
Abstract
The recognition of immune epitopes is an important molecular mechanism of the vertebrate immune system to discriminate between self and non-self. Increasing amounts of data on immune epitopes are becoming available due to technological advances in epitope-mapping techniques and the availability of genomic information for pathogens. Organizing this data poses a challenge that is similar to the successful effort that was required to organize genomic data, which needed the establishment of centralized databases that complement the primary literature to make the data readily accessible and searchable by researchers. As described in this Innovation article, the Immune Epitope Database and Analysis Resource aims to achieve the same for the more complex and context-dependent information on immune epitopes, and to integrate this data with existing and emerging knowledge resources.
Similar content being viewed by others

Main
In this age of information- and technology-driven research, keeping up with the large amounts of published data is overwhelming for any researcher, particularly in areas not related to their primary expertise. To benefit from published data, it is increasingly stored in dedicated searchable databases. There is a family of such established databases, which includes SwissProt, the Research Collaboratory for Structural Bioinformatics (RCSB) Protein Data Bank (PDB) and the National Center for Biotechnology Information (NCBI) databases PubMed, GenBank and Taxonomy Browser (Box 1), all of which can claim to be widely used as the major source of information in their domains. Today, researchers are much more likely to retrieve a protein sequence from SwissProt or GenBank, rather than look up these sequences in the primary publication. For new references, such information would not even be found in the primary literature, as it is now mandatory to deposit sequences in a database before publication. Importantly, these knowledge resources can easily be interlinked, making it possible to proceed from a protein sequence to its crystal structure, to its related source organism and to the literature references that describe it. With the emergence and consolidation of new databases, this information will expand to include single-nucleotide polymorphisms (SNPs), biomedical imaging and disease association, as well as immune epitope data, such as in the Immune Epitope Database and Analysis Resource (IEDB), which is the focus of this article.
Several databases devoted to immune-epitope-related information have been established before the recently developed IEDB, such as SYFPEITHI1, the International ImMunoGeneTics information system (IMGT)2, AntiJen3, φIMM4, MHCBN5 and HLA Ligand Database6, as well as pathogen-specific resources, such as the HIV database7 and the HCV database8 hosted at the Los Alamos National Laboratory, New Mexico, USA (see Further Information for web sites). Clearly, the design of the IEDB9,10 would not have been possible without this pioneering work of others. The development and application of immuno-informatic databases and tools continues to be a very active and exciting field of research, as evidenced by two recent reviews of the field11,12. Although the focus of this Innovation article is on the IEDB, we want to acknowledge that its success is based and is dependent on contributions of a much larger scientific community.
The IEDB is part of the National Institute of Allergy and Infectious Diseases (NIAID) biodefence programme. The IEDB is designed to organize the ever-growing body of information related to immune epitopes that are recognized by T cells and antibodies from humans, non-human primates and laboratory animals. The current focus of the database is on NIAID Category A, B and C Priority Pathogens13, which include influenza A virus, and emerging and re-emerging infectious pathogens, such as Bacillus anthracis, Ebola virus, West Nile virus, Nipah virus and severe acute respiratory syndrome (SARS)-associated coronavirus. Epitopes from other infectious pathogens, allergens and those involved in autoimmunity are also within the scope of the IEDB. The users of the database, which range from clinicians and vaccinologists to basic researchers, are able to freely access all of the information related to each epitope. This includes not only structural information related to the chemical nature of the epitope, but also taxonomic information related to the source of the epitope and contextual information related to the host recognizing the epitope, the conditions associated with immunization and the type of assay used to detect the responses. This rich description of the multiple contexts (each corresponding to a record) in which each epitope was reported to be recognized is important because it allows the researcher to ask specific questions. For example, which T-cell epitopes were recognized in macaques, are derived from SARS and are associated with interferon release, after recognition of infected cells in vitro? Currently, the IEDB contains data derived from over 3,200 published papers, relating to approximately 26,000 different epitopes, and approximately 40 new papers are added weekly14.
In addition to hosting data, the IEDB also hosts a collection of bioinformatics tools that can be used to predict B-cell and T-cell epitopes and to analyse responses. For example, the degree of conservation of a given epitope in different pathogen isolates can be examined, or the three-dimensional structure of epitopes in their native antigen can be visualized. In the design and implementation of the IEDB, new problems were encountered and new solutions devised. This Innovation article discusses the background and rationale of the development of the IEDB, illustrates how different disciplines have come together in its design and implementation, and illustrates its potential use for immunological and biological scientists.
Developing a formal ontology
The IEDB is the first epitope-related database that attempts to capture the context of immune recognition in a detailed, searchable way. We accomplished this by using several hundred different fields encompassing the database, grouped into several main classes or categories, such as the literary reference, the structure of the epitope, the source organism of the epitope and information on the context of epitope recognition, such as the host species, immunization strategy and the type of assay used to detect a response. The complexity of the data captured in the IEDB makes it difficult to ensure consistent annotation of the data by curators and accurate interpretation of the data by users. This common problem in developing a knowledge resource is addressed by developing a formal ontology. Ontologies provide exact definitions of the terms used in annotating the data, as well as their relationships, and they facilitate the integration of data from different sources. The recent creation of The National Center for Biomedical Ontologies15 will centralize and improve efforts in this area.
It is difficult to underestimate the importance of developing a formal ontology for biological processes in general and, in our case, for immune epitopes in particular. Until now, formal ontologies for host–pathogen interactions had not been developed, and accessible formal ontologies for immune-based databases had been limited to a few examples16. Developing a complete ontology requires exhaustive information on the kinds of data present in the knowledge domain. The gathering of such data must be done in a formal way, again requiring an ontology. To escape an infinite loop, it is necessary to start with an incomplete ontology that is updated over time as deficiencies become apparent as more data are collected. This is what we have done by creating the first version of an ontology of immune epitopes17, which is now progressing towards a more formal ontology (Fig. 1). This effort involves the collaboration of a consortium of groups who are working towards the development of an integrated ontology for the description of biological and medical experiments and investigations — the Ontology for Biomedical Investigations (OBI; formerly known as the Functional Genomics Investigation Ontology (FuGO) project18). In addition, we have contributed to, and benefited from, a large-scale revision of immunology-related terms in the Gene Ontology database19,20.

The initial ontology of the IEDB described all elements of the database as classes with associated characteristics (left panel shows this for the immunogen class). In the development process towards a formal ontology, these elements are placed in a hierarchy (simplified view depicted on the right), in which relationships between the different classes are made explicit. For example, the previously separate and unconnected classes Antigen, Immunogen and Adjuvant are now recognized as being objects (for example, Proteins), which participate in a certain role (as Immunogens) in a specific process (such as Immunization).
Collaboration in ontology development between different scientific communities is the key to integrate different biomedical knowledge resources. Different scientific communities often use the same term for different purposes, or different terms for the same concept, so that development of an ontology is necessary to ensure that a given term has the same meaning and associated attributes in all databases that use it, so that researchers can navigate the already immense and ever-growing body of biological data with confidence, accuracy and ease.
Automating the extraction of data
Text mining21 might at first seem to be an improbable ally for the bench immunologist. Nevertheless, we believe that this field will have a dramatic impact in immunobioinformatics and systems biology22. The field originates from the need to automate the extraction of meaning from massive amounts of text. Several of the pioneering data-mining applications were developed for non-biological purposes, such as security projects sponsored by various intelligence agencies, and for the purpose of mining data in patent applications. Over the years the field has become increasingly sophisticated. The basic premise is that of a program that scans through text and extracts data in a defined form (that is, a format that can be recognized by a computer, placed in a correct ontological format, and used by a database). In its basic, but already highly useful, form, a text is classified into one of several categories. For the IEDB, we are using such categorizations to identify abstracts listed in the PubMed database that probably contain epitope-related information — a similar approach to that which has been successfully applied by others23,24.
Ideally, one would like to go further and train a knowledge extraction algorithm to recognize complex immunological information from text, such as 'T-cell epitopes were recognized in macaques, derived from SARS and associated with interferon release'. The challenge has, however, been that currently available text-recognition programs tend to lose efficacy when interpreting complex sentences, not to mention when gathering information distributed throughout an article (for example, the fact that the immune response was observed in macaques may be found in the materials and methods, whereas the actual data may be found in a figure several pages away from the methods section). The lack of available training sets has been a significant stumbling block in progressive training towards accomplishing more complex tasks. However, the IEDB curation may offer an opportunity to make advances in the field, because thousands of different manuscripts are being curated. This provides a comprehensive set of immunological papers and matching records of curated information, which includes categories such as where in the manuscript the information was gathered from. Such datasets have proven to be invaluable in deriving ever-more potent text-mining tools25.
The IEDB
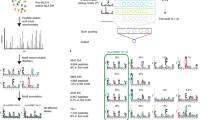
Uses and features. There are numerous ways of accessing the data in the IEDB that are tailored to different user groups. Searches come in three types: quick, simple and advanced. The quick search scans the entire text of a curated record for any occurrence of the specified search term. The simple search allows for more targeted queries without overwhelming the user with choices. It allows for the most-commonly desired types of query, such as epitopes that are recognized by T cells restricted by HLA-A*0201. In the advanced query, values for all of the more than 300 database fields can be specified, and it is also possible to customize the reporting format (Fig. 2). In addition to the search interfaces, it is also possible to browse for epitopes through the IEDB records by their MHC restriction or source species. Finally, the entire content of the IEDB is fully downloadable as a single file in XML format.

Three steps in an advanced query for B-cell epitopes are illustrated. First, criteria are specified to query for epitopes that are recognized in mice, where the immunogen applied was the epitope source species and the species is selected to be severe acute respiratory syndrome (SARS)-associated coronavirus (a). On submitting this query, a summary of epitope records matching these criteria are displayed (b). This includes information on the curated reference, epitope structure, epitope source, and assay used. When choosing the 'Details' link for a specific epitope, the complete curated information is displayed (c).
Tools and tool evaluations: outreach. Numerous tools have been developed to predict the presence of epitopes in protein sequences (see for example the listings in Ref. 11), and several groups have used them successfully to map new epitopes and for other applications (reviewed in Ref. 26). The IEDB provides several tools to predict peptide binding to MHC class I molecules, and these were recently compared to the large set of tools that are available elsewhere on the internet27. Such a large-scale comparison is meant to inform tool users of the current state of the art. For tool developers, this comparison provides a set of benchmark data with which to evaluate newly developed tools against, and it instructs them on which approaches have proven to be successful. In combination with predicting the ability of a particular peptide sequence to bind MHC class I molecules, predictions of its processing by the proteasome and transport by the transporter associated with antigen processing are also made available. These can be used to further narrow the set of candidate T-cell epitopes from a protein sequence. There is an ongoing formal evaluation of these tools that takes advantage of the data collected in the IEDB, as well as an evaluation of MHC-class-II-binding predictions. In addition to evaluations of existing servers, we also plan to hold prediction contests in which interested scientists can submit their predictions for a set of targets. Such contests have had a tremendous positive impact in the evaluation and prediction of protein structure28.
Compared with the T-cell-epitope predictions, the state of antibody-epitope predictions is widely considered to be suboptimal29. In a recent workshop sponsored by the NIAID that brought together many experts from the antibody-epitope-prediction community, this concern was widely shared, and the steps needed to improve this situation were discussed30. There was wide agreement that the field could greatly benefit from community-assembled datasets that clarify what types of epitope should be included in an evaluation and that this should be dependent on the intended use of a prediction. Similarly, the metrics used to quantify the success of predictions should be commonly agreed on for a better comparison between different studies. This effort to establish community-accepted datasets and metrics will aid in the acceptance of a newly emerging second generation of antibody-epitope prediction tools, many of which take advantage of the three-dimensional structures that are available for antibody–antigen binding.
Additional tools are provided to analyse already-identified responses. The conservancy tool (Epitope Conservancy Analysis) of the IEDB calculates the level of sequence identity with which a set of epitopes occurs across different strains of a pathogen. The population-coverage tool (Population Coverage Calculation) estimates the frequencies of responses to a set of T-cell epitopes with known MHC restrictions in different sets of populations with known MHC allele frequencies31. Finally, the epitope-homology mapping tool (Homology Mapping) visualizes the location of epitopes within the three-dimensional structure of their source antigen using a customized epitope viewer32. This mapping is done through a screening of available structures in the PDB, from which proteins are selected that closely resemble the epitope source antigen and specifically conserve the sequence of the epitope itself (Fig. 3).

For a given epitope and its source protein, the homology tool of the Immune Epitope Database and Analysis Resource identifies homologous proteins with known three-dimensional structures, and maps the location of epitopes in these structures. In this example, the peptide NTNSGPDDQIGYYRRATR (shown in blue), which is recognized by antibodies from mice immunized with inactivated severe acute respiratory syndrome (SARS)-associated coronavirus (SARS-CoV)34, is mapped to an X-ray structure of the SARS-CoV nucleocapsid protein. The arrow indicates a section of the epitope that is exposed at the surface of the virus, making it a candidate binding site for the antibody in the native protein structure.
Curation of literature on influenza virus
As an illustration of the usefulness and power of the compilation of immune-epitope-related data, we have recently curated and analysed all the published data on influenza A virus antibody and T-cell epitopes33. This effort has resulted in an inventory of the existing knowledge related to this field, and will also allow us to start probing possible crossreactivities among H5N1 avian and human influenza virus strains. The analysis revealed over 600 different influenza virus epitopes, derived from 58 different strains and 10 viral proteins. As all the data are freely available online, this effort translates into a single resource for researchers that allows access to most existing epitope data for influenza virus. For example, because of the capacity to extract data related to specific contexts, an interested scientist can selectively view epitopes that are known to be associated with protection from challenge with influenza virus. By the use of the epitope analytical tools provided by the IEDB, the degree of conservation of various epitopes in a representative set of influenza-virus sequences was determined. Several interesting, highly conserved epitopes were identified by this analysis. At the same time, the analysis can be used to probe for potential gaps in our knowledge relating to influenza-virus epitopes. Indeed, significant gaps in the current knowledge were revealed, including a paucity of antibody epitopes in comparison to T-cell epitopes (Fig. 4), a limited number of epitopes reported for avian influenza virus strains and/or subtypes, and a limited number of epitopes reported from proteins other than haemagglutinin and nucleocapsid protein. These gaps in our collective knowledge should inspire directions for further study of immunity against the influenza A virus.

After curating all journal articles published with immune epitope information, several summary analyses could be carried out. This pie chart illustrates the relative number of antibody and T-cell epitopes identified. Surprisingly, although protective immunity against the influenza virus is known to be largely mediated by antibodies, this chart reveals that much more data is available on T-cell epitopes for this virus.
Outlook and conclusions
In this Innovation article, we present the experience gained so far in a cutting edge project, which involves the extraction of complex immunological data from the literature, making it available to the scientific community and integrating it in the emerging web of biomedical knowledge resources. The issues and solutions to the challenges that have been encountered in the development of this project are of relevance in the context of the general trend of initiatives that are aimed at collecting and displaying large amounts of data of genomic and proteomic origin, and in the context of host–pathogen interactions in general. The initial phase of building and starting to populate the database with epitope information is completed. The focus now is on enhancing the value of this information to the user community. An important component of the rationale for writing this article is to involve the scientific community at large in the realisation of the best possible epitope database by stimulating debate and feedback on these issues, as changes and improvements are continuously considered and contemplated.
References
Rammensee, H., Bachmann, J., Emmerich, N. P., Bachor, O. A. & Stevanovic, S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50, 213–219. (1999).
Giudicelli, V. et al. IMGT/LIGM-DB, the IMGT comprehensive database of immunoglobulin and T cell receptor nucleotide sequences. Nucleic Acids Res. 34, D781–D784 (2006).
Toseland, C. P. et al. AntiJen: a quantitative immunology database integrating functional, thermodynamic, kinetic, biophysical, and cellular data. Immunome Res. 1, 4 (2005).
Schonbach, C., Koh, J. L., Flower, D. R. & Brusic, V. An update on the functional molecular immunology (FIMM) database. Appl. Bioinformatics 4, 25–31 (2005).
Bhasin, M., Singh, H. & Raghava, G. P. MHCBN: a comprehensive database of MHC binding and non-binding peptides. Bioinformatics 19, 665–666 (2003).
Sathiamurthy, M. et al. Population of the HLA ligand database. Tissue Antigens 61, 12–19 (2003).
HIV Molecular Immunology 2005 (eds Bette T. M. et al.) LA-UR 06–0036 (Los Alamos National Laboratory, Theoretical Biology and Biophysics, Los Alamos, New Mexico, 2005).
Yusim, K. et al. Los Alamos hepatitis C immunology database. Appl. Bioinformatics 4, 217–225 (2005).
Peters, B. et al. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol. 3, e91 (2005).
Peters, B. et al. The design and implementation of the immune epitope database and analysis resource. Immunogenetics 57, 326–336 (2005).
Korber, B., LaBute, M. & Yusim, K. Immunoinformatics comes of age. PLoS Comput. Biol. 2, e71 (2006).
Braga-Neto, U. M. & Marques, E. T. Jr. From functional genomics to functional immunomics: new challenges, old problems, big rewards. PLoS Comput. Biol. 2, e81 (2006).
NIAID Category A, B and C Priority Pathogens. [online], http://www3.niaid.nih.gov/Biodefense/PDF/cat.pdf, (2007).
Vita, R. et al. Curation of complex, context-dependent immunological data. BMC Bioinformatics 7, 341 (2006).
Rubin, D. L. et al. National Center for Biomedical Ontology: advancing biomedicine through structured organization of scientific knowledge. Omics 10, 185–198 (2006).
Lefranc, M. P. et al. IMGT-ONTOLOGY for immunogenetics and immunoinformatics. In Silico Biol. 4, 17–29 (2004).
Sathiamurthy, M. et al. An ontology for immune epitopes: application to the design of a broad scope database of immune reactivities. Immunome Res. 1, 2 (2005).
Whetzel, P. L. et al. Development of FuGO: an ontology for functional genomics investigations. Omics 10, 199–204 (2006).
Harris, M. A. et al. The Gene Ontology (GO) database and informatics resource. Nucleic Acids Res. 32, D258–D261 (2004).
Diehl, A. D., Lee, J. A., Scheuermann, R. H. & Blake, J. A. Ontology development for biological systems: Immunology. Bioinformatics 31 January 2007 (doi: 10.1093/bioinformatics/btm029).
Cohen, A. M. & Hersh, W. R. A survey of current work in biomedical text mining. Brief Bioinform. 6, 57–71 (2005).
Jensen, L. J., Saric, J. & Bork, P. Literature mining for the biologist: from information retrieval to biological discovery. Nature Rev. Genet. 7, 119–129 (2006).
Miotto, O., Tan, T. W. & Brusic, V. Supporting the curation of biological databases with reusable text mining. Genome Inform. 16, 32–44 (2005).
Donaldson, I. et al. PreBIND and Textomy—mining the biomedical literature for protein-protein interactions using a support vector machine. BMC Bioinformatics 4, 11 (2003).
Yeh, A. S., Hirschman, L. & Morgan, A. A. Evaluation of text data mining for database curation: lessons learned from the KDD Challenge Cup. Bioinformatics 19 (Suppl. 1), 331–339 (2003).
De Groot, A. S. Immunomics: discovering new targets for vaccines and therapeutics. Drug Discov. Today 11, 203–209 (2006).
Peters, B. et al. A community resource benchmarking predictions of peptide binding to MHC-I molecules. PLoS Comput. Biol. 2, e65 (2006).
Moult, J. A decade of CASP: progress, bottlenecks and prognosis in protein structure prediction. Curr. Opin. Struct. Biol. 15, 285–289 (2005).
Blythe, M. J. & Flower, D. R. Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci. 14, 246–248 (2005).
Greenbaum, J. A. et al. Towards a consensus on datasets and evaluation metrics for developing B-cell epitope prediction tools. J. Mol. Recognit. 20, 75–82 (2007).
Bui, H. H. et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics 7, 153 (2006).
Beaver, J. E., Bourne, P. E. & Ponomarenko, J. V. EpitopeViewer: a Java application for the visualization and analysis of immune epitopes in the Immune Epitope Database and Analysis Resource (IEDB). Immunome Res. 3, 3 (2007).
Bui, H. H., Peters, B., Assarsson, E., Mbawuike, I. & Sette, A. Ab and T cell epitopes of influenza A virus, knowledge and opportunities. Proc. Natl Acad. Sci. USA 104, 246–251 (2007).
He, Y. et al. Mapping of antigenic sites on the nucleocapsid protein of the severe acute respiratory syndrome coronavirus. J. Clin. Microbiol. 42, 5309–5314 (2004).
Acknowledgements
We thank J. Ponomarenko and P. Bourne at the San Diego Supercomputer Center, who developed the Homology Mapping tool. This work was supported by the National Institutes of Health, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Rights and permissions
About this article
Cite this article
Peters, B., Sette, A. Integrating epitope data into the emerging web of biomedical knowledge resources. Nat Rev Immunol 7, 485–490 (2007). https://doi.org/10.1038/nri2092
Issue Date:
DOI: https://doi.org/10.1038/nri2092
This article is cited by
-
Design of Vaccine Targeting Zika Virus Polyprotein by Immunoinformatics Technique
International Journal of Peptide Research and Therapeutics (2022)
-
Design of Monovalent and Chimeric Tetravalent Dengue Vaccine Using an Immunoinformatics Approach
International Journal of Peptide Research and Therapeutics (2021)
-
Proteasomes generate spliced epitopes by two different mechanisms and as efficiently as non-spliced epitopes
Scientific Reports (2016)
-
Computational approaches to identify common subunit vaccine candidates against bacterial meningitis
Interdisciplinary Sciences: Computational Life Sciences (2013)
-
Computer aided subunit vaccine design against pathogenic Leptospira serovars
Interdisciplinary Sciences: Computational Life Sciences (2012)
