfer.
Dudley, M. E. et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J. Clin. Oncol. 23, 2346–2357 (2005).
Paulos, C. M. et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J. Clin. Invest. 117, 2197–2204 (2007).
Gattinoni, L. et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J. Exp. Med. 202, 907–912 (2005).
Badovinac, V. P., Messingham, K. A., Jabbari, A., Haring, J. S. & Harty, J. T. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nature Med. 11, 748–756 (2005).
Wrzesinski, C. et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J. Clin. Invest. 117, 492–501 (2007). An important observation indicating the clinical benefit expected when combining vaccination and adoptive cell transfer after lymphodepletion.
Turk, J. L. & Parker, D. Effect of cyclophosphamide on immunological control mechanisms. Immunol. Rev. 65, 99–113 (1982).
Lutsiak, M. E. et al. Inhibition of CD4+25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 105, 2862–2868 (2005).
Schiavoni, G. et al. Cyclophosphamide induces type I interferon and augments the number of CD44hi T lymphocytes in mice: implications for strategies of chemoimmunotherapy of cancer. Blood 95, 2024–2030 (2000). The first demonstration that low doses of cyclophosphamide can promote type I IFN secretion.
Glaser, M. Regulation of specific cell-mediated cytotoxic response against SV40-induced tumor associated antigens by depletion of suppressor T cells with cyclophosphamide in mice. J. Exp. Med. 149, 774–779 (1979).
Sutmuller, R. P. et al. Synergism of cytotoxic T lymphocyte-associated antigen 4 blockade and depletion of CD25+ regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J. Exp. Med. 194, 823–832 (2001).
Shimizu, J., Yamazaki, S. & Sakaguchi, S. Induction of tumor immunity by removing CD25+CD4+ T cells: a common basis between tumor immunity and autoimmunity. J. Immunol. 163, 5211–5218 (1999).
MacLean, G. D., Miles, D. W., Rubens, R. D., Reddish, M. A. & Longenecker, B. M. Enhancing the effect of THERATOPE STn–KLH cancer vaccine in patients with metastatic breast cancer by pretreatment with low-dose intravenous cyclophosphamide. J. Immunother. Emphasis Tumor. Immunol. 19, 309–316 (1996).
Berd, D., Maguire, H. C. Jr & Mastrangelo, M. J. Induction of cell-mediated immunity to autologous melanoma cells and regression of metastases after treatment with a melanoma cell vaccine preceded by cyclophosphamide. Cancer Res. 46, 2572–2577 (1986).
Berd, D. & Mastrangelo, M. J. Effect of low dose cyclophosphamide on the immune system of cancer patients: depletion of CD4+, 2H4+ suppressor-inducer T-cells. Cancer Res. 48, 1671–1675 (1988).
Ghiringhelli, F. et al. CD4+CD25+ regulatory T cells inhibit natural killer cell functions in a transforming growth factor-β-dependent manner. J. Exp. Med. 202, 1075–1085 (2005). The first demonstration of the inhibitory effect of T Reg cells on NK-cell responses in mice and humans in vitro and in vivo.
Ghiringhelli, F. et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 56, 641–648 (2007). A clinical protocol testing the dose of oral cyclophosphamide needed to control end-stage tumours associated with restoration of T-cell and NK-cell functions in patients.
Orsini, F., Pavelic, Z. & Mihich, E. Increased primary cell-mediated immunity in culture subsequent to adriamycin or daunorubicin treatment of spleen donor mice. Cancer Res. 37, 1719–1726 (1977). A pioneering study highlighting the indirect role of anthracyclines in boosting cellular immunity.
Arinaga, S., Akiyoshi, T. & Tsuji, H. Augmentation of the generation of cell-mediated cytotoxicity after a single dose of adriamycin in cancer patients. Cancer Res. 46, 4213–4216 (1986).
Nigam, A. et al. Immunomodulatory properties of antineoplastic drugs administered in conjunction with GM-CSF-secreting cancer cell vaccines. Int. J. Oncol. 12, 161–170 (1998).
Zagozdzon, R. et al. Effective chemo-immunotherapy of L1210 leukemia in vivo using interleukin-12 combined with doxorubicin but not with cyclophosphamide, paclitaxel or cisplatin. Int. J. Cancer 77, 720–727 (1998).
Casares, N. et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 202, 1691–1701 (2005).
Byrd-Leifer, C. A., Block, E. F., Takeda, K., Akira, S. & Ding, A. The role of MyD88 and TLR4 in the LPS-mimetic activity of Taxol. Eur. J. Immunol. 31, 2448–2457 (2001).
Machiels, J. P. et al. Cyclophosphamide, doxorubicin, and paclitaxel enhance the antitumor immune response of granulocyte/macrophage-colony stimulating factor-secreting whole-cell vaccines in HER-2/neu tolerized mice. Cancer Res. 61, 3689–3697 (2001). Interesting and elegant work providing evidence for the synergistic antitumour effects of genetically modified tumour vaccines and chemotherapy, both overcoming tumour-induced tolerance.
Yu, B. et al. Effective combination of chemotherapy and dendritic cell administration for the treatment of advanced-stage experimental breast cancer. Clin. Cancer Res. 9, 285–294 (2003).
Chu, Y. et al. Efficacy of GM-CSF-producing tumor vaccine after docetaxel chemotherapy in mice bearing established Lewis lung carcinoma. J. Immunother. 29, 367–380 (2006).
Carson, W. E. 3rd, Shapiro, C. L., Crespin, T. R., Thornton, L. M. & Andersen, B. L. Cellular immunity in breast cancer patients completing taxane treatment. Clin. Cancer Res. 10, 3401–3409 (2004).
Nowak, A. K., Robinson, B. W. & Lake, R. A. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemo-immunotherapy. Cancer Res. 62, 2353–2358 (2002).
Qin, Z. et al. B cells inhibit induction of T cell-dependent tumor immunity. Nature Med. 4, 627–630 (1998).
Suzuki, E., Kapoor, V., Jassar, A. S., Kaiser, L. R. & Albelda, S. M. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin. Cancer Res. 11, 6713–6721 (2005).
Nowak, A. K. et al. Induction of tumor cell apoptosis in vivo increases tumor antigen cross-presentation, cross-priming rather than cross-tolerizing host tumor-specific CD8 T cells. J. Immunol. 170, 4905–4913 (2003).
Nowak, A. K., Robinson, B. W. & Lake, R. A. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. 63, 4490–4496 (2003). The first study to delineate the idea that chemotherapy-induced cell death can be immunogenic rather than tolerogenic in tumour-bearing mice.
Plate, J. M., Plate, A. E., Shott, S., Bograd, S. & Harris, J. E. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol. Immunother. 54, 915–925 (2005).
Levitt, M. L. et al. Phase I study of gemcitabine given weekly as a short infusion for non-small cell lung cancer: results and possible immune system-related mechanisms. Lung Cancer 43, 335–344 (2004).
Correale, P. et al. Chemo-immunotherapy of metastatic colorectal carcinoma with gemcitabine plus FOLFOX 4 followed by subcutaneous granulocyte macrophage colony-stimulating factor and interleukin-2 induces strong immunologic and antitumor activity in metastatic colon cancer patients. J. Clin. Oncol. 23, 8950–8958 (2005).
Galetto, A. et al. Drug- and cell-mediated antitumor cytotoxicities modulate cross-presentation of tumor antigens by myeloid dendritic cells. Anticancer Drugs 14, 833–843 (2003).
Tanaka, F. et al. Intratumoral injection of dendritic cells after treatment of anticancer drugs induces tumor-specific antitumor effect in vivo. Int. J. Cancer 101, 265–269 (2002).
Correale, P. et al. 5-fluorouracil-based chemotherapy enhances the antitumor activity of a thymidylate synthase-directed polyepitopic peptide vaccine. J. Natl Cancer Inst. 97, 1437–1445 (2005).
Lubbert, M. DNA methylation inhibitors in the treatment of leukemias, myelodysplastic syndromes and hemoglobinopathies: clinical results and possible mechanisms of action. Curr. Top. Microbiol. Immunol. 249, 135–164 (2000).
Serrano, A. et al. Rexpression of HLA class I antigens and restoration of antigen-specific CTL response in melanoma cells following 5-aza-2′-deoxycytidine treatment. Int. J. Cancer 94, 243–251 (2001).
Kozar, K. et al. Interleukin 12-based immunotherapy improves the antitumor effectiveness of a low-dose 5-Aza-2′-deoxycitidine treatment in L1210 leukemia and B16F10 melanoma models in mice. Clin. Cancer Res. 9, 3124–3133 (2003).
Baguley, B. C. Antivascular therapy of cancer: DMXAA. Lancet Oncol. 4, 141–148 (2003).
Jassar, A. S. et al. Activation of tumor-associated macrophages by the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid induces an effective CD8+ T-cell-mediated antitumor immune response in murine models of lung cancer and mesothelioma. Cancer Res. 65, 11752–11761 (2005).
Roberts, Z. J. et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J. Exp. Med. 204, 1559–1569 (2007). References 69 – 71 highlight the unexpected immunological side effects of the flavonoids.
Borg, C. et al. Novel mode of action of c-kit tyrosine kinase inhibitors leading to NK cell-dependent antitumor effects. J. Clin. Invest. 114, 379–388 (2004).
Fernandez, N. C. et al. Dendritic cells directly trigger NK cell functions: cross-talk relevant in innate anti-tumor immune responses in vivo. Nature Med. 5, 405–411 (1999).
Taieb, J. et al. A novel dendritic cell subset involved in tumor immunosurveillance. Nature Med. 12, 214–219 (2006). References 72 and 74 highlight the role of imatinib mesylate in enhancing innate immune responses, leading to the regression of NK-cell-dependent tumours.
Lake, R. A. & Robinson, B. W. Immunotherapy and chemotherapy — a practical partnership. Nature Rev. Cancer 5, 397–405 (2005).
Somersan, S. et al. Primary tumor tissue lysates are enriched in heat shock proteins and induce the maturation of human dendritic cells. J. Immunol. 167, 4844–4852 (2001).
Srivastava, P. K. Immunotherapy for human cancer using heat shock protein-peptide complexes. Curr. Oncol. Rep. 7, 104–108 (2005).
Binder, R. J. & Srivastava, P. K. Peptides chaperoned by heat-shock proteins are a necessary and sufficient source of antigen in the cross-priming of CD8+ T cells. Nature Immunol. 6, 593–599 (2005).
Li, Z. et al. Combination of imatinib mesylate with autologous leukocyte-derived heat shock protein and chronic myelogenous leukemia. Clin. Cancer Res. 11, 4460–4468 (2005).
Spisek, R. et al. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood 109, 4839–4845 (2007).
Gardai, S. J. et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334 (2005).
Obeid, M. et al. Calreticulin exposure is required for the immunogenicity of γ-irradiation and UVC light-induced apoptosis. Cell Death Differ. 14, 1848–1850 (2007).
Obeid, M. et al. Ecto-calreticulin in immunogenic chemotherapy. Immunol. Rev. 220, 22–34 (2007)
Apetoh, L. et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nature Med. 13, 1050–1059 (2007). The first demonstration of a role for TLR4 and HMGB1 in the antitumour effects that are mediated by chemotherapy and radiotherapy.
Apetoh L et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol. Rev. 220, 47–59 (2007).
Shiratsuchi, A., Watanabe, I., Takeuchi, O., Akira, S. & Nakanishi, Y. Inhibitory effect of Toll-like receptor 4 on fusion between phagosomes and endosomes/lysosomes in macrophages. J. Immunol. 172, 2039–2047 (2004).
Antonia, S. J. et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin. Cancer Res. 12, 878–887 (2006).
Gribben, J. G. et al. Unexpected association between induction of immunity to the universal tumor antigen CYP1B1 and response to next therapy. Clin. Cancer Res. 11, 4430–4436 (2005).
Arlen, P. M. et al. A randomized phase II study of concurrent docetaxel plus vaccine versus vaccine alone in metastatic androgen-independent prostate cancer. Clin, Cancer Res. 12, 1260–1269 (2006).
Noguchi, M. et al. Immunological evaluation of individualized peptide vaccination with a low dose of estramustine for HLA-A24+ HRPC patients. Prostate 63, 1–12 (2005).
Sun, T. et al. FASL -844C polymorphism is associated with increased activation-induced T cell death and risk of cervical cancer. J. Exp. Med. 202, 967–974 (2005).
Lech-Maranda, E. et al. Interleukin-10 gene promoter polymorphisms influence the clinical outcome of diffuse large B-cell lymphoma. Blood 103, 3529–3534 (2004).
Bushley, A. W. et al. Polymorphisms of interleukin (IL)-1α, IL-1β, IL-6, IL-10, and IL-18 and the risk of ovarian cancer. Gynecol. Oncol. 95, 672–679 (2004).
Nimmerjahn, F. & Ravetch, J. V. Antibodies, Fc receptors and cancer. Curr. Opin. Immunol. 19, 239–245 (2007).
Kroemer, G. et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 12 (Suppl. 2), 1463–1467 (2005).
Zitvogel, L. et al. Immune response against dying tumor cells. Adv. Immunol. 84, 131–179 (2004).
Claude, L. et al. Lymphopenia: a new independent prognostic factor for survival in patients treated with whole brain radiotherapy for brain metastases from breast carcinoma. Radiother. Oncol. 76, 334–339 (2005).
Schuerwegh, A. J., van Offel, J. F., Bridts, C. H., Stevens, W. J. & De Clerck, L. S. Influence of longterm therapy with methotrexate and low dose corticosteroids on type 1 and type 2 cytokine production in CD4+ and CD8+ T lymphocytes of patients with rheumatoid arthritis. J. Rheumatol. 28, 1793–1799 (2001).
Panici, P. B. et al. Systematic aortic and pelvic lymphadenectomy versus resection of bulky nodes only in optimally debulked advanced ovarian cancer: a randomized clinical trial. J. Natl Cancer Inst. 97, 560–566 (2005).
Chan, O. T. & Yang, L. X. The immunological effects of taxanes. Cancer Immunol. Immunother. 49, 181–185 (2000).
Chakraborty, M. et al. External beam radiation of tumors alters phenotype of tumor cells to render them susceptible to vaccine-mediated T-cell killing. Cancer Res. 64, 4328–4337 (2004).
Cho, B. K., Rao, V. P., Ge, Q., Eisen, H. N. & Chen, J. Homeostasis-stimulated proliferation drives naive T cells to differentiate directly into memory T cells. J. Exp. Med. 192, 549–556 (2000).
Ge, Q., Hu, H., Eisen, H. N. & Chen, J. Different contributions of thymopoiesis and homeostasis-driven proliferation to the reconstitution of naive and memory T cell compartments. Proc. Natl Acad. Sci. USA 99, 2989–2994 (2002).
Ghiringhelli, F. et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 34, 336–344 (2004).
Mullins, D. W., Burger, C. J. & Elgert, K. D. Paclitaxel enhances macrophage IL-12 production in tumor-bearing hosts through nitric oxide. J. Immunol. 162, 6811–6818 (1999).
Yuan, L. et al. Restoration of macrophage tumoricidal activity by bleomycin correlates with the decreased production of transforming growth factor β in rats bearing KDH-8 hepatoma cells. Cancer Immunol. Immunother. 45, 71–76 (1997).
Hornung, R. L., Young, H. A., Urba, W. J. & Wiltrout, R. H. Immunomodulation of natural killer cell activity by flavone acetic acid: occurrence via induction of interferon α/β. J. Natl Cancer Inst. 80, 1226–1231 (1988).
Keilholz, U. et al. Dacarbazine, cisplatin, and interferon-α2b with or without interleukin-2 in metastatic melanoma: a randomized phase III trial (18951) of the European Organisation for Research and Treatment of Cancer Melanoma Group. J. Clin. Oncol. 23, 6747–6755 (2005).
Bajetta, E. et al. Multicenter phase III randomized trial of polychemotherapy (CVD regimen) versus the same chemotherapy (CT) plus subcutaneous interleukin-2 and interferon-α2b in metastatic melanoma. Ann. Oncol. 17, 571–577 (2006).
Massacesi, C., Burattini, L., Marcucci, F. & Bonsignori, M. Short communication: the efficacy of fixed dose rate infusion of gemcitabine combined with IFN-α2a in patients with advanced refractory renal cell carcinoma. J. Interferon Cytokine Res. 25, 165–168 (2005).
Atzpodien, J. et al. Interleukin-2- and interferon α2a-based immunochemotherapy in advanced renal cell carcinoma: a prospectively randomized trial of the German Cooperative Renal Carcinoma Chemoimmunotherapy Group (DGCIN). J. Clin. Oncol. 22, 1188–1194 (2004).
Parra, H. S. et al. Combined regimen of cisplatin, doxorubicin, and α-2b interferon in the treatment of advanced malignant pleural mesothelioma: a Phase II multicenter trial of the Italian Group on Rare Tumors (GITR) and the Italian Lung Cancer Task Force (FONICAP). Cancer 92, 650–656 (2001).
Ishida, A. et al. Intrapleural cisplatin and OK432 therapy for malignant pleural effusion caused by non-small cell lung cancer. Respirology 11, 90–97 (2006).
Kasamon, Y. L. et al. Phase I study of low-dose interleukin-2, fludarabine, and cyclophosphamide for previously untreated indolent lymphoma and chronic lymphocytic leukemia. Clin. Cancer Res. 11, 8413–8417 (2005).
Gomez, G. G., Hutchison, R. B. & Kruse, C. A. Chemo-immunotherapy and chemo-adoptive immunotherapy of cancer. Cancer Treat. Rev. 27, 375–402 (2001).
Ehrlich, P. Über den jetztigen Stand der Karzinomforschung. Ned.Tijdschr. Geneeskd. 5, 273–290, 1909 (in German).
Burnet, M. Cancer; a biological approach. I. The processes of control. BMJ 1, 779–786 (1957).
van den Broek, M. E. et al. Decreased tumor surveillance in perforin-deficient mice. J. Exp. Med. 184, 1781–1790 (1996).
Shankaran, V. et al. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 410, 1107–1111 (2001).
Galon, J. et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964 (2006).
Acknowledgements
The authors are supported by grants from the Ligue Nationale contre le Cancer (L.Z., G.K. and L.A.), the European Union (ALLOSTEM, DC-THERA; L.Z.), Cancéropôle Île-de-France, Institut National du Cancer and Agence Nationale pour la Recherche (G.K.). F.G. is supported by a Poste d'acceuil INSERM.
Author information
Authors and Affiliations
Corresponding authors
Related links
FURTHER INFORMATION
Glossary
- Cancer stem cells
-
A small population of undifferentiated cells from which the differentiating cancer cells originate. These cells are suspected to account for relapse after conventional therapy.
- Oncogene
-
A gene of which the overexpression or gain-of-function mutation contributes to oncogenesis.
- Tumour-suppressor gene
-
A gene that, when eliminated or inactivated, is permissive for the development of cancers. These genes often determine cell-cycle checkpoints or facilitate induction of programmed cell death.
- Genome-stability genes
-
Genes that control cell-cycle advancement and/or DNA repair to allow for the maintenance of genome stability.
- Neoplasia
-
From the Greek for 'new formations'. New growths or tumours, which can be malignant.
- Pattern-recognition receptors
-
Host receptors (such as Toll-like receptors) that are able to sense pathogen-associated molecular patterns and initiate signalling cascades (involving activation of nuclear factor-κB) that lead to an innate immune response.
- TH1 cells
-
(T helper 1 cells). Among the two well-described subsets of activated CD4+ T cells, TH1 cells produce interferon-γ and tumour-necrosis factor and enhance cell-mediated immunity. TH2 cells produce interleukin-4 (IL-4), IL-5 and IL-13, supporting humoral immunity and counteracting TH1-cell responses.
- NKG2D
-
(Natural-killer group 2, member D). A lectin-type activating receptor encoded by the natural killer (NK)-cell gene complex and expressed on the surface of NK, NKT, γδ T cells and some cytolytic CD8+ αβ T cells. NKG2D ligands are MHC-class-I-polypeptide-related sequence A (MICA) and MICB in humans, as well as retinoic acid early transcript 1 (RAE1) and H60 in mice. Such ligands are generally expressed at the cell surface of infected, stressed or transformed cells.
- Endoplasmic reticulum stress
-
(ER stress). A response by the ER that results in the disruption of protein folding and in the accumulation of unfolded proteins in the ER.
- Eat-me signal
-
A signal emitted by dying cells to facilitate their recognition and phagocytosis by neighbouring healthy cells.
- Isolated limb perfusion
-
(ILP). A surgical technique consisting of injection of chemotherapeutic agents into the artery of an extremity while the venous outflow is recovered, thus avoiding systemic drug effects.
- p53
-
A major transcription factor that is activated by numerous genotoxic insults to induce cell-cycle arrest, cellular senescence or apoptosis. p53 is frequently mutated or functionally inactivated in cancer.
- DNA-damage response
-
A cellular response that is usually elicited by DNA-damaging agents (such as ionizing irradiation or mutagenic chemicals) and involves the activation of DNA-damage foci (with phosphorylated histone H2AX as a hallmark). The DNA-damage response elicits cell-cycle arrest, DNA repair or apoptosis.
- NKT cells
-
(Natural-killer T cells). A heterogeneous subset of T cells that are characterized by the co-expression of semi-invariant T-cell receptor (TCR) α-chains together with NK-cell markers.
- Senescence
-
A nearly irreversible stage of permanent G1 cell-cycle arrest, linked to morphological changes (flattening of the cells), metabolic changes and changes in gene expression (with expression of senescence-associated β-galactosidase), the induction of which depends on p53 and cell-cycle-blockers such as p21 and p16.
- Abscopal effects
-
Distant antitumour effects seen after local radiation therapy.
- TReg cell
-
(Regulatory T cell). A specialized type of CD4+ T cell that can suppress the responses of other T cells. These cells provide a crucial mechanism for the maintenance of peripheral self-tolerance and are characterized by the expression of CD25 (also known as the α-chain of the interleukin-2 receptor) and the transcription factor forkhead box P3 (FOXP3).
- HER2
-
(Human epidermal growth-factor receptor 2). A receptor protein-tyrosine kinase that is overexpressed in a subset of human breast cancers.
- 3LL tumour model
-
3LL is a non-small-cell lung cancer cell line that grows in vivo after injection into C57BL/6 syngenic hosts.
- Myeloid suppressor cells
-
A group of immature CD11b+GR1+ cells (which include precursors of macrophages, granulocytes, dendritic cells and myeloid cells) that are produced in response to various tumour-derived cytokines. These cells have been shown to induce tumour-associated antigen-specific CD8+ T-cell tolerance.
- Myelodysplastic syndrome
-
A pre-neoplastic syndrome characterized by a hypercellular bone marrow with reduced haematopoietic capacity.
- Vascular-disrupting agents
-
In contrast to anti-angiogenic approaches, which aim to prevent the neovascularization processes in tumours, vascular-disrupting agents aim to cause the rapid and selective shutdown of the established tumour vasculature, leading to secondary tumour-cell death.
- Angiogenesis
-
The development of new blood vessels from existing blood vessels. It is frequently associated with tumour development and inflammation.
- IKDCs
-
(Interferon-producing killer dendritic cells). Isolated in mouse models only, these cells express both NK-cell and B-cell markers but lack plasmacytoid DC- and T-cell-specific, and co-stimulatory molecules. They react to a large variety of tumour cells by producing interferon-γ and killing the tumour cells without exogenous stimulation.
- Death receptors
-
A family of cell-surface receptors capable of mediating cell death on ligand-induced trimerization. The best-studied members include tumour-necrosis factor receptor 1 (TNFR1), FAS (or CD95, which binds FAS ligand) and two receptors for TNF-related apoptosis-inducing ligand (TRAILR1 and TRAILR2).
- Cross-priming
-
Initiation of a CD8+ T-cell response against an antigen that is not present in antigen-presenting cells (APCs). The antigen must be taken up by APCs and then re-routed to the MHC class I presentation pathway.
- Small interfering RNAs
-
(siRNA). Synthetic RNA molecules of 19–23 nucleotides that are used to 'knock down' (that is, to silence the expression of) a specific gene. This is known as RNA interference (RNAi) and is mediated by the sequence-specific degradation of mRNA.
- Neoadjuvant therapy
-
Radiotherapy and chemotherapy before surgical resection of the tumour.
- Adjuvant therapy
-
Radiotherapy and chemotherapy after surgical resection of the primary tumour.
Rights and permissions
About this article
Cite this article
Zitvogel, L., Apetoh, L., Ghiringhelli, F. et al. Immunological aspects of cancer chemotherapy. Nat Rev Immunol 8, 59–73 (2008). https://doi.org/10.1038/nri2216
Issue Date:
DOI: https://doi.org/10.1038/nri2216
This article is cited by
-
Tumor-infiltrating lymphocytes in HER2-positive breast cancer treated with neoadjuvant chemotherapy and dual HER2-blockade
npj Breast Cancer (2024)
-
Pruritus related to trastuzumab and pertuzumab in HER2 + breast cancer patients
Breast Cancer Research and Treatment (2024)
-
STING-targeted PET tracer for early assessment of tumor immunogenicity in colorectal cancer after chemotherapy
European Journal of Nuclear Medicine and Molecular Imaging (2024)
-
Carbon nanoparticles-Fe(II) complex for efficient theranostics of xenografted colonic tumor
Cancer Nanotechnology (2023)
-
Clinical implications of aberrant PD-1 expression for acute leukemia prognosis
European Journal of Medical Research (2023)
