
Key Points
-
Cancer vaccines can both generate new antigen-specific T cell responses and amplify existing responses and thereby focus the host immune response against tumour cells. Because of this ability to induce and direct a potent and tumour-specific immune response while minimizing autoimmunity, a cancer vaccine may be an effective combinatorial partner with immune checkpoint blockade or other immune therapeutics.
-
A successful cancer vaccine has four key components, each of which is crucial to its effectiveness: tumour antigens, formulations, immune adjuvants and delivery vehicles. For each of these components, various approaches have been tested preclinically and in hundreds of clinical trials in patients, with limited therapeutic success so far.
-
Neoantigens have emerged as targets of effective tumour-directed T cell responses. Increased neoantigen load is associated with improved patient outcomes, and neoantigen-reactive T cell populations expand in settings of effective immunotherapy and mediate tumour cell killing in preclinical models and patients.
-
Three clinical trials of neoantigen-based vaccines in patients with melanoma, using dendritic cells loaded with short peptides, long peptides or RNA, have shown the safety, feasibility and robust immunogenicity of this approach.
-
A crucial aspect of a vaccine targeting neoantigens is the selection of epitopes that can be presented in vivo by tumour or antigen-presenting cells. HLA-binding prediction, high-resolution mass spectrometry and understanding of antigen processing are important research areas for further discovery.
-
Optimal neoantigen delivery — use of the most effective formulations, immune adjuvants, delivery vehicles and dosing — in combination with complementary therapies will be crucial for maximum therapeutic effectiveness.
Abstract
Cancer vaccines, which are designed to amplify tumour-specific T cell responses through active immunization, have long been envisioned as a key tool of effective cancer immunotherapy. Despite a clear rationale for such vaccines, extensive past efforts were unsuccessful in mediating clinically relevant antitumour activity in humans. Recently, however, next-generation sequencing and novel bioinformatics tools have enabled the systematic discovery of tumour neoantigens, which are highly desirable immunogens because they arise from somatic mutations of the tumour and are therefore tumour specific. As a result of the diversity of tumour neoepitopes between individuals, the development of personalized cancer vaccines is warranted. Here, we review the emerging field of personalized cancer vaccination and discuss recent developments and future directions for this promising treatment strategy.
Similar content being viewed by others
Main
Methods to harness the exquisite specificity of the immune system to eliminate tumours have been under development since the start of the 20th century1,2,3. Several effective strategies have emerged over the past decade, such that immunotherapy is now widely considered to be an important additional tool for the treatment of individuals with cancer. One powerful approach is adoptive cell transfer (ACT)4,5. Autologous tumour-reactive T cells derived from tumour-infiltrating lymphocytes (TILs) and genetically engineered lymphocytes that express highly active T cell receptors (TCRs) or chimeric antigen receptors (CARs) have shown potent antitumour activity6. CAR–T cell therapy targeting the B cell antigen CD19 was approved by the US Food and Drug Administration (FDA) for childhood acute lymphoblastic leukaemia in 2017. Immune checkpoint blockade (CPB) has emerged as another clinically effective approach7,8. Monoclonal antibodies directed against the programmed cell death protein 1 (PD1) and cytotoxic T lymphocyte antigen 4 (CTLA4) signalling pathways have shown clinical efficacy in a wide range of solid and haematological malignancies, which has led to approvals by the FDA for the treatment of a rapidly growing list of tumour types7,8. The distinct immune-based strategies of ACT and CPB illustrate the promise of how cancer can be eradicated when active T cells recognize their cognate antigens.
Nevertheless, both ACT and CPB have limitations, which are reflected by the restricted patient populations that benefit from either therapy. By design, CAR–T cell therapy is directed against a single antigen target, and clinical efficacy has thus far been achieved primarily in individuals with B cell tumours, in which tumour cells are mostly uniform and express a common dominant antigen (such as CD19). Solid tumours typically lack a common surface antigen target, which poses a considerable challenge for this approach. Similarly, despite some promising results from CPB, the objective response rate (ORR) of single-agent CPB is limited to 30% in most tumour types for which activity has been shown9,10,11 (exceptions include microsatellite-instable tumours12, Merkel cell carcinoma13 and Hodgkin lymphoma14, for which ORRs of CPB are 50–80%). Furthermore, antitumour activity of CPB has been reported as being absent to minimal in several types of malignancy, including microsatellite-stable colorectal cancer15 and pancreatic cancer16.
Therefore, increasing research attention has shifted to understanding the biological basis of these variable responses and to identifying biomarkers that can predict which patients are likely to respond or not respond to these therapies. Currently, a large number of clinical trials assessing combination therapies, which are typically based on PD1 targeting, are under way17. For CPB, growing evidence supports the idea that individuals with tumours that lack pre-existing TILs are less likely to respond to therapy. However, the presence of pre-existing T cell responses at the site of the tumour does not guarantee an antitumour response18. One approach to increasing the effectiveness of CPB is the combined administration of PD1-blocking antibodies and CTLA4-blocking antibodies, which has been approved for the treatment of melanoma and is being investigated in many other malignancies. Although this approach can increase response rates in individuals with advanced melanoma, it also leads to substantially higher toxicity19,20. Clearly, alternative approaches are needed to achieve maximal benefit from these agents while minimizing toxicity.
A rational approach to achieve this goal is to combine CPB with a therapy that can presensitize the host immune system to the tumour. Given their potential to both generate new antigen-specific T cell responses against tumour cells and amplify existing responses, cancer vaccines may be an effective combinatorial partner with CPB (Fig. 1). By selecting suitable antigen targets, a potent and tumour-specific immune response can be induced while minimizing autoimmunity. Recent studies have shown that tumour neoantigens are key targets for ACT, CPB and therapeutic vaccination21,22,23,24,25,26,27,28. In this Review, we focus on the current development of neoantigen-based, personalized, tumour-specific, therapeutic vaccines for cancer.

a | Cancer vaccines can select suitable antigen targets to generate new antigen-specific T cell responses against tumour cells. b | Cancer vaccines can also amplify existing tumour-specific T cell responses. c | Finally, cancer vaccines can increase the breadth and diversity of the tumour-specific T cell response. Together, these effects can result in regression of tumours.
Building cancer vaccines
Increased knowledge of methods for antigen discovery and of soluble immunomodulatory factors has led to our current understanding of the key cellular components that are involved in mounting effective antitumour immune responses, as extensively reviewed elsewhere29,30. Fundamental to the generation of an antitumour immune response is the activation or priming of naive antigen-specific T cells by professional antigen-presenting cells (APCs), such as dendritic cells (DCs), which requires interactions between diverse cell types and distinct tissue compartments31 (Fig. 2a). In both mice and humans, there has been a growing appreciation of the various defects at the level of antigen processing and presentation that occur in tumour-bearing hosts32, as well as of the impaired functionality of tumour-specific T cells within the immunosuppressive tumour microenvironment due to various mechanisms of primary immune resistance and adaptive immune resistance33. Tumour heterogeneity between and within tumours34 is also a major challenge to the development of cancer immunotherapy; a crucial factor in this regard is the clonal evolution of tumour cells arising from selective pressure, which leads some mutant clones to expand, whereas it leads others to become extinct35.

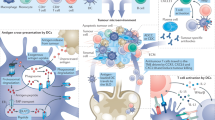
a | The tumour antigen presentation process. As a first step, antigen encounter by antigen-presenting cells (APCs) such as dendritic cells (DCs) occurs at the injection site (or, in the case of DC vaccines, antigens may be exogenously loaded on APCs before injection). The antigen-loaded APCs traffic through the lymphatics to the draining lymph nodes, which are the primary site of T cell priming. In the lymph node, mature DCs present the tumour-derived peptides on MHC class I molecules and MHC class II molecules to CD8+ and CD4+ T cells, respectively, of both naive and memory phenotypes. The generation of tumour-specific T cell responses is promoted by the delivery of a costimulatory 'signal 2' to T cells, such as through CD80–CD28, CD86–CD28, CD70–CD27 and CD40–CD40 ligand (CD40L) interactions. Costimulation is increased by IL-12 and type I interferons (IFNs) produced by DCs. Together, these interactions promote the generation and expansion of activated tumour-specific CD4+ and CD8+ T cell populations. CD4+ and CD8+ T cells traffic to the tumour site, and upon encountering their cognate antigens, they can kill tumour cells through cytotoxicity and the production of effector cytokines, such as IFNγ and tumour necrosis factor (TNF). In turn, the lysed tumour cells release tumour antigens that can again be captured, processed and presented by APCs to induce polyclonal T cell responses, thereby increasing the antigenic breadth of the antitumour immune response and leading to the process of epitope spreading. b | There are four key components of cancer vaccines: tumour antigens, formulations, immune adjuvants and delivery vehicles. CpG ODN, CpG oligodeoxynucleotide; GM-CSF, granulocyte–macrophage colony-stimulating factor; MPL, monophosphoryl lipid A; poly-ICLC, polyinosinic–polycytidylic acid with polylysine and carboxymethylcellulose; STING, stimulator of interferon genes protein; TCR, T cell receptor; TLR, Toll-like receptor.
In this context, an open question remains regarding whether therapeutic cancer vaccines designed to activate APCs can overcome these challenges to facilitate tumour antigen presentation and to promote an efficient antitumour immune response. On the basis of our understanding of how productive antigen-specific immune responses are generated, the active ingredients of cancer vaccines comprise four key components: tumour antigens, formulations, immune adjuvants and delivery vehicles, which are described below (Fig. 2b). As we discuss, despite the large number of antigens, adjuvants, delivery strategies and formulations that have been tested so far, comparative data on these different approaches, particularly in humans, are scarce. Therefore, fundamental questions such as the most effective type of adjuvant (including different adjuvants for different types of antigen), delivery approach, dose, route of administration and schedule remain unanswered.
Tumour antigens
The identification of the optimal antigen to target for a particular tumour type has long been a priority for the field of cancer immunotherapy (Boxes 1,2). Tumour antigens can generally be categorized as being tumour associated or tumour specific. As described below, accumulating experimental evidence supports tumour-specific neoantigens as being bona fide tumour rejection antigens and favours them as highly suitable antigens for cancer vaccination.
Tumour-associated antigens. Decades of effort in antigen discovery have led to the identification of a broad category of tumour-associated antigens (TAAs), which include those that are overexpressed, involved in tissue differentiation or preferentially expressed by cancer cells but not normal tissues (except for fetal or immune-privileged tissues). Salient examples of overexpressed tumour antigens include human epidermal growth factor receptor 2 (HER2; also known as ERBB2), human telomerase reverse transcriptase (TERT) and antiapoptotic proteins (such as survivin (also known as BIRC5))36. Tissue differentiation antigens are encoded by genes that are expressed by the specific cell lineages from which a tumour and its corresponding normal tissue arise but that are not expressed more widely. Examples of this class of TAA include mammaglobin-A, which is expressed in the mammary gland and overexpressed in breast cancer; prostate-specific antigen (PSA), which is expressed in the prostate gland and prostate cancer; and melanoma antigen recognized by T cells 1 (MART1; also known as melan-A), melanocyte protein PMEL and tyrosinase, which are expressed by normal melanocytes and melanoma cells37. For both overexpressed and tissue differentiation antigens, an antitumour immune response is presumably induced when high levels of expression of these proteins reach the threshold for T cell recognition, thereby breaking immunological tolerance. However, this carries the risk of inducing autoimmunity against the corresponding normal tissues38. Moreover, as these antigens are also expressed in healthy tissue, natural T cell recognition is often of low affinity as a result of negative selection of high-affinity T cells in the thymus39,40. Cancer–testis antigens (CTAs) are a specialized subset of TAAs that are thought to provide higher tumour specificity, as they are not expressed in normal adult tissues, except by germline and trophoblastic cells, but are highly expressed across cancers. More than 60 genes encoding CTAs have been identified, the best studied of which are the melanoma-associated antigen (MAGE) family, sarcoma antigen 1 (SAGE1) and cancer–testis antigen 1 (CTAG1A; commonly known and referred to here as NY-ESO-1)1,41. Similar to CTAs, oncofetal antigens (such as 5T4 oncofetal antigen (also known as TPBG)) are thought to be specific to tumours, as they are present during fetal development with generally limited expression in adult tissues but are upregulated in cancerous somatic cells42. Of note, however, all of the above-described TAAs are subject to some degree of central tolerance and lack complete specificity to the tumour.
Tumour-specific antigens. Oncogenic viral antigens have been identified in virus-induced cancers such as human papillomavirus (HPV)-associated cervical cancer, hepatitis B virus-associated hepatocellular carcinoma and human herpesvirus 8-associated Kaposi sarcoma43. Given that these antigens are foreign to the body (and therefore not subject to central tolerance) and expressed only by cancer cells (and therefore specific for the tumour), they are highly suitable for use in a cancer vaccine. In fact, vaccines using these antigens have already shown efficacy in both preventive and therapeutic settings for HPV-associated cancers43,44.
Tumour neoantigens are generated as products of somatic mutations, and hence they are not only exquisitely tumour specific but also highly immunogenic on the basis of lack of central tolerance. Although tumour neoantigens have long been conceptualized as ideal antigen targets, with numerous anecdotal accounts of their involvement as targets of tumour rejection in various cancers, their commonplace identification was not feasible until the recent availability of next-generation sequencing45. Now, through integration of tumour sequencing with the prediction of MHC-binding epitopes, it is possible to identify candidate tumour neoantigens on a per patient basis. Growing evidence supports their immunogenicity and their use for developing personalized vaccines26,46.
Several lines of evidence support neoantigens as being important targets of effective antitumour immune responses (Fig. 3). First, many studies have found that higher neoantigen load is associated with stronger T cell responses and better clinical outcome in patients with cancer. On the basis of RNA-sequencing (RNA-seq) data from thousands of samples across 18 solid tumour types from The Cancer Genome Atlas (TCGA), the number of neoantigens per tumour type was shown to correlate positively with a gene expression signature of T cell cytolytic activity47. A comparatively higher burden of mutated predicted immunogenic epitopes was associated with improved patient survival in a study that assessed 515 tumours with 6 different histologies from TCGA48. Consistent with these findings, analysis of whole-exome sequencing of 619 colorectal cancers showed that high neoantigen load is associated with increased numbers of TILs and improved survival49. The association between neoantigen load and numbers of TILs has also been shown in endometrial cancers50. In addition, associations between mutational load and clinical response to CPB have been shown in patients with melanoma25,51, non-small-cell lung cancer (NSCLC)24 and colorectal cancer15.

a | Several lines of evidence support neoantigens as being crucial targets of antitumour T cell responses. b | Potential antigens for use in cancer vaccines differ in terms of tumour specificity and vaccine personalization. Neoantigens are optimal targets for personalized, tumour-specific cancer vaccines. CTL, cytotoxic T lymphocyte; DC, dendritic cell; TCR, T cell receptor.
Second, neoantigen-specific T cell populations are expanded in settings of effective antitumour immunity. This has been shown in patients with melanoma who have clinical responses following ipilimumab (anti-CTLA4) treatment25,52 and in patients with NSCLC treated with pembrolizumab (anti-PD1)24. Furthermore, CD4+ and CD8+ TILs that mediate tumour regression upon adoptive transfer in patients with solid tumours have been characterized to have specificity for neoantigens53,54,55. In the setting of haematopoietic stem cell transplantation (HSCT) for chronic lymphocytic leukaemia, patients who experienced long-term disease-free survival after HSCT had circulating neoantigen-specific CD8+ T cells with cytotoxicity against autologous tumour56.
Third, animal experiments and human studies have directly shown that neoantigen-specific T cells are cytolytic for tumour cells that present mutated peptides, and they can contribute to complete or partial tumour regression. In both a transplantable chemically induced sarcoma model57 and an inducible sarcoma model expressing transgenic immunodominant antigens58, the epitopes that were recognized by CD8+ T cells in rejected tumours were identified as neoantigens. These preclinical models have also shown a potential role for the therapeutic targeting of neoantigens. In both a melanoma model and a transplantable colon cancer model, vaccination with neoantigen peptides elicited T cell responses and mediated antitumour activity in prophylactic and therapeutic settings28,59. In a chemically induced sarcoma model, a neoantigen long-peptide vaccine (capable of stimulating both CD4+ and CD8+ T cell responses) induced tumour rejection comparable to that induced by CPB therapy22. Likewise, neoantigen vaccines, delivered as poly-neoepitope mRNA that could target both MHC class I-restricted and MHC class II-restricted neoepitopes, induced potent tumour-specific immune responses and led to rejection of established melanoma and colon cancer in mice23. In humans, adoptively transferred neoepitope-specific CD4+ T cells mediated tumour regression in a patient with cholangiocarcinoma, thus providing direct evidence for the antitumour activity of neoantigen-specific T cells21.

Formulation
Consistent with the complexity of cancer genomes and the high degree of genetic variability between individuals, a cancer vaccine formulation should ideally capture the breadth and highly personal nature of the antigen content of the original tumour target. To this end, diverse antigen formulations have been tested, each of which has addressed this challenge with a varying degree of success. In general, the two broad categories of vaccine formulation are those that are complex in format, thereby constituting all antigen targets within a tumour (such as whole tumour cell-based vaccines), and those that are tumour antigen specific. The effectiveness of the whole tumour cell-based vaccines may be compromised by dilution of the most immunogenic tumour antigens with all of the other self-antigens that are also present in normal cells, thus essentially mimicking endogenous presentation of tumour antigens to the immune system. By contrast, tumour-specific formulations focus on specific immunogens by use of various pharmacological compounds, including proteins, peptides and nucleic acids. The most systematic efforts to test diverse vaccine formulations have been carried out using NY-ESO-1 as a uniform model antigen (Box 2).
Whole tumour cell-based vaccines. These vaccines have been generated from irradiated intact tumour cells or tumour cell lysates, derived from autologous tumour tissues60 or established heterologous tumour cell lines61, and have been genetically modified to secrete granulocyte–macrophage colony-stimulating factor (GM-CSF)62,63. One example of a cell line-based vaccine was the use of an allogeneic GM-CSF-secreting pancreatic tumour cell line as a priming vaccine followed by a boost vaccine with CRS-207, a Listeria monocytogenes-based vaccine engineered to secrete mesothelin (a TAA that is overexpressed in most pancreatic ductal adenocarcinomas); the combined strategy resulted in improved survival64. Autologous tumour cells provide the possibility of inducing an immune response against tumour antigens specific to that individual cancer. Indeed, whole-cell vaccines generated from irradiated autologous tumour cells from patients with chronic lymphocytic leukaemia — administered with irradiated bystander cells secreting GM-CSF in the setting of early immune reconstitution following allogeneic HSCT — were shown to generate tumour-specific (rather than purely alloimmune) reactivity that was associated with prolonged clinical responses65. Ongoing studies are exploring the effect of GM-CSF-secreting autologous tumour cells in the post-transplant setting following HSCT for acute myeloid leukaemia (ClinicalTrials.gov identifier: NCT01773395)66. In addition, DC–tumour cell hybrid vaccines can be generated by the fusion of DCs with tumour cells, resulting in a product that confers not only the antigen-presenting functionality of the DCs but also a continuous source of endogenous tumour antigens67. Indeed, this approach has shown promising activity in patients with acute myeloid leukaemia68.
Tumour antigen-specific vaccines. These vaccines differ from whole-cell vaccines by containing only the antigenic parts of the tumour cells that are necessary to elicit an immune response. Protein vaccines are composed of recombinant or purified proteins, with peptides being the most commonly tested antigen format for vaccination69. Multipeptide vaccines have shown improved efficacy compared with single-peptide formulations, as they are less likely to trigger immune escape70,71. In addition, long peptides (defined as 20–30-mers), rather than the 8–10-mers that are predicted to bind to MHC class I molecules, have gained attention, as they require internalization and processing by professional APCs before MHC-restricted presentation, thereby providing optimal activation of T cells. Long peptides also generally contain MHC class II-restricted peptides and thus have the potential to stimulate both CD4+ and CD8+ T cells44,72.
A specialized variant of peptide-based and protein-based vaccines are anti-idiotype antibody-based vaccines and heat shock protein–peptide complex (HSPPC) vaccines. Anti-idiotype vaccines contain an antibody directed against idiotypes (antigenic determinants of the variable region of an antibody) of another antibody that recognizes a particular tumour antigen; the anti-idiotype antibody is used as an antigen surrogate for vaccination rather than as passive antibody therapy. This approach has been tested in multiple clinical trials across various tumour types73. Racotumomab is an anti-idiotype monoclonal antibody vaccine that mimics Neu-glycolyl-GM3 ganglioside (NeuGcGM3), which is overexpressed in several solid tumours, and has shown modest clinical activity for patients with advanced NSCLC74. Heat shock proteins function as intracellular chaperones and can bind and present tumour antigens on professional APCs through MHC class I and class II molecules, leading to the activation of antitumour T cells75. HSPPCs isolated from autologous tumour cells contain a broad spectrum of patient-specific, tumour-derived peptides, which minimizes the risk of immune evasion, and they deliver antigens directly to APCs. Autologous tumour-derived HSPPCs have been evaluated in multiple clinical studies and have been shown to generate tumour-specific immunity, although clinical efficacy has been minimal76.
For nucleic acid-based vaccines, DNA-based and mRNA-based vaccines are being developed as a means to encode antigenic proteins and provide adjuvant function. Double-stranded and unmethylated-CG-rich plasmid DNA leads to stimulation of the innate immune system, providing a built-in immune adjuvant77. DNA vaccines have been evaluated in several types of cancer, including melanoma, breast cancer, colorectal cancer, prostate cancer and cervical cancer78. Nevertheless, the immunogenicity and clinical activity of DNA cancer vaccines have so far been modest, and few DNA vaccines have progressed beyond phase I clinical trial evaluation79. Vaccination based on mRNA is another delivery platform that combines the potential of mRNA to encode almost any protein with an excellent safety profile, flexibility and adjuvant ability80. Intradermal inoculation with an mRNA-based vaccine encoding several TAAs for renal cell carcinoma resulted in T cell responses and modest antitumour activity in several patients in a phase I/II trial81. Recently, intranodal vaccination with a personalized mRNA mutanome vaccine showed that an RNA-based poly-neoepitope approach can elicit potent antitumour immunity against neoantigens in patients with melanoma, as described in more detail below82.
An alternative strategy to deliver antigens in vivo is to use viral vectors. A prominent example of such a strategy and arguably (as it functions as an in situ vaccine in addition to its directly cytotoxic effect) the second cancer vaccine that has received approval by the FDA is talimogene laherparepvec (T-VEC) for the treatment of advanced melanoma. This is an intralesionally delivered oncolytic immunotherapy comprised of a genetically engineered attenuated herpes simplex virus type 1 (HSV-1) encoding GM-CSF83. Another example is the prostate cancer vaccine Prostvac (ProstVac-VF; Bavarian Nordic), which consists of a recombinant vaccinia virus vector as a priming vaccination, followed by a series of booster vaccinations containing a recombinant fowlpox vector. Both viral vectors contain transgenes encoding PSA and three immune costimulatory molecules (CD80, intercellular adhesion molecule 1 (ICAM1) and lymphocyte function-associated antigen 3 (LFA3)). This vaccine induced modest cellular immune responses in patients with metastatic prostate cancer and showed an overall survival benefit in a randomized phase II study, leading to its investigation in an ongoing phase III trial (ClinicalTrials.gov identifier: NCT01322490)84.
Various strategies have been developed to effectively load tumour antigens onto DCs, including the introduction of antigenic proteins or peptides, whole tumour cells, DNA or mRNA encoding tumour antigens or recombinant virus expressing tumour antigens85. For ex vivo-generated DC vaccines, DCs are first differentiated and activated with cytokines and subsequently loaded with antigens. Clinical trials testing vaccination with ex vivo-generated DCs have been carried out in multiple malignant tumours, including prostate cancer, melanoma, renal cell carcinoma, malignant glioma and colon cancer86.
Taken together, the data show that of the various antigen formulations each has inherent advantages and limitations. For example, both nucleic acid-based and peptide-based vaccines can be produced fairly easily and induce minimal if any toxic effects. The built-in adjuvant function of single-stranded RNA through Toll-like receptor 7 (TLR7) and TLR8 stimulation (together with the relative ease of encoding multiple vaccine epitopes on the same RNA molecule) suggests its superiority as a cancer vaccine formulation. However, the application of an RNA vaccine is limited by the fact that it cannot be as flexibly combined with different, potentially more effective, immune adjuvants as can, for example, a peptide vaccine. In the absence of randomized studies, there is currently no consensus as to whether some vaccine formulations are superior to others with regard to immunogenicity.
Adjuvants and delivery
Antigens presented by immature DCs in the steady state — in the absence of inflammatory and/or microbial stimuli — induce tolerance rather than immunity87, which indicates that antigens alone are poor inducers of adaptive immunity and that effective vaccination will therefore require coadministration of an immune adjuvant. In addition, optimal methods for vaccine delivery should protect vaccine antigens from degradation and bring them in contact with APCs for the most effective priming of T cells.
TLR agonists, which can initiate inflammatory responses by mimicking microbial stimulation, have emerged as a class of effective vaccine adjuvants88. Examples of TLR agonists that have been tested in cancer vaccine trials include the TLR3 agonist poly-ICLC (polyinosinic–polycytidylic acid with polylysine and carboxymethylcellulose), the TLR4 agonist monophosphoryl lipid A (MPL), the TLR7 agonist imiquimod, the TLR7 and TLR8 agonist resiquimod and the TLR9 agonist CpG oligodeoxynucleotide (CpG ODN)88,89. Poly-ICLC was shown to induce similar innate immune signalling pathways to the highly immunogenic yellow fever vaccine90, and it markedly increased the immunogenicity of a peptide vaccine in patients with ovarian cancer91.
DCs are an important bridge between innate and adaptive immune responses, and hence the direct targeting of antigens to DCs is a promising strategy to generate more specific and potent immune responses. Monoclonal antibodies targeting DEC205 (also known as LY75), a DC endocytosis receptor, increased the efficiency of antigen presentation92 and improved the immunogenicity and antitumour activity of vaccination in mice93. Indeed, a phase I clinical trial of the vaccine CDX-1401, which is composed of a human monoclonal antibody specific for DEC205 fused to the full-length tumour antigen NY-ESO-1, induced humoral and cellular immune responses to NY-ESO-1 as well as modest antitumour activity in patients with advanced malignancies89. Directing cancer vaccine antigens specifically to early endosomes of DCs by stimulating CD40 mediates efficient cross-presentation of antigens in vitro94. Antibody-mediated stimulation of CD40 may therefore be an attractive vaccine adjuvant, although this has not been studied extensively in clinical trials (CD40-specific agonistic antibodies have been evaluated in clinical trials independent of a vaccine in several types of tumour)95. Notably, CD40 agonists have been shown to synergize effectively with TLR agonists for cancer vaccination in mouse models96,97.
ISCOMATRIX is a saponin-based immune adjuvant composed of cholesterol, phospholipid and saponin that induces rapid antigen translocation into the cytosol, thus promoting efficient endocytosis by DCs98 and facilitating cross-priming of CD8+ T cells99. Various ISCOMATRIX-based vaccines have been tested in animal models and clinical trials, showing the safety of this approach and induction of a T cell response100. Another saponin-based adjuvant is QS-21, which has been used as an immunopotentiator in many clinical studies, and vaccines containing QS-21 are under development for several types of cancer101.
GM-CSF has emerged as a key immunostimulatory factor62 and has been incorporated in numerous clinical studies of cancer vaccines3. Most commonly, GM-CSF has been injected subcutaneously together with antigens. Other delivery formats are through viral transduction (such as retroviral or adenoviral transduction of whole tumour cells such as in the GVAX) or through cell-based delivery (for example, using bystander cells such as the GM-CSF-secreting cell line GM-K562)60,65. Stimulator of interferon genes protein (STING) is a transmembrane protein that mediates innate immune signalling by inducing type I interferon production in response to intracellular DNA102. STING agonists such as synthetic cyclic dinucleotide derivatives and cyclic di-guanosine monophosphate have shown antitumour activity in mice103,104 and can boost antitumour immune responses when used as a vaccine adjuvant in mice with metastatic breast cancer104. In addition, stimulation with a potent recall antigen may also be used to induce strong immune responses. For example, in a study of patients with glioblastoma, preconditioning the vaccine site with tetanus–diphtheria toxoid (to which most people have been exposed during childhood vaccinations) improved DC migration to the draining lymph nodes of the vaccine site following vaccination against the tumour antigen105.
One widely used strategy to increase the immunogenicity of a vaccine is through the generation of a local inflammatory response at the vaccine site, which results in trafficking of APCs to the site, where they can capture and process the vaccine antigens for presentation or cross-presentation to T cells in the draining lymph node. To this end, emulsions and aluminium salts can generate a depot at the site of the injection (by trapping the soluble antigen), which prevents the antigen from immediately trafficking to the lymph drainage system, leading to inflammation and allowing gradual antigen release. Aluminium salts have been successfully used in vaccination for almost a century; however, they predominantly induce humoral, rather than cell-mediated, immunity106,107. More widely used delivery vehicles for cancer vaccines in clinical trials are the water-in-oil emulsions, such as Montanide ISA-720 and Montanide ISA-51 (Refs 44,108,109). In a clinical trial of patients with high-risk vulvar intraepithelial neoplasia, a pre-malignant condition, vaccination with long peptides of the oncoproteins E6 and E7 with Montanide ISA-51 resulted in CD4+ and CD8+ T cell responses to these antigens and complete clinical response in 47% of patients44.
Liposomes are synthetic phospholipid vehicles that preferentially distribute via the lymph and can reach local lymphoid organs. They can deliver antigens into the endosomal and cytosolic processing pathways of APCs and have been used to direct vaccine antigens and immunomodulatory molecules to draining lymph nodes110,111. Synthetic high-density lipoprotein nanodiscs are an alternative to nanoparticles to codeliver peptides and the TLR9 agonist CpG and were shown to stimulate substantially higher numbers of antigen-specific cytotoxic CD8+ T cells in a mouse model112. Virosomes are a variation of conventional liposomes that include fusogenic viral proteins113; they have been tested in patients with metastatic breast cancer114. In addition, polymer scaffolds have been shown to increase the effectiveness of vaccine adjuvants; for example, particle formation by polymer–TLR7 or polymer–TLR8 increased the magnitude and duration of innate immune cell activation and vaccine immunogenicity115.
Towards increasingly personalized vaccines
Given the lack of absolute tumour specificity inherent to TAAs, the concept of targeting multiple neoantigens, which are truly tumour specific, has been recognized as a desirable cancer vaccine strategy. However, until recently, the technical and cost barriers to discovering personal tumour antigens in real time for individual patients precluded efforts to use them in personalized cancer vaccines. Moreover, the practical appeal of targeting TAAs that are overexpressed routinely in a particular type of cancer, and the possibility of developing a common vaccine per tumour type, led to efforts focused on targeting tumour antigens with shared expression across cancers, such as MAGE1 and NY-ESO-1 (Ref. 116).
Older studies and more recent data over the past decade, generated from the analysis of large-scale cancer-sequencing data sets, have provided irrefutable evidence of the vast genetic heterogeneity of tumour cells between individuals with the same type of cancer and even within individual tumours117,118,119 (Box 3). Furthermore, because of the complexity and diversity of HLA molecules, the spectrum of presented peptides derived from the same tumour antigen may be fairly diverse between individuals. Moreover, extensive empirical clinical experience has suggested that vaccines targeting single tumour antigens are inadequate for addressing tumour heterogeneity and for meeting the challenge of clonal evolution and immune escape by the tumour120. Compelling evidence therefore suggests that an effective cancer vaccine needs to target multiple neoantigens and be personalized to an individual's tumour.
Towards the realization of this goal, computational pipelines have been generated to identify personal candidate neoantigens in real time (Fig. 4a). Comprehensive mutational analysis is carried out through whole-exome sequencing and neoepitopes encoded by somatic mutations in the tumour are selected that have the highest probability of being presented by the individual's MHC molecules on the basis of affinity predictions47,52,53,56,57,59. The most commonly used prediction algorithm for MHC class I binding is NetMHCpan, which is based on neural networks121. Matched RNA-seq is used to confirm with high confidence the mutations that are likely to be expressed by tumour cells. Using this approach, three independent phase I clinical trials have evaluated the feasibility, safety and immunogenicity of neoantigen-based cancer vaccines82,122,123 (Fig. 4b).

a | The typical workflow for neoepitope selection and vaccine manufacture. DNA and RNA are extracted from single-cell suspensions of tumour cells and matched normal tissue cells. Somatic mutations of tumour cells are discovered by whole-exome sequencing (WES). RNA sequencing (RNA-seq) narrows the focus to mutations of expressed genes. Clinical HLA typing is carried out on DNA from normal tissue. The potential antigenicity of neoepitopes identified by WES and RNA-seq is assessed by predicting the affinity of the neoepitopes for binding to the HLA type of that individual (using NetMHCpan), thereby generating candidate vaccine epitopes. Validated epitopes are selected for incorporation into the personalized cancer vaccine, which is administered to patients in combination with an immune adjuvant. b | The schema of three phase I clinical trials of personalized neoantigen vaccines in patients with melanoma. These trials have used dendritic cells (DCs) pulsed with short HLA-A2-restricted neoantigen peptides122 (ClinicalTrials.gov identifier: NCT00683670) (top); synthetic long peptides targeting neoantigens admixed with poly-ICLC (polyinosinic–polycytidylic acid with polylysine and carboxymethylcellulose) (NeoVax)123 (ClinicalTrials.gov identifier: NCT01970358) (middle); or neoantigen-targeting mRNA (IVAC MUTANOME)82 (ClinicalTrials.gov identifier: NCT02035956) (bottom). These studies show that vaccination is feasible, safe and able to induce robust neoepitope-specific T cell responses. c | Strategies to improve personalized neoantigen vaccines for cancer.
Melanoma was selected for these initial studies because of its well-established immunogenicity and high mutational load82,122,123. The first clinical trial of a neoantigen-based vaccine was carried out in three patients with advanced melanoma who had previously been treated with ipilimumab. In this study, DCs pulsed with HLA-A2-defined 8–10-mer neoantigen peptides were vaccinated intravenously122 (Fig. 4b; top). We carried out a clinical trial in patients with high-risk melanoma by use of a peptide vaccine (NeoVax) based on the neoantigen discovery pipeline. Long peptides (15–30-mers) representing up to 20 neoantigens specific to each patient's tumour and admixed with poly-ICLC were injected subcutaneously on a prime–boost schedule (ClinicalTrials.gov identifier: NCT01970358)123,124 (Fig. 4b; middle). Of the six individuals who were vaccinated, four who had enrolled with stage III melanoma showed no signs of tumour recurrence for up to 32 months after vaccination. Two patients who had stage IV melanoma showed disease recurrence shortly after vaccination but experienced complete tumour regression after receiving anti-PD1 therapy. A clinical trial of a neoantigen-based mRNA vaccine IVAC MUTANOME was also carried out in 13 patients with melanoma (ClinicalTrials.gov identifier: NCT02035956)82 (Fig. 4b; bottom). Patients with NY-ESO-1-positive and/or tyrosinase-positive melanoma were vaccinated intranodally with one dose of an off-the-shelf mRNA vaccine encoding these TAAs — while the neoantigen mRNA was being prepared (median of 103 days) — before receiving a personalized vaccine consisting of up to 10 neoantigen mRNAs. Eight patients who had no detectable disease at the time of vaccination remained free of tumours throughout a follow-up period of 12–23 months. Of the five patients who had metastatic disease at the time of vaccination, two experienced a vaccine-related objective response and one had a complete response in combination with anti-PD1 therapy.
Immune analysis in all three studies has shown that the neoantigen-based cancer vaccines induced robust T cell responses in all vaccinated patients82,122,123. Two studies82,123 independently reached the similar conclusion that neoantigen-based vaccines induced robust de novo CD4+ and CD8+ T cell responses in all vaccinated patients, that these T cell responses were polyfunctional and that, overall, 60–70% of the predicted immunizing neoantigens were recognized. In the majority of cases, neoantigen-specific T cells could discriminate mutated antigens from wild-type antigens in vitro; in some patients, these T cells recognized autologous tumour cells. We reported that the repertoire of neoantigen-specific T cells expanded and persisted for more than 1 year in the setting of ongoing complete clinical response following the administration of PD1 blockade after vaccination123. In both studies, more neoepitope-specific CD4+ T cell responses than CD8+ T cell responses were detected. One possible structural explanation for the predominance of CD4+ T cell responses is that whereas MHC class I-binding epitopes fit precisely within an MHC class I binding pocket, MHC class II-binding epitopes are typically longer and extend out of the core binding site125,126. In addition, the somewhat promiscuous binding properties of peptides to MHC class II proteins enable a larger number of peptides to function as epitopes. Moreover, because cross-presenting C-type lectin domain family 9 member A (CLEC9A)+ DCs are fairly rare, many more APCs are likely to be available for MHC class II-restricted antigen presentation to CD4+ T cells127. A recent mouse study showed that DCs that activate CD8+ T cells are in the deep paracortex of the lymph node where antigen quantities are reduced, whereas those that activate CD4+ T cells were mostly in the periphery of the lymph node regions, resulting in greater activation of CD4+ T cells128. Thus, a combination of structural, cellular and other factors is likely to explain the predominance of CD4+ T cell responses.
Taken together, these three studies show that personalized neoantigen-based vaccination is feasible, safe and able to induce robust and broadened neoepitope-specific T cell responses in patients with melanoma. These data, coupled with the observation of T cell repertoire expansion upon CPB129,130, provide a strong rationale for combining personalized neoantigen vaccines with CPB. Clinical trials are ongoing to assess the above-described neoantigen long-peptide vaccine in combination with locally administered ipilimumab (ClinicalTrials.gov identifier: NCT02950766) and systemic nivolumab (ClinicalTrials.gov identifier: NCT02897765) as well as to test a personalized mRNA mutanome vaccine in combination with the PD1 ligand 1 (PDL1)-directed antibody atezolizumab (ClinicalTrials.gov identifier: NCT03289962).
Clinical trials of neoantigen-based personalized cancer vaccines are now taking place in multiple tumour types and are using various adjuvants and delivery approaches131. These include peptide-based vaccines for glioblastoma and breast, pancreatic, paediatric brain and hepatocellular cancers132 (ClinicalTrials.gov identifiers: NCT02287428, NCT02510950, NCT02427581, NCT02600949 and NCT03068832), poly-epitope-encoding RNA-based or DNA-based vaccines for breast and pancreatic cancers (ClinicalTrials.gov identifiers: NCT02316457, NCT02348320 and NCT03122106) and a peptide-loaded DC vaccine for colorectal cancer (ClinicalTrials.gov identifier: NCT01885702). Other ongoing projects include the Glioma Actively Personalized Vaccine Consortium (GAPVAC) and the Cancer Vaccine development for Hepatocellular Carcinoma Consortium (HEPAVAC), which are conducting phase I clinical trials using off-the-shelf peptides matched to highly overexpressed antigens in the tumours of individual patients as well as personalized mutated peptides in glioblastoma (ClinicalTrials.gov identifier: NCT02149225) and hepatocellular cancer132.
Perspective
The coming of age of cancer immunotherapy has largely been driven by the broad clinical antitumour activity of ACT and CPB. Recent successes in these areas provide a strong rationale to revisit the full arsenal of immunotherapeutic approaches to cancer therapy. Given their ability to steer the host immune response directly to the tumour, cancer vaccines have great potential. Clear evidence supports neoantigens as being crucial targets of the antitumour immune response, and strong neoantigen-specific T cell responses have been observed in the recent initial series of vaccine trials. Notably, despite the use of distinct neoantigen-based vaccination approaches (synthetic long peptides versus RNA) by these independent studies, similar results were observed with regards to safety, immunogenicity and early signs of clinical activity. These data have led to fast-paced development of neoantigen-based personalized vaccines for patients with multiple tumour types, with ongoing clinical trials using peptide-based, DNA-based, RNA-based and DC-based approaches.
Although the number and quality of tumour neoantigens required for the generation of an effective antitumour immune response are not known, identification of neoepitopes that have the greatest probability of generating effective immunity is important, particularly for tumours in which the number of mutations encoding such neoantigens is limiting (tumours with low mutation rates). Further optimization of the neoepitope selection process is therefore imperative. Another important question is how to best address the immunosuppressive tumour microenvironment, which will limit the effectiveness of even the best neoantigen-based vaccine approach. A separate line of questions relates to optimization of vaccination dose, route and schedule. Now, with the ability to identify this new class of promising tumour-specific antigens, we should be able to address these questions. We propose that there are at least four areas in which we can expect improvements in the near term (Fig. 4c).
Improving antigen prediction. Improvement of HLA-binding algorithms provides an opportunity to increase the likelihood of targeting neoantigens that are expressed by a patient's cancer cells. Mass spectrometry-based approaches can identify peptides that are processed and presented by the tumour cell, and these have been used in combination with exome sequencing followed by the use of MHC class I-binding prediction algorithms. However, mass spectrometry has conventionally required large numbers of tumour cells and is labour intensive. Recently, new prediction models trained on MHC class I-binding peptides eluted from single HLA-allele-expressing cell lines and detected by mass spectrometry suggest that further improvements in the accuracy of these algorithms can be achieved by incorporating antigen-processing information and through the de novo discovery of new peptide motifs133. Furthermore, CD4+ T cells are essential for the formation of memory CD8+ T cell populations134, and CD4+ T cell-mediated antitumour immune responses have been increasingly recognized. Therefore, accurate algorithms to predict MHC class II binding will be needed. The quality of algorithms predicting MHC class II binding is lower than that of those predicting MHC class I binding owing to the relatively higher promiscuity of peptide binding to MHC class II molecules135. It can be anticipated that future improvement in prediction algorithms to identify neoantigens arising from gene fusions, splice variants and errors in translation will increase the number of targetable neoantigens, which is particularly important for tumours with lower mutation load.
Developing combination therapy. Strategies such as targeting multiple neoepitopes and combining personalized vaccines with complementary therapies such as CPB will be important to prevent immune escape. For example, recent mouse studies have shown that the combination of four components (a tumour antigen-targeting antibody, IL-2, anti-PD1 therapy and a T cell vaccine) could eliminate large tumour burdens136. Complementary therapies to reverse immune suppression in the tumour microenvironment — such as depleting regulatory cells, inhibiting regulatory molecules or blocking metabolic suppression — will be important to unleash the full potential of a neoantigen-based cancer vaccine. Several studies have suggested potential additive or synergistic effects between a cancer vaccine and CPB137,138,139,140,141,142. The above-described ongoing trials of personalized neoantigen vaccines plus ipilimumab, nivolumab or atezolizumab are examples of such a complementary approach that are already being tested in the clinic. Targeting of other inhibitory receptors such as lymphocyte activation gene 3 protein (LAG3) and T cell immunoglobulin and mucin domain-containing protein 3 (TIM3; also known as HAVR2) is being actively tested in preclinical and clinical studies. The activation of costimulatory receptors, such as CD137 (also known as TNFRSF9), tumour necrosis factor receptor (TNFR) superfamily member 4 (TNFRSF4), glucocorticoid-induced TNFR-related protein (GITR; also known as TNFRSF18) and CD27, is also being evaluated in clinical trials143. Thus, we anticipate future studies in which these and other complementary therapies will be tested in combination with personalized cancer vaccines.
Developing preclinical models. To optimize dosing, scheduling and administration routes of personalized cancer vaccines, careful investigation in early-phase clinical trials, in addition to the use of preclinical models, will be important. For example, a recent study highlighted the importance of dose schedule: the activation of DCs after tumoricidal activity improved tumour control by multiple distinct combination immunotherapies, whereas the activation of DCs too early resulted in less antigen capture and therefore less priming of CD8+ T cells144. Preclinical models are practical tools for studying the effects of differences in vaccination approach as well as for identifying the mechanism of the underlying immune response. To increase the immunogenicity of a neoantigen-based vaccine, there is also a crucial need for optimized immune adjuvants and novel vaccine delivery approaches, such as scaffold and nanoparticle technologies. There is already substantial preclinical evidence that these novel delivery approaches can markedly increase the immunogenicity of cancer vaccines, including neoantigen-based vaccines112,145,146. In the clinical realm, there is no clear consensus with regards to an optimal vaccination schedule, dosing or clinically available immune adjuvant. Randomized controlled larger studies assessing promising new technologies may be needed.
Improving manufacturing practices. Producing a personalized vaccine might be expected to be more costly and time consuming than off-the-shelf therapeutic agents. Personalized production requires a drastic change in standard manufacturing practices, which until now have focused on optimization of typically single uniform products at large scale rather than smaller-scale complex, flexibly formulated products. The increasing speed and decreasing cost of sequencing, the economies of scale resulting from higher volume and more automated peptide or DNA production and advanced sample tracking systems will all substantially lower the cost and production time of personalized vaccines. More disruptive technologies are on the horizon, such as automated flow peptide synthesis to enable vastly accelerated peptide manufacture147.
Conclusion
The ability of the immune system to specifically attack cancer cells coupled with its ability to adapt to an evolving tumour and its built-in function of memory make it arguably the most powerful weapon to control cancer in the long term. Neoantigens, which are highly specific to the tumour and distinct from self, now provide the long-elusive antigen target for effective cancer vaccination. Personalized vaccines targeting these antigens, in combination with innovative immune adjuvants and effective complementary immune-modulating therapies such as CPB, should therefore provide a powerful tool to unleash the full potential of a patient's immune response and direct it specifically against their cancer.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Coulie, P. G., Van den Eynde, B. J., van der Bruggen, P. & Boon, T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat. Rev. Cancer 14, 135–146 (2014).
Bachireddy, P., Burkhardt, U. E., Rajasagi, M. & Wu, C. J. Haematological malignancies: at the forefront of immunotherapeutic innovation. Nat. Rev. Cancer 15, 201–215 (2015).
Wong, K. K., Li, W. A., Mooney, D. J. & Dranoff, G. Advances in therapeutic cancer vaccines. Adv. Immunol. 130, 191–249 (2016).
Restifo, N. P., Dudley, M. E. & Rosenberg, S. A. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol. 12, 269–281 (2012).
Rosenberg, S. A. & Restifo, N. P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 348, 62–68 (2015).
Fesnak, A. D., June, C. H. & Levine, B. L. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer 16, 566–581 (2016).
Sharma, P. & Allison, J. P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214 (2015).
Whiteside, T. L., Demaria, S., Rodriguez-Ruiz, M. E., Zarour, H. M. & Melero, I. Emerging opportunities and challenges in cancer immunotherapy. Clin. Cancer Res. 22, 1845–1855 (2016).
Topalian, S. L. et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 366, 2443–2454 (2012).
Topalian, S. L. et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 32, 1020–1030 (2014).
McDermott, D. F. et al. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J. Clin. Oncol. 33, 2013–2020 (2015).
Le, D. T. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Nghiem, P. T. et al. PD-1 blockade with pembrolizumab in advanced Merkel-cell carcinoma. N. Engl. J. Med. 374, 2542–2552 (2016).
Ansell, S. M. et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N. Engl. J. Med. 372, 311–319 (2015).
Le, D. T. et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015).
Brahmer, J. R. et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 366, 2455–2465 (2012).
Ott, P. A., Hodi, F. S., Kaufman, H. L., Wigginton, J. M. & Wolchok, J. D. Combination immunotherapy: a road map. J. Immunother. Cancer 5, 16 (2017).
Hegde, P. S., Karanikas, V. & Evers, S. The where, the when, and the how of immune monitoring for cancer immunotherapies in the era of checkpoint inhibition. Clin. Cancer Res. 22, 1865–1874 (2016).
Larkin, J. et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 373, 23–34 (2015).
Postow, M. A. et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 372, 2006–2017 (2015).
Tran, E. et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). This study shows that neoantigen-specific CD4+ TILs can result in tumour regression in vivo in a human.
Gubin, M. M. et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014). This study shows that long-peptide vaccination against tumour neoepitopes can result in tumour control to the same degree as CPB in a mouse model.
Kreiter, S. et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 520, 692–696 (2015). This study shows that both MHC class II-restricted and MHC class I-restricted neoepitopes delivered as poly-neoepitope mRNA can induce potent tumour-specific immune responses and lead to rejection of tumour in mouse models.
Rizvi, N. A. et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Snyder, A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 371, 2189–2199 (2014). References 15, 24 and 25 show a direct association between the number of mutations in a tumour and the effectiveness of CPB therapy.
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
Desrichard, A., Snyder, A. & Chan, T. A. Cancer neoantigens and applications for immunotherapy. Clin. Cancer Res. 22, 807–812 (2016).
Yadav, M. et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). This study developed an approach that combines whole-exome and transcriptome sequencing analysis with mass spectrometry to identify neoantigens in mouse models. Vaccination with neoantigens resulted in tumour control.
Chen, D. S. & Mellman, I. Oncology meets immunology: the cancer-immunity cycle. Immunity 39, 1–10 (2013).
Chen, D. S. & Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 541, 321–330 (2017).
Joffre, O. P., Segura, E., Savina, A. & Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 12, 557–569 (2012).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016).
Sharma, P., Hu-Lieskovan, S., Wargo, J. A. & Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168, 707–723 (2017).
Meacham, C. E. & Morrison, S. J. Tumour heterogeneity and cancer cell plasticity. Nature 501, 328–337 (2013).
Greaves, M. & Maley, C. C. Clonal evolution in cancer. Nature 481, 306–313 (2012).
Novellino, L., Castelli, C. & Parmiani, G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol. Immunother. 54, 187–207 (2005).
Buonaguro, L., Petrizzo, A., Tornesello, M. L. & Buonaguro, F. M. Translating tumor antigens into cancer vaccines. Clin. Vaccine Immunol. 18, 23–34 (2011).
Dudley, M. E. et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298, 850–854 (2002).
Theobald, M. et al. Tolerance to p53 by A2.1-restricted cytotoxic T lymphocytes. J. Exp. Med. 185, 833–841 (1997).
Thaxton, J. E. & Li, Z. To affinity and beyond: harnessing the T cell receptor for cancer immunotherapy. Hum. Vaccin. Immunother. 10, 3313–3321 (2014).
Chen, Y. T. et al. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl Acad. Sci. USA 94, 1914–1918 (1997).
Stern, P. L. & Harrop, R. 5T4 oncofoetal antigen: an attractive target for immune intervention in cancer. Cancer Immunol. Immunother. 66, 415–426 (2017).
Melief, C. J., van Hall, T., Arens, R., Ossendorp, F. & van der Burg, S. H. Therapeutic cancer vaccines. J. Clin. Invest. 125, 3401–3412 (2015).
Kenter, G. G. et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 361, 1838–1847 (2009). This study shows that for patients with pre-malignant vulvar intraepithelial neoplasia, therapeutic vaccination using synthetic long peptides directed against HPV antigens has clinical benefit.
Fritsch, E. F. et al. HLA-binding properties of tumor neoepitopes in humans. Cancer Immunol. Res. 2, 522–529 (2014).
Yarchoan, M., Johnson, B. A. 3rd, Lutz, E. R., Laheru, D. A. & Jaffee, E. M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 17, 209–222 (2017).
Rooney, M. S., Shukla, S. A., Wu, C. J., Getz, G. & Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61 (2015). This systematic analysis across tumour types reveals genomic correlates that are related to variation in natural tumour immunity and immune escape, and it shows that tumours shape and are shaped by their immune microenvironment.
Brown, S. D. et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 24, 743–750 (2014).
Giannakis, M. et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Res. 15, 857–865 (2016).
Howitt, B. E. et al. Association of polymerase e-mutated and microsatellite-instable endometrial cancers with neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 1, 1319–1323 (2015).
Van Allen, E. M. et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211 (2015).
van Rooij, N. et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 31, e439–e442 (2013).
Robbins, P. F. et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat. Med. 19, 747–752 (2013).
Tran, E. et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 350, 1387–1390 (2015).
Linnemann, C. et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat. Med. 21, 81–85 (2015).
Rajasagi, M. et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 124, 453–462 (2014).
Matsushita, H. et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 482, 400–404 (2012).
DuPage, M., Mazumdar, C., Schmidt, L. M., Cheung, A. F. & Jacks, T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 482, 405–409 (2012).
Castle, J. C. et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 72, 1081–1091 (2012). References 57–59 show that neoantigen-specific T cells have an important role in the control of cancer in mouse models.
Soiffer, R. et al. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J. Clin. Oncol. 21, 3343–3350 (2003).
Vaishampayan, U., Abrams, J., Darrah, D., Jones, V. & Mitchell, M. S. Active immunotherapy of metastatic melanoma with allogeneic melanoma lysates and interferon alpha. Clin. Cancer Res. 8, 3696–3701 (2002).
Dranoff, G. et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl Acad. Sci. USA 90, 3539–3543 (1993).
Jaffee, E. M. et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J. Clin. Oncol. 19, 145–156 (2001).
Le, D. T. et al. Safety and survival with GVAX pancreas prime and Listeria Monocytogenes-expressing mesothelin (CRS-207) boost vaccines for metastatic pancreatic cancer. J. Clin. Oncol. 33, 1325–1333 (2015).
Burkhardt, U. E. et al. Autologous CLL cell vaccination early after transplant induces leukemia-specific T cells. J. Clin. Invest. 123, 3756–3765 (2013).
Ho, V. T. et al. Biologic activity of irradiated, autologous, GM-CSF-secreting leukemia cell vaccines early after allogeneic stem cell transplantation. Proc. Natl Acad. Sci. USA 106, 15825–15830 (2009).
Phan, V. et al. A new genetic method to generate and isolate small, short-lived but highly potent dendritic cell-tumor cell hybrid vaccines. Nat. Med. 9, 1215–1219 (2003).
Rosenblatt, J. et al. Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Sci. Transl Med. 8, 368ra171 (2016).
Melero, I. et al. Therapeutic vaccines for cancer: an overview of clinical trials. Nat. Rev. Clin. Oncol. 11, 509–524 (2014).
Walter, S. et al. Multipeptide immune response to cancer vaccine IMA901 after single-dose cyclophosphamide associates with longer patient survival. Nat. Med. 18, 1254–1261 (2012).
Slingluff, C. L. Jr. et al. Immunologic and clinical outcomes of a randomized phase II trial of two multipeptide vaccines for melanoma in the adjuvant setting. Clin. Cancer Res. 13, 6386–6395 (2007).
Melief, C. J. & van der Burg, S. H. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat. Rev. Cancer 8, 351–360 (2008).
Ladjemi, M. Z. Anti-idiotypic antibodies as cancer vaccines: achievements and future improvements. Front. Oncol. 2, 158 (2012).
Alfonso, S. et al. A randomized, multicenter, placebo-controlled clinical trial of racotumomab-alum vaccine as switch maintenance therapy in advanced non-small cell lung cancer patients. Clin. Cancer Res. 20, 3660–3671 (2014).
Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2, 185–194 (2002).
Ampie, L. et al. Heat shock protein vaccines against glioblastoma: from bench to bedside. J. Neuro. Oncol. 123, 441–448 (2015).
Hemmi, H. et al. A Toll-like receptor recognizes bacterial DNA. Nature 408, 740–745 (2000).
Yang, B., Jeang, J., Yang, A., Wu, T. C. & Hung, C. F. DNA vaccine for cancer immunotherapy. Hum. Vaccin. Immunother. 10, 3153–3164 (2014).
Zahm, C. D., Colluru, V. T. & McNeel, D. G. DNA vaccines for prostate cancer. Pharmacol. Ther. 174, 27–42 (2017).
Heil, F. et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529 (2004).
Rittig, S. M. et al. Intradermal vaccinations with RNA coding for TAA generate CD8+ and CD4+ immune responses and induce clinical benefit in vaccinated patients. Mol. Ther. 19, 990–999 (2011).
Sahin, U. et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547, 222–226 (2017).
Ott, P. A. & Hodi, F. S. Talimogene laherparepvec for the treatment of advanced melanoma. Clin. Cancer Res. 22, 3127–3131 (2016).
Kantoff, P. W. et al. Overall survival analysis of a phase II randomized controlled trial of a Poxviral-based PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 28, 1099–1105 (2010).
Pyzer, A. R., Avigan, D. E. & Rosenblatt, J. Clinical trials of dendritic cell-based cancer vaccines in hematologic malignancies. Hum. Vaccin. Immunother. 10, 3125–3131 (2014).
Palucka, K. & Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 12, 265–277 (2012).
Mellman, I. & Steinman, R. M. Dendritic cells: specialized and regulated antigen processing machines. Cell 106, 255–258 (2001).
Maisonneuve, C., Bertholet, S., Philpott, J. & De Gregorio, E. Unleashing the potential of NOD- and Toll-like agonists as vaccine adjuvants. Proc. Natl Acad. Sci. USA 111, 12294–12299 (2014).
Dhodapkar, M. V. et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci. Transl Med. 6, 232ra51 (2014).
Caskey, M. et al. Synthetic double-stranded RNA induces innate immune responses similar to a live viral vaccine in humans. J. Exp. Med. 208, 2357–2366 (2011). This study shows that poly-ICLC induces innate immune signalling pathways similar to those induced by the highly immunogenic yellow fever vaccine.
Sabbatini, P. et al. Phase I trial of overlapping long peptides from a tumor self-antigen and poly-ICLC shows rapid induction of integrated immune response in ovarian cancer patients. Clin. Cancer Res. 18, 6497–6508 (2012).
Jiang, W. et al. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature 375, 151–155 (1995).
Bonifaz, L. C. et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J. Exp. Med. 199, 815–824 (2004).
Cohn, L. et al. Antigen delivery to early endosomes eliminates the superiority of human blood BDCA3+ dendritic cells at cross presentation. J. Exp. Med. 210, 1049–1063 (2013).
Vonderheide, R. H. & Glennie, M. J. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 19, 1035–1043 (2013).
McWilliams, J. A., Sanchez, P. J., Haluszczak, C., Gapin, L. & Kedl, R. M. Multiple innate signaling pathways cooperate with CD40 to induce potent, CD70-dependent cellular immunity. Vaccine 28, 1468–1476 (2010).
Nimanong, S. et al. CD40 signaling drives potent cellular immune responses in heterologous cancer vaccinations. Cancer Res. 77, 1918–1926 (2017).
Schnurr, M. et al. ISCOMATRIX adjuvant induces efficient cross-presentation of tumor antigen by dendritic cells via rapid cytosolic antigen delivery and processing via tripeptidyl peptidase II. J. Immunol. 182, 1253–1259 (2009).
Wilson, N. S. et al. ISCOMATRIX vaccines mediate CD8+ T-cell cross-priming by a MyD88-dependent signaling pathway. Immunol. Cell Biol. 90, 540–552 (2012).
Davis, I. D. et al. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4+ and CD8+ T cell responses in humans. Proc. Natl Acad. Sci. USA 101, 10697–10702 (2004).
Ragupathi, G., Gardner, J. R., Livingston, P. O. & Gin, D. Y. Natural and synthetic saponin adjuvant QS-21 for vaccines against cancer. Expert Rev. Vaccines 10, 463–470 (2011).
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Corrales, L. et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 11, 1018–1030 (2015).
Chandra, D. et al. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer Immunol. Res. 2, 901–910 (2014).
Mitchell, D. A. et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519, 366–369 (2015).
Reed, S. G., Orr, M. T. & Fox, C. B. Key roles of adjuvants in modern vaccines. Nat. Med. 19, 1597–1608 (2013).
Hogenesch, H. Mechanism of immunopotentiation and safety of aluminum adjuvants. Front. Immunol. 3, 406 (2012).
Aucouturier, J., Dupuis, L., Deville, S., Ascarateil, S. & Ganne, V. Montanide ISA 720 and 51: a new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev. Vaccines 1, 111–118 (2002).
Fenstermaker, R. A. et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol. Immunother. 65, 1339–1352 (2016).
Alving, C. R. Liposomes as carriers of antigens and adjuvants. J. Immunol. Methods 140, 1–13 (1991).
Neelapu, S. S. et al. Human autologous tumor-specific T-cell responses induced by liposomal delivery of a lymphoma antigen. Clin. Cancer Res. 10, 8309–8317 (2004).
Kuai, R., Ochyl, L. J., Bahjat, K. S., Schwendeman, A. & Moon, J. J. Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 16, 489–496 (2017).
Moser, C., Muller, M., Kaeser, M. D., Weydemann, U. & Amacker, M. Influenza virosomes as vaccine adjuvant and carrier system. Expert Rev. Vaccines 12, 779–791 (2013).
Wiedermann, U. et al. A virosomal formulated Her-2/neu multi-peptide vaccine induces Her-2/neu-specific immune responses in patients with metastatic breast cancer: a phase I study. Breast Cancer Res. Treat. 119, 673–683 (2010).
Lynn, G. M. et al. In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol. 33, 1201–1210 (2015).
Gjerstorff, M. F., Andersen, M. H. & Ditzel, H. J. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget 6, 15772–15787 (2015).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Vogelstein, B. et al. Cancer genome landscapes. Science 339, 1546–1558 (2013).
Lawrence, M. S. et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013).
Rosenberg, S. A., Yang, J. C. & Restifo, N. P. Cancer immunotherapy: moving beyond current vaccines. Nat. Med. 10, 909–915 (2004).
Nielsen, M. & Andreatta, M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 8, 33 (2016).
Carreno, B. M. et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science 348, 803–808 (2015).
Ott, P. A. et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 547, 217–221 (2017). References 82, 122 and 123 show that personalized neoantigen vaccines are safe and feasible and can induce robust neoantigen-specific T cell responses in patients with melanoma.
Fritsch, E. F., Hacohen, N. & Wu, C. J. Personal neoantigen cancer vaccines: The momentum builds. Oncoimmunology 3, e29311 (2014).
Stern, L. J. et al. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature 368, 215–221 (1994).
Rossjohn, J. et al. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 33, 169–200 (2015).
Mildner, A. & Jung, S. Development and function of dendritic cell subsets. Immunity 40, 642–656 (2014).
Gerner, M. Y., Casey, K. A., Kastenmuller, W. & Germain, R. N. Dendritic cell and antigen dispersal landscapes regulate T cell immunity. J. Exp. Med. 214, 3105–3122 (2017).
Robert, L. et al. CTLA4 blockade broadens the peripheral T-cell receptor repertoire. Clin. Cancer Res. 20, 2424–2432 (2014).
Cha, E. et al. Improved survival with T cell clonotype stability after anti-CTLA-4 treatment in cancer patients. Sci. Transl Med. 6, 238ra70 (2014).
Tureci, O. et al. Targeting the heterogeneity of cancer with individualized neoepitope vaccines. Clin. Cancer Res. 22, 1885–1896 (2016).
Buonaguro, L. Developments in cancer vaccines for hepatocellular carcinoma. Cancer Immunol. Immunother. 65, 93–99 (2016).
Abelin, J. G. et al. Mass spectrometry profiling of HLA-associated peptidomes in mono-allelic cells enables more accurate epitope prediction. Immunity 46, 315–326 (2017). This study establishes new epitope prediction models trained on MHC class I-binding peptides eluted from single HLA-allele-expressing cell lines and detected by mass spectrometry.
Laidlaw, B. J., Craft, J. E. & Kaech, S. M. The multifaceted role of CD4+ T cells in CD8+ T cell memory. Nat. Rev. Immunol. 16, 102–111 (2016).
Nielsen, M., Lund, O., Buus, S. & Lundegaard, C. MHC class II epitope predictive algorithms. Immunology 130, 319–328 (2010).
Moynihan, K. D. et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med. 22, 1402–1410 (2016).
van Elsas, A., Hurwitz, A. A. & Allison, J. P. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J. Exp. Med. 190, 355–366 (1999).
Duraiswamy, J., Kaluza, K. M., Freeman, G. J. & Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 73, 3591–3603 (2013).
Ali, O. A., Lewin, S. A., Dranoff, G. & Mooney, D. J. Vaccines combined with immune checkpoint antibodies promote cytotoxic T-cell activity and tumor eradication. Cancer Immunol. Res. 4, 95–100 (2016).
Hodi, F. S. et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl Acad. Sci. USA 100, 4712–4717 (2003). This study shows that antibody-mediated CTLA4 blockade increases tumour immunity in patients who were previously vaccinated with autologous GM-CSF-secreting tumour cells.
Soares, K. C. et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J. Immunother. 38, 1–11 (2015).
Fu, J. et al. STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockade. Sci. Transl Med. 7, 283ra52 (2015).
Melero, I. et al. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 15, 457–472 (2015).
Tzeng, A. et al. Temporally programmed CD8α+ DC activation enhances combination cancer immunotherapy. Cell Rep. 17, 2503–2511 (2016).
Kim, J. et al. Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat. Biotechnol. 33, 64–72 (2015).
Ali, O. A. et al. Identification of immune factors regulating antitumor immunity using polymeric vaccines with multiple adjuvants. Cancer Res. 74, 1670–1681 (2014).
Mijalis, A. J. et al. A fully automated flow-based approach for accelerated peptide synthesis. Nat. Chem. Biol. 13, 464–466 (2017).
Coley, W. B. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin. Orthop. Relat. Res. 262, 3–11 (1991).
Coley, W. B. The treatment of inoperable sarcoma by bacterial toxins (the mixed toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc. R. Soc. Med. 3, 1–48 (1910).
Hoover, H. C. et al. Prospectively randomized trial of adjuvant active-specific immunotherapy for human colorectal cancer. Cancer 55, 1236–1243 (1985).
van der Bruggen, P. et al. A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643–1647 (1991).
Cox, A. L. et al. Identification of a peptide recognized by five melanoma-specific human cytotoxic T cell lines. Science 264, 716–719 (1994).
Steinman, R. M. & Cohn, Z. A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 137, 1142–1162 (1973).
Hsu, F. J. et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat. Med. 2, 52–58 (1996).
Murphy, G., Tjoa, B., Ragde, H., Kenny, G. & Boynton, A. Phase I clinical trial: T-cell therapy for prostate cancer using autologous dendritic cells pulsed with HLA-A0201-specific peptides from prostate-specific membrane antigen. Prostate 29, 371–380 (1996).
Nestle, F. O. et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat. Med. 4, 328–332 (1998).
Kantoff, P. W. et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 363, 411–422 (2010).
Cebon, J., Knights, A., Ebert, L., Jackson, H. & Chen, W. Evaluation of cellular immune responses in cancer vaccine recipients: lessons from NY-ESO-1. Expert Rev. Vaccines 9, 617–629 (2010).
Valmori, D. et al. Vaccination with NY-ESO-1 protein and CpG in Montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc. Natl Acad. Sci. USA 104, 8947–8952 (2007).
Landau, D. A. et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726 (2013).
Greenman, C. et al. Patterns of somatic mutation in human cancer genomes. Nature 446, 153–158 (2007).
Segal, N. H. et al. Epitope landscape in breast and colorectal cancer. Cancer Res. 68, 889–892 (2008).
George, S. et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity 46, 197–204 (2017).
Verdegaal, E. M. et al. Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 536, 91–95 (2016).
Gerlinger, M. et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892 (2012).
Acknowledgements
This work is supported by grants from the US National Institutes of Health (NCI-1RO1CA155010-02 and NHLBI-5R01HL103532-03) (to C.J.W), the Francis and Adele Kittredge Family Immuno-Oncology and Melanoma Research Fund (to P.A.O.), the Faircloth Family Research Fund (to P.A.O.) and the Dana-Farber Cancer Institute Center for Cancer Immunotherapy Research fellowship (to Z.H.). C.J.W. is a Scholar of the Leukemia and Lymphoma Society.
Author information
Authors and Affiliations
Contributions
Z.H., P.A.O. and C.J.W. contributed to researching data for the article and discussing the content in addition to writing, reviewing and editing the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
C.J.W. is a founder of Neon Therapeutics and member of its scientific advisory board. P.A.O. has advised Neon Therapeutics. Patent applications have been filed on aspects of the described work entitled as follows: Compositions and Methods for Personalized Neoplasia Vaccines (C.J.W.), Methods for Identifying Tumour Specific Neo-Antigens (C.J.W.) and Combination Therapy for Neoantigen Vaccine (C.J.W.). Z.H. declares no competing financial interests.
Related links
Glossary
- Adoptive cell transfer
-
(ACT). A form of immunotherapy in which naturally occurring or genetically engineered tumour-specific lymphocytes, which may be autologous (patient's own) or allogeneic (donor), are activated and selected in vitro for tumour reactivity and expanded to reach high numbers for reinfusion into the patient.
- Chimeric antigen receptors
-
(CARs). Engineered receptors that are formed by fusing the antigen-recognition domain of a specific antibody to an intracellular signalling domain. They can be used to modify effector T cells or natural killer cells.
- Immune checkpoint blockade
-
(CPB). The reversion of T cell exhaustion using monoclonal antibodies that inhibit the function of inhibitory immune receptors on the cell surface, such as programmed cell death protein 1 (PD1), PD1 ligand 1 (PDL1) and cytotoxic T lymphocyte antigen 4 (CTLA4).
- Objective response rate
-
A common efficacy end point used in clinical trials of cancer therapies in solid tumours; it is usually defined as the percentage of patients who experience at least a 30% decrease in tumour diameter on an imaging scan.
- Neoantigens
-
Antigens arising from mutation of the tumour genome that causes tumour cells to express specific proteins that are not expressed on normal cells.
- Adaptive immune resistance
-
A mechanism of resistance to the antitumour immune response whereby cancer cells are recognized by the immune system but protect themselves by adapting to the immune attack.
- Tumour heterogeneity
-
Cancer cells within the same tumour tissue can have distinct phenotypical and functional profiles as a consequence of genetic changes, environmental differences and reversible changes in cell properties.
- Epitope spreading
-
Broadening of the immune response from the initially targeted epitope to other epitopes on the same antigen or different antigens.
- Central tolerance
-
The clonal deletion of autoreactive B cells and T cells in the thymus during ontogenesis to create a state in which immune cells are unresponsive to autoantigens.
- Whole-exome sequencing
-
Sequencing of the expressed genes in a genome (known as the exome) using high-throughput DNA sequencing technology.
- Immune escape
-
The escape of a tumour from attack by the immune system through various mechanisms such as antigen loss.
- Immune adjuvant
-
A component that is designed to improve the immune response to a vaccine antigen.
Rights and permissions
About this article

Cite this article
Hu, Z., Ott, P. & Wu, C. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat Rev Immunol 18, 168–182 (2018). https://doi.org/10.1038/nri.2017.131
Published:
Issue Date:
DOI: https://doi.org/10.1038/nri.2017.131
This article is cited by
-
Screening of novel tumor-associated antigens for lung adenocarcinoma mRNA vaccine development based on pyroptosis phenotype genes
BMC Cancer (2024)
-
Tumour circular RNAs elicit anti-tumour immunity by encoding cryptic peptides
Nature (2024)
-
Computational immunogenomic approaches to predict response to cancer immunotherapies
Nature Reviews Clinical Oncology (2024)
-
Virus-like particle-mediated delivery of structure-selected neoantigens demonstrates immunogenicity and antitumoral activity in mice
Journal of Translational Medicine (2024)
-
Distinctly altered lipid components in hepatocellular carcinoma relate to impaired T cell-dependent antitumor immunity
Hepatology International (2024)
