
Key Points
-
DNA sequence variation that influences gene expression is an important source of variation in higher-order organismal traits.
-
Variants that influence transcription factor binding often affect multiple levels of chromatin organization and gene expression.
-
Genetic variation in protein levels is studied much less frequently than that in mRNA levels. Based on current knowledge, most loci that influence a given mRNA also seem to affect the abundance of the corresponding protein.
-
Model organisms show that the interplay between regulatory variation and higher-order traits can be highly complex.
-
Rapidly growing data sets and detailed functional follow-up studies show that regulatory variation contributes to disease risk in humans, sometimes in surprisingly complex ways.
Abstract
We are in a phase of unprecedented progress in identifying genetic loci that cause variation in traits ranging from growth and fitness in simple organisms to disease in humans. However, a mechanistic understanding of how these loci influence traits is lacking for the majority of loci. Studies of the genetics of gene expression have emerged as a key tool for linking DNA sequence variation to phenotypes. Here, we review recent insights into the molecular nature of regulatory variants and describe their influence on the transcriptome and the proteome. We discuss conceptual advances from studies in model organisms and present examples of complete chains of causality that link individual polymorphisms to changes in gene expression, which in turn result in physiological changes and, ultimately, disease risk.
Similar content being viewed by others

Main
Propelled by technological advances, we will soon have essentially complete catalogues of all but the rarest genetic variants in humans and several other species. Individuals in most species differ from each other by thousands to millions of DNA sequence variants. Some of this variation contributes to observable phenotypic differences in traits ranging from morphology, physiology and behaviour to predisposition to many human diseases. While the identification of variants that affect phenotypes is rapidly progressing, the fundamental challenge now is to understand how these variants exert their effects.
An important class of variants, termed expression quantitative trait loci (eQTLs), influence the expression level of genes (Box 1). The genetics of expression variation of single genes has been studied since at least 1962 (Ref. 1). Genome-wide eQTL mapping was proposed in 2001 (Ref. 2) and first carried out in its modern form at about the same time in a cross between two yeast strains3. Brem et al.3 used microarrays to measure variation in mRNA abundance for all expressed genes among recombinant offspring of these two parent strains. Relating this variation to the alleles that each offspring inherited from either parent allowed the identification of regions in the genome that harbour sequence variants that influence gene expression. The now-ubiquitous term eQTL for such regions was coined shortly thereafter4, although the related term protein quantity loci (pQLs; now more commonly known as protein QTLs (pQTLs)) was used in an examination of the genetics of protein levels for a limited number of genes in 1994 (Ref. 5). Since then, there has been tremendous progress in the study of regulatory variation. Maps of eQTLs are being built in increasingly large-scale studies in humans6,7,8,9,10,11,12 (see Ref. 13 for earlier landmark studies) (Table 1), rodents4,14,15,16,17,18,19,20, flies21,22, plants4,23,24,25,26,27,28,29,30, worms31,32 and other species. The early observations in yeast of local and distant eQTLs, eQTL hot spots, a complex genetic basis of expression traits, and connections between expression and organismal phenotypes3,33 (see below) have since been found to hold in other species.
Beyond ever larger catalogues of eQTLs, our understanding is now being expanded in two directions. Although eQTLs were typically identified as 'loci' — that is, statistical associations between regions of the genome and the expression of genes — the identity of the precise causal variants and their molecular mode of action are coming into increasingly sharper view. Additionally, there is a growing understanding of the consequences of variation in gene expression levels for organisms. This second aspect is especially important because a crucial rationale for large eQTL studies is that they can help to prioritize likely causal variants among the multiple polymorphisms within the regions identified by genome-wide association studies (GWASs), as well as to reveal the precise biological mechanisms through which DNA differences influence organismal traits. For example, the majority of loci identified in human GWASs are found in non-coding regions that are not in linkage disequilibrium with coding exons and must therefore reflect the effects of regulatory variation34.
Previous reviews have covered the various types of eQTLs and the ways in which they can be identified and fine-mapped13,35,36,37, the rich variety of molecular traits that can be assayed along the cascade of gene expression regulation38,39 and the ways to integrate these molecular traits in a systems genetics perspective40. Here, we review new insights into the molecular basis of eQTLs and the genetics of mRNA versus protein levels. We then present recent discoveries into the causal links between eQTLs and higher-order organismal phenotypes, such as physiology and disease. We describe recent experimental insights into eQTL causality (many of which were derived from model organisms) and close by presenting an overview of the emerging evidence for eQTL causality in human disease.

What are eQTLs?
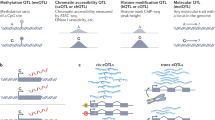
eQTLs contain sequence variants that affect the expression of a gene. They are similar to other QTLs that can influence any given trait of interest (for example, height, growth rate and disease risk) except that the trait under study is gene expression. eQTLs are identified by measuring gene expression in panels of genetically different, genotyped individuals13,36 (Boxes 1,2). These panels can be designed experimental crosses, existing pedigrees or families9 or unrelated individuals from natural populations — the most common eQTL study design in humans6,11 (Fig. 1A). eQTLs are often classified according to the relative locations of the eQTLs and the gene or genes that they influence, and according to the type of mechanism through which they affect expression (Fig. 1C).

Molecular variation is mapped in genetically variable populations. Aa | These populations can be generated through designed crosses in model organisms. For example, the genetic backgrounds of a set of yeast strains are reshuffled by mating followed by meiosis, resulting in a set of recombinant offspring. Ab | Alternatively, outbred populations can be used that carry genetic variation which was spread and recombined by historical genetic processes (illustrated by the genetic history of a hypothetical region of the genome). This is the most popular design for expression quantitative trait locus (eQTL) mapping in humans. Pedigrees or families of related individuals can also be used (not shown). B | The molecular quantity of interest is measured in each individual in the study panel. The figure illustrates the results for two individuals that differ in the expression of a certain gene. To map the loci involved (marked by the star), this molecular variation is compared to genetic variation among the individuals (Box 1). Many of the steps along the gene expression cascade can be studied in this way, including DNA methylation (Me), histone modifications, transcription factor (TF) binding, active transcription, mRNA levels (resulting in eQTLs), translation and protein levels (resulting in protein QTLs (pQTLs)). In the example, the altered protein level due to the genetic polymorphism influences disease risk, an organismal trait. C | eQTLs can be classified according to their location (local or distant to the gene they influence) and according to their mode of action (cis or trans). MRCA, most recent common ancestor.
Local eQTLs. An early observation was that some eQTLs are located near the genes they influence, whereas others are located elsewhere in the genome3. The former have been called 'local' eQTLs13. Local eQTLs can influence gene expression by two different mechanisms. Most obviously, they can act in cis and affect expression in an allele-specific manner. By definition, each allele of such eQTLs affects only the expression of the copy of the gene that is located on the same physical chromosome with it and not the expression of the copy on the homologous chromosome. Therefore, cis-eQTLs can be detected in heterozygous individuals by quantifying the relative expression levels of the two alleles, for example, by counting the number of times each allele is observed in RNA sequencing data. If there is an imbalance in the expression levels of the two alleles, then the gene is affected by a cis-eQTL41,42,43,44,45,46.
Local eQTLs do not always act in cis13,41,45 but can also act in trans. Trans-eQTLs are due to polymorphisms that alter the structure, function or expression of a diffusible factor (Fig. 1C). The resulting differential activity or abundance of this factor alters expression levels of the genes that are influenced by the trans-eQTL. As the diffusible intermediate is equally available to both alleles of a target gene, trans-eQTLs do not lead to allele-biased expression in heterozygous individuals. Furthermore, trans-eQTLs can be located anywhere in the genome relative to the genes they regulate. If they happen to be close to the given gene, then they will appear as local but not cis-acting eQTLs. An extreme example of a trans-acting local eQTL is a single amino acid substitution in the yeast AMN1 gene, which results in differential regulation of AMN1 itself through a regulatory feedback loop that involves several additional factors41. Although it has become common to use the terms 'local eQTLs' and 'cis-eQTLs' interchangeably in human eQTL studies, we advocate using the appropriate precise terminology to clearly delineate relative position from mode of action.
Local eQTLs are abundant in all species studied so far. In humans, nearly 80% of expressed genes in whole blood had a local eQTL in a recent survey of nearly 1,000 individuals47. In yeast, ~25% of genes had a local eQTL in a comparison of two different isolates41, and many more local regulatory variants are expected to exist in additional yeast strains that have not been studied so far. Indeed, a population genetic extrapolation predicted that most or all yeast genes will have local eQTLs across the global yeast diversity48.
Distant eQTLs. Distant eQTLs are defined as loci that are located further away from the genes they influence. The precise distance required for an eQTL to be distant is arbitrary and can be defined in physical or genetic distance; consequently, it differs between studies. For example, such a distance can range from 10 kb in yeast3 to 2 Mb in humans7; some studies even require distant eQTLs to be located on different chromosomes from the genes they influence49. Distant eQTLs usually act in trans. The number of distant eQTLs that have been identified so far is much more variable between species than that of local eQTLs. In yeast3,50,51, the nematode Caenorhabditis elegans31, the plant Arabidopsis thaliana23,25 and rodents16,52,53,54, there are multiple strong trans-acting-eQTL hot spots that can each affect the expression of up to hundreds of genes. In yeast, many of the expression effects at some of these hot spots are caused by variation in single genes33,50. Other yeast hot spots may contain multiple causal genes55,56 — a finding also seen in mouse mapping panels16,57. Studies of model organism panels also routinely identify large numbers of distant eQTLs that do not fall into hot spots31,50.
In contrast to these findings in model organisms, distant eQTLs have been harder to find in human population-based samples, and family-based analyses have provided mixed support for distant-eQTL hot spots9,58. The apparent difference in the prevalence of distant eQTLs between species is most likely due to the fact that the detection of distant eQTLs is more difficult in human populations than in experimental crosses. In crosses, few alleles segregate at high frequencies at each locus, and linkage blocks are relatively large, resulting in high statistical power at any position in the genome. In human population samples, there are multiple haplotypes at most positions in the genome, multiple variants per region (most of which are at low frequencies) and shorter linkage blocks. Together, these features necessitate many more association tests. The strict significance thresholds required to correct for this large number of tests result in low statistical power, making distant eQTLs harder to find. Indeed, to reduce the multiple-testing burden, most human eQTL searches have been restricted to local variation around each gene, such that distant eQTLs cannot be discovered by design. Distant eQTLs also have smaller effect sizes59 and seem to be more tissue-specific than local eQTLs59,60, which further complicates their detection.
Several recent studies estimated the relative importance of local and distant genetic variation on human mRNA abundance variation61,62,63. This can be achieved even without knowing the identity of individual eQTLs. Genome-wide genotyping data can be used to partition the variance in a trait into various sources such as genetic or environmental variation64,65. The genetic component (that is, the 'heritability') of the gene expression variance can be further partitioned into the contribution of the genomic region centred on the gene itself and the contribution of the remainder of the genome. This distant contribution accounts for the majority (60–75%) of the heritability in human gene expression61,62,63. Thus, distant eQTLs clearly exist in humans just as they do in model organisms. Indeed, as sample sizes increase in human studies, more distant eQTLs are being discovered62,63,66,67,68,69,70,71,72,73. In both yeast50 and humans67,74, the effects of distant eQTLs can change considerably in different environments. Based on the evidence so far, the trans-acting component in human populations seems to be more dispersed across many loci across the genome than the major hot spots seen in model organisms. It remains unclear whether this is due to the comparatively low power in human studies, a result of the fact that more variation is examined in human population studies than in crosses in model organisms, or a true reflection of biological differences.
The molecular chain of causality
The molecular nature of cis- eQTLs. Regulatory variation can affect organisms by interfering with any of the steps along the gene expression cascade from DNA to protein (Fig. 1B). For many years, technology largely limited eQTL studies to measures of mRNA abundance. Now, new technologies fuelled by the advances in massively parallel sequencing enable detailed examination of how sequence variation influences the individual steps of gene expression. The first wave of these studies examined transcription factor (TF) binding75,76,77,78,79, chromatin accessibility80,81, DNA methylation82,83,84,85,86, alternative splicing43,44,87,88,89, small RNAs88,90,91, large intergenic non-coding RNAs (lincRNAs)92,93, RNA editing89 and mRNA degradation94. It is now clear that all of these types of transcripts and processes can be affected by regulatory variants39. There is emerging evidence that much of the variation at multiple genomic levels is orchestrated through cis-acting sequence differences that affect TF binding.
A major advance in understanding the nature of the causal DNA variants that underlie eQTLs is the growing availability of whole-genome sequences88,95. All sequence variants are essentially known in these studies, so that the causal eQTL variants themselves (rather than a linked single-nucleotide polymorphism (SNP) on a genotyping array) will often show the highest association with gene expression. For example, a recent eQTL mapping of 462 fully sequenced human individuals found that short insertions and deletions (indels) are more likely to result in local eQTLs than SNPs88. Causal eQTL variants are further enriched in DNase I-hypersensitive sites, in regions annotated96 as active promoters and strong enhancers, and in TF binding sites88.
Investigation of allele-specific histone modifications, TF binding and mRNA levels in human parent–offspring trios has provided additional support for the importance of variation in TF binding97,98,99. Sequence variants that are located in TF binding sites are correlated not only with variation in TF binding itself but also with differential histone modifications, mRNA levels and DNA methylation84. A parsimonious explanation for these observations is that differential TF binding is the primary molecular change. In turn, the TFs then direct the changes in histone modifications, DNA methylation and mRNA expression.
These global analyses complement multiple examples of individual human cis-eQTLs that are caused by sequence differences in TF binding sites49,74,100,101,102,103. However, only 25–35% of genetically variable TF binding events are associated with a known sequence variant within the corresponding TF binding core motif77,79,98,99. Some of the remaining cases may be due to missed motif variants. Alternatively, causal sequence variants outside core motifs may influence TF binding, perhaps by altering the local shape of the DNA or by influencing the binding of other TFs that form complexes with the assayed TF. An interesting variation on this theme is a 2-bp deletion in a promoter that segregates among yeast strains and causes variation in the expression of the ERG28 gene104. The deletion allele does not disrupt a TF binding motif but instead moves two neighbouring TF binding sites closer together, resulting in reduced binding by both factors. Experimental dissection of additional cis-eQTLs will reveal the full spectrum of their molecular causes.
The molecular nature of trans- eQTLs. Trans-eQTLs can be due to a diverse set of molecular causes. They can be coding variants in regulatory genes or local eQTLs of such genes. Work in yeast showed that the regulatory genes that act as trans factors themselves have diverse functions. Proteins encoded by trans-acting yeast eQTLs are not enriched for TFs33. Instead, the functions they encode range from RNA-binding proteins (such as MKT1)55,105,106 to members of signalling cascades (such as IRA2 (Ref. 50) and GPA1 (Ref. 33)) and modifiers of nucleosome composition (such as swc5 in Schizosaccharomyces pombe51).
Recent work in humans is beginning to reveal similar molecular diversity among trans-eQTLs66,67,68,70,74. For example, IRF7 (which encodes interferon regulatory factor 7, a transcription factor) is influenced by a local eQTL in activated dendritic cells, a type of immune cell. The same eQTL SNP is associated with the expression of a set of genes in trans74. Experimental overexpression of IRF7 influences the same set of genes, demonstrating that the altered expression of IRF7 caused by the local eQTL is responsible for driving further expression changes74. Adding another level of complexity, in human monocytes IRF7 is influenced by another trans-acting locus that maps to a local eQTL in EBI2 (also known as GPR183, which encodes a G protein-coupled receptor)69. Other trans-eQTLs in humans include an amino acid substitution in a cytochrome P450 enzyme67 that reduces the half-life of the protein107, a local eQTL for the LYZ gene encoding the secreted enzyme lysozyme66,67,68, and multiple associations within the highly variable human major histocompatibility complex (MHC) region66,67. When more human trans-eQTLs have been fine-mapped, it will be interesting to examine whether certain types of molecular causes (coding versus regulatory) or genes (encoding TFs, signalling molecules or others) are more likely to result in trans-acting variation.
From the transcriptome to the proteome. The vast majority of current studies use mRNA rather than protein abundance as the measure of gene expression. However, coding genes ultimately function through their protein products. The experimental preference for mRNA is because transcript levels can be measured more easily than protein levels. Whereas eQTL studies can rely on standardized methods that readily quantify most of the transcripts in the genome108, the few studies that have examined proteome variation used a wide range of assays, including commercial antibody kits for certain blood proteins109, 2D difference gel electrophoresis110, microwestern arrays111 and different types of mass spectrometry112,113,114,115,116,117,118. Many of these studies were limited in the number of samples and/or proteins.
Two questions arise when comparing genetic influences on the transcriptome and the proteome. Does a typical eQTL feed forward into variation in protein levels? Conversely, are most pQTLs simply a reflection of the underlying mRNA variation, or do they arise from genetic influences on post-transcriptional mechanisms? These questions are an important area of active research, and a consistent picture has yet to emerge. The first direct comparisons of eQTLs and pQTLs obtained by mass spectrometry in model organisms57,119,120 suggested that most eQTLs did not seem to influence protein levels and, conversely, that most pQTLs seemed to arise without corresponding differences in mRNA abundance.
More recent proteome-wide studies reported better agreement between eQTLs and pQTLs. For example, an analysis of 22 diverse yeast strains found that most of the identified eQTLs had concordant effects on protein levels118. Studies in human cell lines have not only reported substantial overlap between eQTLs and pQTLs but also detected a number of protein-specific pQTLs without apparent effects on mRNA111,112,117. The effects of eQTLs seemed to be attenuated at the protein level relative to their effects on mRNA levels112. These studies used modest numbers of individuals (<100) so that only local loci with large effects could be examined. Some of the 'missing' eQTLs or pQTLs might therefore have been due to low statistical power.
To overcome the limitation of sample size, a novel experimental design was recently introduced in yeast. The approach allows the detection of distant and local pQTLs by comparing the genetic make-up of pools of single cells with very high protein levels to those with very low protein levels from populations of hundreds of thousands of genetically different cells121,122. Both studies showed that more than half of the known distant eQTLs that previously seemed to be mRNA-specific do in fact have concordant pQTLs. Importantly, the high detection power also revealed that a typical protein is affected by several times more distant pQTLs than seen before. It remains to be seen whether the many newly discovered pQTLs only arise at the protein level or whether they reflect eQTLs with small effects that have not been detected so far.
Protein translation may provide a plausible target for sequence variants that influence protein, but not mRNA, levels123,124. However, recent studies that used ribosome profiling125 in yeast126,127 and in humans112 found strong concordance between eQTLs and ribosome occupancy QTLs, suggesting that protein-specific genetic effects are likely to act beyond the stage of translation.
Lessons from model organisms
Testing causality of human variants. Model organisms can provide insights into the consequences of sequence variants that are suspected to be important in human disease. For instance, it is possible to engineer the genome and observe the effects of a given sequence variant on the whole organism rather than on a molecular or cellular trait. A regulatory element upstream of the MYC oncogene (which encodes a TF), for example, contains a SNP that is associated with cancer in humans. The SNP alleles lead to differential expression of MYC128, but the association between differential expression of MYC and cancer was inconclusive. To test this, transgenic mice were generated in which the mouse orthologue of the SNP-containing cis-regulatory element (CRE) was deleted129. Ablation of the CRE resulted in modestly reduced Myc expression levels. Crucially, the CRE-deleted mice were also more resistant to tumorigenesis. However, although this work shows that the CRE is an important determinant of cancer risk, it does not prove that the SNP within the element contributes to this risk through gene expression. To formally achieve this, it would be necessary to generate transgenic mice that differ in the SNP alleles.
The Myc study in mice was facilitated by the fact that the orthologous CRE actually exists in mice, in spite of the fact that individual regulatory elements turn over rapidly in mammalian evolution130. Many other human CREs would have to be inserted into the mouse genome in order to study their sequence variation. For example, transgenic mice were created that carried a human CRE that is not present in mice and that regulates the KIT ligand (Kitl) gene131. The mice were engineered to carry either of the alleles of a human SNP inside the CRE. The SNP was suspected to contribute to blond hair in humans. The two CRE alleles drove differential expression of Kitl in the skin and also resulted in a difference in mouse coat colour, suggesting that this variant may influence human physical appearance through altered gene regulation.
Genetic associations shared between species. The powerful mapping panels in model organisms are designed to maximize statistical power to detect genetic associations from sets of defined genetic backgrounds. The panels can be used to identify novel links between sequence variants, expression and disease40. Although the individual causal DNA variants are most probably not the same as those in humans, the same genes, pathways and networks may nevertheless harbour important genetic variation in several species. For example, eQTL maps from a panel of genetically heterogeneous rats revealed a network of immune-related genes69 centred on the TF-encoding gene Irf7. In turn, the expression of Irf7 is influenced in trans by a cis-acting eQTL for the Ebi2 gene. Remarkably, an expression network with a similar structure to that in rats was also found in human monocytes69. As in rats, the human network was also influenced in trans by a cis-eQTL for EBI2. One of the human EBI2 cis-eQTL SNPs was found to be associated with the autoimmune disease type I diabetes, an association that had been missed previously.
Conceptual insights into causality. In addition to applications with direct relevance for human disease, model organisms can provide powerful conceptual insights into the relationship between regulatory variation and organismal traits. Genetic tools that allow precise genome engineering and experimental control of gene expression make yeast an especially suitable organism for such studies. These advantages allowed the first identification of a trans-acting eQTL that causes trait variation through expression changes. The protein encoded by the AMN1 gene differs between a common laboratory budding yeast strain and other strains at two amino acids33. The Amn1 protein is a negative regulator of ACE2, which encodes a TF132. In turn, Ace2 activates CTS1 (Ref. 133), which encodes chitinase, an enzyme required for cell separation134. The laboratory strain allele of Amn1 fails to repress ACE2, resulting in upregulation of CTS1 and separation between mother and daughter cells. In other strains, CTS1 expression is lower, and budding cells stick together to result in 'clumpy' growth33.
Recent work in yeast has provided further insights into how genetic effects on expression shape phenotypes (Fig. 2). Rest et al.135 showed that effects on fitness of the expression level of the essential gene LCB2 are nonlinear and depend on both the environment and the genetic background (Fig. 2A). They replaced the native LCB2 promoter with a promoter system that can be used to finely control LCB2 expression and found that the reduction in expression level compared with the wild-type level led to a sharp decrease in fitness. By contrast, overexpression had only minor fitness effects. Growth in the presence of osmotic stress preserved the shape of the expression–fitness curve but shifted it such that higher expression of LCB2 was required for a given fitness level. Wild yeast strains had lower expression levels of LCB2 than those seen in the laboratory strain, suggesting that the expression–fitness curve is also shifted by different genetic backgrounds135.

A | Fitness consequences of experimentally altered expression of the LCB2 gene are shown135. Note that even low levels of upregulation or downregulation can have drastically different consequences on fitness, depending on the baseline expression level and the environment. B | Genetic effects on gene expression caused by four well-characterized single-nucleotide polymorphisms (SNPs) that had been originally identified through their effects on yeast sporulation efficiency137. The genes differ from each other in the degree to which their expression is influenced by the four SNPs and in the relative importance of epistatic effects compared with additive effects. For comparison, the effects of the four SNPs on sporulation efficiency are shown in the bar furthest to the right. The impact of these SNPs on the organismal trait (sporulation) cannot be simply explained by their effect on the expression of any one gene. C | Gagneur et al.136 studied the possible flows of causality from DNA variation between two yeast strains (Y and S) to gene expression and yeast growth rate (part Ca). The correspondence of expression quantitative trait locus (eQTL) hot spots (black peaks) and growth QTLs (coloured bars) varies substantially among different environments (part Cb). Part A reproduced from Rest, J. S. et al. Nonlinear fitness consequences of variation in expression level of a eukaryotic gene. Mol. Biol. Evol. (2013) 30(2), 448–456, by permission of Oxford University Press. Parts B and C from Ref. 137 and Ref. 136, respectively.
Yeast eQTL hot spots often overlap with loci that influence growth rates. This observation could arise when gene expression differences cause growth differences or vice versa, as well as when expression and growth do not affect each other but are both caused by the same locus or by different loci in close proximity (Fig. 2C). To distinguish among these scenarios, Gagneur et al.136 mapped yeast eQTLs in five environmental conditions and compared the eQTLs to loci that influence growth in the respective conditions. Whenever a QTL affected growth in a given condition, this locus also affected the expression of multiple genes in that condition; that is, the locus was an eQTL hot spot. However, some QTLs affected growth in only a few conditions but remained eQTL hot spots even in conditions in which they did not affect growth. When a growth QTL was detected in several conditions, it was also an eQTL for some genes in all of these conditions but for other genes in only some of the conditions. The authors used a statistical model to show that those genes influenced by a hot spot irrespective of growth condition were more likely to cause growth differences than genes with condition-dependent eQTLs136.
Two studies used allelic engineering to show that four nucleotide changes in three TFs together explain nearly all of the variation in sporulation efficiency between two yeast isolates137,138 (Fig. 2B). By contrast, these same four variants accounted for much less variation in gene expression at the time point at which cells switch to the meiotic state137. The effects of these SNPs on gene expression had different magnitudes and different degrees of additive versus epistatic contributions compared with those on sporulation, suggesting that there is no simple direct relationship between the effects of these specific nucleotide variants on gene expression and on the cellular phenotype. Together, these yeast studies show that the relationship between gene expression variation and higher-order traits can be highly complex, and they provide context for interpreting links between eQTLs and disease in humans.
The role of eQTLs in human disease
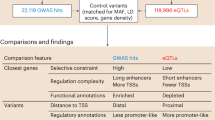
A region identified by GWASs as associated with disease typically contains more than one gene and multiple sequence variants that are in linkage disequilibrium with each other. The next task is to find the causal genes and sequence variants, and to understand how they affect the disease. Although variants that alter coding sequences are obvious candidates, most human GWAS hits fall far from coding regions of genes and are overrepresented in regulatory elements34,139. Therefore, most causal variants probably influence traits by altering gene expression. A GWAS hit typically contains multiple regulatory elements, and these elements can influence genes at some distance140. It is therefore not easy to pinpoint the causal variants and to discern the genes that they affect. eQTLs can provide the crucial link between the variants in a GWAS region and the biological processes they affect.
Bigger data, better maps. When a GWAS hit is also an eQTL for a given gene, this provides the hypothesis that the expression of this gene influences the disease. It is now firmly established that GWAS hits for common diseases are enriched for eQTLs, and vice versa. This enrichment was first noted in a comparison of associations for traits such as body mass index to eQTLs in adipose tissues and blood10, and was later observed in comparisons of GWAS results and eQTLs mapped in immortalized cell lines141,142. There have since been many eQTL studies in tissues with relevance for a given disease, providing numerous cases of GWAS–eQTL pairs that suggest plausible causal mechanisms (Table 1).
To maximize the chance of finding GWAS–eQTL links, eQTLs need to be mapped in additional tissues143,144. For example, Kapoor et al.145 identified a causal regulatory variant that influences heart function by altering an enhancer in heart tissue; this variant had been missed in previous eQTL studies of different tissues. Reference panels of eQTLs identified in multiple human tissues146 will be useful in this regard, as are current efforts to map eQTLs in purified primary individual cell types (such as certain subtypes of immune cells66,67,68,73,74,147,148,149,150,151) rather than in tissues that contain a mixture of cell types (such as whole blood). Such analyses of purified cell types are especially important because eQTL architectures can change dynamically during developmental differentiation of related cell types, as demonstrated in the haematopoietic cell lineage in mice152,153. For tissues that are difficult to obtain from primary sources, induced pluripotent stem cells (iPSCs)154, which can be differentiated into various cell types, provide a promising alternative. As a crucial prerequisite for using iPSCs, it was recently shown that gene expression differences between iPSCs derived from several donors are larger than the technical variation induced by cellular reprogramming and cell culture155.
Even in tissues and cell types that have been studied, the currently available maps are incomplete. This is partly because eQTL architectures can change considerably in cells exposed to different growth conditions, as initially demonstrated in yeast50,106,136. A rapidly growing set of studies is mapping eQTLs in human cells exposed to physiologically relevant stimuli. So far, the main focus of this work is on comparing eQTLs in unstimulated immune cells66,68,73,147,148,149,151 to eQTLs that are only seen after these cells have been activated by triggering the immune response16,67,74,150,156,157. Immune cell activation can reveal large numbers of eQTLs that are hidden in resting cells158. Stimulus-dependent eQTLs in whole blood also help to explain individual differences in the transcriptional response to vaccination159.
Many eQTLs are missed owing to low statistical power in small samples. As sample sizes increase towards thousands of individuals, eQTL catalogues have grown remarkably. Recent studies of a thousand or more individuals report eQTLs for the majority of genes expressed in a given tissue47,63,70. Many of these eQTLs have small effects that were beyond the detection limit of the earlier, smaller panels, and there are likely to be thousands more eQTLs beyond our current statistical reach.
Analytical challenges and opportunities. A common approach for prioritizing likely causal variants among the variants that are linked to a genomic region implicated by GWASs is to focus on variants that are eQTLs in a published data set (Table 1) and on variants that are located in functional elements such as promoters or enhancers160,161,162,163. Similar to eQTLs, maps of such regulatory features are available in a growing number of tissues and cell types96,161. As both eQTL catalogues and maps of regulatory features continue to grow, more and more eQTLs will be found to colocalize with regulatory regions, requiring decisions on which overlaps are the most informative. Furthermore, GWAS signals would ideally be compared to eQTLs and regulatory features obtained from the 'causal' cell type in which the given disease emerges. However, for many diseases and traits it is not a priori clear which cell type is the most relevant.
Recently, integrative Bayesian methods have been developed to identify sets of functional sequence annotations (for example, from sets of regulatory elements from many different cell lines) that are the most relevant for the given trait in an unbiased manner139,164,165,166,167,168. These methods complement similar approaches to fine-map the causal sites within eQTLs themselves47,105,169,170, to jointly analyse eQTLs across different tissues171 and to predict expression levels from genotypes172. When annotations inferred to be important for the trait come from a certain tissue, this suggests that the tissue is biologically important for the trait. Not only can these links support known connections (for example, between high-density lipoprotein (HDL) levels and regulatory elements in liver cells164), but they can also generate new hypotheses. For example, GWAS hits for platelet volume were enriched in regulatory elements in the spleen, which is not obviously connected to this trait164. In turn, the annotations inferred to be the most important can then be used to select the most promising variants for future study.
The approaches show promise in fine-mapping individual GWAS regions165 but often still result in sets of multiple potentially causal variants. A natural extension of these methods is to also consider whether each variant is an eQTL166,168. For example, in one analysis eQTLs were found to be the most important source of information when prioritizing among GWAS hits for autoimmune disorders166. Further information can be gained by including published functional knowledge about the gene that the eQTL regulates and by studying whether it is known to have functions related to the disease173. This type of information will be particularly useful when a given eQTL influences the expression of multiple genes, perhaps in different tissues. Other Bayesian approaches formally test whether an eQTL and a GWAS hit are due to the same causal variant rather than to two closely linked variants174, and may gain predictive power by considering functional annotations. As the diverse genomic data sets continue to grow, rigorous integration across them will be crucial.
Bridges across the causality gap. Several examples of causal links between human regulatory variants and disease have been uncovered in recent years49,101,102,103,145,175,176 (see Ref. 177 for a review of earlier work). A prominent example is a common SNP at 1p13, a locus associated with the risk of myocardial infarction100 (Fig. 3). Remarkably, although the SNP is located in the 3′ untranslated region of a gene, its causal effect on infarction is not mediated through this gene. Instead, the minor SNP allele, which is associated with reduced risk, creates a binding site for a TF that is preferentially expressed in the liver100. As a consequence, the sortilin 1 (SORT1) gene, which is ~40 kb away and separated from the causal SNP by 2 additional genes, is upregulated specifically in the liver. Knockdown and overexpression studies in mouse liver confirmed that higher expression of the sortilin protein results in lower levels of low-density lipoprotein cholesterol (LDL-C). In turn, LDL-C is a well-known risk factor for myocardial infarction, providing the final link in the causal connection between this eQTL and a major human disease.

a | The minor allele of a non-coding single-nucleotide polymorphism (SNP) in the 3′ untranslated region (3′UTR) of the CELSR2 (cadherin, EGF LAG seven-pass G-type receptor 2) gene creates a transcription factor binding site for CCAAT/enhancer-binding protein (C/EBP), to which the major allele does not bind100. Binding of C/EBP at this site leads to increased expression of the sortilin 1 (SORT1) gene in liver cells. b | In mice, overexpression of Sort1 in the liver reduces low-density lipoprotein cholesterol (LDL-C) levels (calculated using fractions 10–26). c | Small interfering RNA (siRNA)-mediated knockdown of Sort1 increases LDL-C levels in mice. As LDL-C, in turn, is a known risk factor for myocardial infarction, this work provides a complete causal path from a non-coding variant to altered risk for a major human disease. SORT1 is separated from the causal SNP by two additional genes, and the causal effect on LDL-C is not mediated through CELSR2, although the causal SNP is in the 3′UTR of this gene. Chr1, chromosome 1; MYBPHL, myosin-binding protein H-like; PSMA5, proteasome (prosome, macropain) subunit, alpha type, 5; PSRC1, proline/serine-rich coiled-coil 1; SARS, seryl-tRNA synthetase; SYPL2, synaptophysin-like 2. Figure adapted from Ref. 100, Nature Publishing Group.
A second case involves SNPs associated with obesity that are located within introns of the fat mass and obesity-associated (FTO) gene178. As knockout of Fto results in leaner mice179, it had been suspected (but not demonstrated) that the causal variants might act through expression levels of FTO. However, recent work showed that the genomic region containing the variants in the obesity-associated region is in physical contact with the iroquois homeobox 3 (IRX3) gene, which is located at a distance of ~500 kb from the variants180. SNPs in the obesity-associated region also showed association with the expression of IRX3 in human cerebellum, albeit with only nominal significance. Phenotypes of mouse knockout models of Irx3 are consistent with a causal role of IRX3 in obesity. More work will be needed to identify the precise causal variant or variants within the obesity-associated region and to determine whether their effects on obesity are mediated primarily by IRX3 expression variation.
Notably, these examples both involve SNPs in regulatory sequences that are located some distance away from the genes they influence. Such long-range regulatory interactions are usually mediated by physical contact between the regulatory DNA and the regulated gene. Targeted181,182,183,184,185 and global186 methods for mapping the physical interactions of GWAS hits will be useful to systematically dissect the regulatory consequences of disease-associated variants, as recently demonstrated for several breast cancer risk loci184.
Conclusions and perspectives
The work we have reviewed here demonstrates that eQTLs have important roles in influencing downstream traits ranging from yeast growth and fitness to human disease. The connections along the causal chain from DNA variant to altered expression and trait variation can be surprisingly complex. Dissection of the functional impact of regulatory variation will continue to require careful experimental follow-up work. The existing and rapidly growing catalogues of eQTLs will enable more precise targeting of these efforts. Large sample sizes will be crucial to ensure that no relevant eQTLs are missed, especially the more elusive trans-acting variants. To ensure that the relevant biology is captured, eQTL maps need to be constructed in a wide range of tissues and cell types, and under a variety of physiologically important conditions. When tissues are difficult to obtain or when they represent complex mixtures of cells (such as brain tissues), differentiated iPSCs187 provide promising alternatives. Methods to study expression patterns in single cells obtained directly from dissociated complex tissues188,189 might also be useful in this regard. Prioritizing putative causal links between DNA variation, expression and phenotypes will require further development of sophisticated data integration techniques.
Furthermore, in addition to studying the downstream effects of eQTLs, it will be equally important to more completely explore the genetic and molecular architecture of gene expression variation. Are expression levels themselves as polygenic as many disease traits, or do they have simpler architectures? How varied are the genetic architectures of the thousands of genes in a genome190? Does the importance of epistatic variance differ between gene expression56,191,192,193,194 and higher-order traits195,196? How plastic are gene expression architectures in different environments50,67,74,106,192,197? A major challenge is the identification of large catalogues of causal variants that underlie eQTLs. Rapid advances in genome editing — for example, using the CRISPR–Cas9 (clustered regularly interspaced short palindromic repeat–CRISPR-associated protein 9) system198,199 — will greatly speed up our ability to experimentally validate and investigate the effects of putative causative variants. Ultimately, a complete genetic and molecular understanding of how genetic variation shapes gene expression and cell biology will be useful for improving predictive models of the consequences of new mutations and for personalized genomic medicine.
References
Schwartz, D. Genetic studies on mutant enzymes in maize. III. Control of gene action in the synthesis of pH 7.5 esterase. Genetics 47, 1609–1615 (1962).
Jansen, R. C. & Nap, J. P. Genetical genomics: the added value from segregation. Trends Genet. 17, 388–391 (2001).
Brem, R. B., Yvert, G., Clinton, R. & Kruglyak, L. Genetic dissection of transcriptional regulation in budding yeast. Science 296, 752–755 (2002). This is the first genome-wide eQTL study, carried out in a cross between two yeast strains.
Schadt, E. E. et al. Genetics of gene expression surveyed in maize, mouse and man. Nature 422, 297–302 (2003). This is the first eQTL study in mammals (mice and humans) and in maize.
Damerval, C., Maurice, A., Josse, J. M. & de Vienne, D. Quantitative trait loci underlying gene product variation: a novel perspective for analyzing regulation of genome expression. Genetics 137, 289–301 (1994).
Stranger, B. E. et al. Genome-wide associations of gene expression variation in humans. PLoS Genet. 1, e78 (2005).
Stranger, B. E. et al. Population genomics of human gene expression. Nature Genet. 39, 1217–1224 (2007).
Dimas, A. S. et al. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science 325, 1246–1250 (2009).
Morley, M. et al. Genetic analysis of genome-wide variation in human gene expression. Nature 430, 743–747 (2004). This is the first large eQTL study in human families.
Emilsson, V. et al. Genetics of gene expression and its effect on disease. Nature 452, 423–428 (2008).
Cheung, V. G. et al. Mapping determinants of human gene expression by regional and genome-wide association. Nature 437, 1365–1369 (2005). References 7 and 11 are the first studies to use GWAS results to map eQTLs in humans.
Stranger, B. E. et al. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science 315, 848–853 (2007).
Rockman, M. V. & Kruglyak, L. Genetics of global gene expression. Nature Rev. Genet. 7, 862–872 (2006). This review presents the conceptual basis of eQTLs and provides a comprehensive overview of the first 5 years of eQTL discovery.
Kelly, S. A., Nehrenberg, D. L., Hua, K., Garland, T. & Pomp, D. Functional genomic architecture of predisposition to voluntary exercise in mice: expression QTL in the brain. Genetics 191, 643–654 (2012).
Heyne, H. O. et al. Genetic influences on brain gene expression in rats selected for tameness and aggression. Genetics 198, 1277–1290 (2014).
Orozco, L. D. et al. Unraveling inflammatory responses using systems genetics and gene-environment interactions in macrophages. Cell 151, 658–670 (2012).
Hubner, N. et al. Integrated transcriptional profiling and linkage analysis for identification of genes underlying disease. Nature Genet. 37, 243–253 (2005).
Aylor, D. L. et al. Genetic analysis of complex traits in the emerging collaborative cross. Genome Res. 21, 1213–1222 (2011).
Chen, Y. et al. Variations in DNA elucidate molecular networks that cause disease. Nature 452, 429–435 (2008).
Schadt, E. E. et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nature Genet. 37, 710–717 (2005).
Massouras, A. et al. Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet. 8, e1003055 (2012).
King, E. G., Sanderson, B. J., McNeil, C. L., Long, A. D. & Macdonald, S. J. Genetic dissection of the Drosophila melanogaster female head transcriptome reveals widespread allelic heterogeneity. PLoS Genet. 10, e1004322 (2014).
West, M. A. et al. Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics 175, 1441–1450 (2006).
Swanson-Wagner, R. A. et al. Paternal dominance of trans-eQTL influences gene expression patterns in maize hybrids. Science 326, 1118–1120 (2009).
Fu, J. et al. System-wide molecular evidence for phenotypic buffering in Arabidopsis. Nature Genet. 41, 166–167 (2009).
Keurentjes, J. J. et al. Regulatory network construction in Arabidopsis by using genome-wide gene expression quantitative trait loci. Proc. Natl Acad. Sci. 104, 1708–1713 (2007).
Cubillos, F. A. et al. Expression variation in connected recombinant populations of Arabidopsis thaliana highlights distinct transcriptome architectures. BMC Genomics 13, 117 (2012).
Fu, J. et al. RNA sequencing reveals the complex regulatory network in the maize kernel. Nature Commun. 4, 2832 (2013).
Holloway, B., Luck, S., Beatty, M., Rafalski, J. A. & Li, B. Genome-wide expression quantitative trait loci (eQTL) analysis in maize. BMC Genomics 12, 336 (2011).
Lowry, D. B. et al. Expression quantitative trait locus mapping across water availability environments reveals contrasting associations with genomic features in Arabidopsis. Plant Cell 25, 3266–3279 (2013).
Rockman, M. V., Skrovanek, S. S. & Kruglyak, L. Selection at linked sites shapes heritable phenotypic variation in C. elegans. Science 330, 372–376 (2010).
Li, Y. et al. Mapping determinants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet. 2, e222 (2006).
Yvert, G. et al. Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nature Genet. 35, 57–64 (2003).
Maurano, M. T. et al. Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012).
Emerson, J. J. & Li, W. H. The genetic basis of evolutionary change in gene expression levels. Phil. Trans. R. Soc. B 365, 2581–2590 (2010).
Skelly, D. A., Ronald, J. & Akey, J. M. Inherited variation in gene expression. Annu. Rev. Genomics Hum. Genet. 10, 313–332 (2009).
Battle, A. & Montgomery, S. B. Determining causality and consequence of expression quantitative trait loci. Hum. Genet. 133, 727–735 (2014).
Dermitzakis, E. T. Cellular genomics for complex traits. Nature Rev. Genet. 13, 215–220 (2012).
Gaffney, D. J. Global properties and functional complexity of human gene regulatory variation. PLoS Genet. 9, e1003501 (2013).
Civelek, M. & Lusis, A. J. Systems genetics approaches to understand complex traits. Nature Rev. Genet. 15, 34–48 (2014).
Ronald, J., Brem, R. B., Whittle, J. & Kruglyak, L. Local regulatory variation in Saccharomyces cerevisiae. PLoS Genet. 1, e25 (2005).
Wittkopp, P. J., Haerum, B. K. & Clark, A. G. Evolutionary changes in cis and trans gene regulation. Nature 430, 85–88 (2004).
Montgomery, S. B. et al. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464, 773–777 (2010).
Pickrell, J. K. et al. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464, 768–772 (2010).
Doss, S., Schadt, E. E., Drake, T. A. & Lusis, A. J. Cis-acting expression quantitative trait loci in mice. Genome Res. 15, 681–691 (2005).
Yan, H., Yuan, W., Velculescu, V. E., Vogelstein, B. & Kinzler, K. W. Allelic variation in human gene expression. Science 297, 1143 (2002).
Battle, A. et al. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 24, 14–24 (2014).
Ronald, J. & Akey, J. M. The evolution of gene expression QTL in Saccharomyces cerevisiae. PLoS ONE 2, e678 (2007).
Göring, H. H. et al. Discovery of expression QTLs using large-scale transcriptional profiling in human lymphocytes. Nature Genet. 39, 1208–1216 (2007).
Smith, E. N. & Kruglyak, L. Gene–environment interaction in yeast gene expression. PLoS Biol. 6, e83 (2008).
Clement-Ziza, M. et al. Natural genetic variation impacts expression levels of coding, non-coding, and antisense transcripts in fission yeast. Mol. Syst. Biol. 10, 764 (2014).
Langley, S. R. et al. Systems-level approaches reveal conservation of trans-regulated genes in the rat and genetic determinants of blood pressure in humans. Cardiovasc. Res. 97, 653–665 (2013).
Bystrykh, L. et al. Uncovering regulatory pathways that affect hematopoietic stem cell function using 'genetical genomics'. Nature Genet. 37, 225–232 (2005).
Chesler, E. J. et al. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nature Genet. 37, 233–242 (2005).
Zhu, J. et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nature Genet. 40, 854–861 (2008).
Brem, R. B., Storey, J. D., Whittle, J. & Kruglyak, L. Genetic interactions between polymorphisms that affect gene expression in yeast. Nature 436, 701–703 (2005).
Ghazalpour, A. et al. Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 7, e1001393 (2011).
Monks, S. A. et al. Genetic inheritance of gene expression in human cell lines. Am. J. Hum. Genet. 75, 1094–1105 (2004).
Petretto, E. et al. Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet. 2, e172 (2006).
van Nas, A. et al. Expression quantitative trait loci: replication, tissue- and sex-specificity in mice. Genetics 185, 1059–1068 (2010).
Price, A. L. et al. Single-tissue and cross-tissue heritability of gene expression via identity-by-descent in related or unrelated individuals. PLoS Genet. 7, e1001317 (2011).
Grundberg, E. et al. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nature Genet. 44, 1084–1089 (2012).
Wright, F. A. et al. Heritability and genomics of gene expression in peripheral blood. Nature Genet. 46, 430–437 (2014).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nature Genet. 42, 565–569 (2010).
Lynch, M. & Walsh, B. Genetics and Analysis of Quantitative Traits (Sinauer Associates, 1998).
Fairfax, B. P. et al. Genetics of gene expression in primary immune cells identifies cell type-specific master regulators and roles of HLA alleles. Nature Genet. 44, 502–510 (2012).
Fairfax, B. P. et al. Innate immune activity conditions the effect of regulatory variants upon monocyte gene expression. Science 343, 1246949 (2014).
Rotival, M. et al. Integrating genome-wide genetic variations and monocyte expression data reveals trans-regulated gene modules in humans. PLoS Genet. 7, e1002367 (2011).
Heinig, M. et al. A trans-acting locus regulates an anti-viral expression network and type 1 diabetes risk. Nature 467, 460–464 (2010). This study reveals a physiologically important regulatory network in monocytes that is conserved between rats and humans, and that is affected by genetic variation in both species.
Westra, H.-J. et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nature Genet. 45, 1238–1243 (2013). With >5,000 samples, this meta-analysis is the largest human eQTL study so far. The authors use the large sample size to identify >100 trans -eQTLs.
Fehrmann, R. S. et al. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 7, e1002197 (2011).
Bryois, J. et al. Cis and trans effects of human genomic variants on gene expression. PLoS Genet. 10, e1004461 (2014).
Raj, T. et al. Polarization of the effects of autoimmune and neurodegenerative risk alleles in leukocytes. Science 344, 519–523 (2014).
Lee, M. N. et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 343, 1246980 (2014).
McDaniell, R. et al. Heritable individual-specific and allele-specific chromatin signatures in humans. Science 328, 235–239 (2010).
Reddy, T. E. et al. Effects of sequence variation on differential allelic transcription factor occupancy and gene expression. Genome Res. 22, 860–869 (2012).
Kasowski, M. et al. Variation in transcription factor binding among humans. Science 328, 232–235 (2010).
Heinz, S. et al. Effect of natural genetic variation on enhancer selection and function. Nature 503, 487–492 (2013).
Ding, Z. et al. Quantitative genetics of CTCF binding reveal local sequence effects and different modes of X-chromosome association. PLoS Genet. 10, e1004798 (2014).
Degner, J. F. et al. DNase I sensitivity QTLs are a major determinant of human expression variation. Nature 482, 390–394 (2012).
Lee, K. et al. Genetic landscape of open chromatin in yeast. PLoS Genet. 9, e1003229 (2013).
Heyn, H. et al. DNA methylation contributes to natural human variation. Genome Res. 23, 1363–1372 (2013).
Bell, J. T. et al. DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol. 12, R10 (2011).
Banovich, N. E. et al. Methylation QTLs are associated with coordinated changes in transcription factor binding, histone modifications, and gene expression levels. PLoS Genet. 10, e1004663 (2014).
Gutierrez-Arcelus, M. et al. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. eLife 2, e00523 (2013).
McRae, A. F. et al. Contribution of genetic variation to transgenerational inheritance of DNA methylation. Genome Biol. 15, R73 (2014).
Pickrell, J. K., Pai, A. A., Gilad, Y. & Pritchard, J. K. Noisy splicing drives mRNA isoform diversity in human cells. PLoS Genet. 6, e1001236 (2010).
Lappalainen, T. et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506–511 (2013). This is the first paper to integrate fully sequenced genomes with RNA sequencing-based transcriptomes in a large human population, which provides many important insights into the functional diversity of the genetics of the transcriptome.
Hassan, M. A., Butty, V., Jensen, K. D. & Saeij, J. P. The genetic basis for individual differences in mRNA splicing and APOBEC1 editing activity in murine macrophages. Genome Res. 24, 377–389 (2014).
Civelek, M. et al. Genetic regulation of human adipose microRNA expression and its consequences for metabolic traits. Hum. Mol. Genet. 22, 3023–3037 (2013).
Parts, L. et al. Extent, causes, and consequences of small RNA expression variation in human adipose tissue. PLoS Genet. 8, e1002704 (2012).
Popadin, K., Gutierrez-Arcelus, M., Dermitzakis, E. T. & Antonarakis, S. E. Genetic and epigenetic regulation of human lincRNA gene expression. Am. J. Hum. Genet. 93, 1015–1026 (2013).
Kumar, V. et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 9, e1003201 (2013).
Pai, A. A. et al. The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet. 8, e1003000 (2012).
Montgomery, S. B., Lappalainen, T., Gutierrez-Arcelus, M. & Dermitzakis, E. T. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 7, e1002144 (2011).
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012).
McVicker, G. et al. Identification of genetic variants that affect histone modifications in human cells. Science 342, 747–749 (2013).
Kilpinen, H. et al. Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science 342, 744–747 (2013).
Kasowski, M. et al. Extensive variation in chromatin states across humans. Science 342, 750–752 (2013).
Musunuru, K. et al. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature 466, 714–719 (2010). This paper provides an example of how a regulatory sequence change leads to gene expression variation at a distant gene, which in turn influences cholesterol levels and the risk for myocardial infarction.
Harismendy, O. et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature 470, 264–268 (2011).
Emison, E. S. et al. Differential contributions of rare and common, coding and noncoding Ret mutations to multifactorial Hirschsprung disease liability. Am. J. Hum. Genet. 87, 60–74 (2010).
De Gobbi, M. et al. A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science 312, 1215–1217 (2006).
Chang, J. et al. The molecular mechanism of a cis-regulatory adaptation in yeast. PLoS Genet. 9, e1003813 (2013).
Lee, S.-I. et al. Learning a prior on regulatory potential from eQTL data. PLoS Genet. 5, e1000358 (2009).
Lewis, J. A., Broman, A. T., Will, J. & Gasch, A. P. Genetic architecture of ethanol-responsive transcriptome variation in Saccharomyces cerevisiae strains. Genetics 198, 369–382 (2014).
Bandiera, S. et al. Proteasomal degradation of human CYP1B1: effect of the Asn453Ser polymorphism on the post-translational regulation of CYP1B1 expression. Mol. Pharmacol. 67, 435–443 (2005).
't Hoen, P. A. et al. Reproducibility of high-throughput mRNA and small RNA sequencing across laboratories. Nature Biotech. 31, 1015–1022 (2013).
Melzer, D. et al. A genome-wide association study identifies protein quantitative trait loci (pQTLs). PLoS Genet. 4, e1000072 (2008).
Garge, N. et al. Identification of quantitative trait loci underlying proteome variation in human lymphoblastoid cells. Mol. Cell. Proteomics 9, 1383–1399 (2010).
Hause, R. J. et al. Identification and validation of genetic variants that influence transcription factor and cell signaling protein levels. Am. J. Hum. Genet. 95, 194–208 (2014).
Battle, A. et al. Impact of regulatory variation from RNA to protein. Science 347, 664–667 (2015). This is the first paper to present an integrated analysis of the genetics of mRNA levels, translation and protein abundance in humans.
Picotti, P. et al. A complete mass-spectrometric map of the yeast proteome applied to quantitative trait analysis. Nature 494, 266–270 (2013).
Lourdusamy, A. et al. Identification of cis-regulatory variation influencing protein abundance levels in human plasma. Hum. Mol. Genet. 21, 3719–3726 (2012).
Holdt, L. M. et al. Quantitative trait loci mapping of the mouse plasma proteome (pQTL). Genetics 193, 601–608 (2012).
Johansson, Å. et al. Identification of genetic variants influencing the human plasma proteome. Proc. Natl Acad. Sci. 110, 4673–4678 (2013).
Wu, L. et al. Variation and genetic control of protein abundance in humans. Nature 499, 79–82 (2013).
Skelly, D. A. et al. Integrative phenomics reveals insight into the structure of phenotypic diversity in budding yeast. Genome Res. 23, 1496–1504 (2013).
Foss, E. J. et al. Genetic basis of proteome variation in yeast. Nature Genet. 39, 1369–1375 (2007).
Foss, E. J. et al. Genetic variation shapes protein networks mainly through non-transcriptional mechanisms. PLoS Biol. 9, e1001144 (2011).
Albert, F. W., Treusch, S., Shockley, A. H., Bloom, J. S. & Kruglyak, L. Genetics of single-cell protein abundance variation in large yeast populations. Nature 506, 494–497 (2014).
Parts, L. et al. Heritability and genetic basis of protein level variation in an outbred population. Genome Res. 24, 1363–1370 (2014). References 121 and 122 pioneer the use of pools of hundreds of thousands of yeast cells to identify pQTLs and provide important insights into the genetics of protein versus mRNA levels.
Schwanhäusser, B. et al. Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011).
Li, J. J., Bickel, P. J. & Biggin, M. D. System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ 2, e270 (2014).
Ingolia, N. T., Ghaemmaghami, S., Newman, J. R. & Weissman, J. S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009).
Albert, F. W., Muzzey, D., Weissman, J. S. & Kruglyak, L. Genetic influences on translation in yeast. PLoS Genet. 10, e1004692 (2014).
Muzzey, D., Sherlock, G. & Weissman, J. S. Extensive and coordinated control of allele-specific expression by both transcription and translation in Candida albicans. Genome Res. 24, 963–973 (2014).
Pomerantz, M. M. et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nature Genet. 41, 882–884 (2009).
Sur, I. K. et al. Mice lacking a MYC enhancer that includes human SNP rs6983267 are resistant to intestinal tumors. Science 338, 1360–1363 (2012).
Villar, D., Flicek, P. & Odom, D. T. Evolution of transcription factor binding in metazoans — mechanisms and functional implications. Nature Rev. Genet. 15, 221–233 (2014).
Guenther, C. A., Tasic, B., Luo, L., Bedell, M. A. & Kingsley, D. M. A molecular basis for classic blond hair color in Europeans. Nature Genet. 46, 748–752 (2014). This study uses transgenic mice to show that a genetic change in a human regulatory element contributes to blond hair.
Wang, Y., Shirogane, T., Liu, D., Harper, J. W. & Elledge, S. J. Exit from exit: resetting the cell cycle through Amn1 inhibition of G protein signaling. Cell 112, 697–709 (2003).
Doolin, M. T., Johnson, A. L., Johnston, L. H. & Butler, G. Overlapping and distinct roles of the duplicated yeast transcription factors Ace2p and Swi5p. Mol. Microbiol. 40, 422–432 (2001).
Kuranda, M. J. & Robbins, P. W. Chitinase is required for cell separation during growth of Saccharomyces cerevisiae. J. Biol. Chem. 266, 19758–19767 (1991).
Rest, J. S. et al. Nonlinear fitness consequences of variation in expression level of a eukaryotic gene. Mol. Biol. Evol. 30, 448–456 (2013).
Gagneur, J. et al. Genotype–environment interactions reveal causal pathways that mediate genetic effects on phenotype. PLoS Genet. 9, e1003803 (2013).
Sudarsanam, P. & Cohen, B. A. Single nucleotide variants in transcription factors associate more tightly with phenotype than with gene expression. PLoS Genet. 10, e1004325 (2014). References 135–137 are examples of how the powerful experimental tools in yeast provide conceptual insights into the relationship between eQTLs and higher-order phenotypes.
Gerke, J., Lorenz, K. & Cohen, B. Genetic interactions between transcription factors cause natural variation in yeast. Science 323, 498–501 (2009).
Gusev, A. et al. Partitioning heritability of regulatory and cell-type-specific variants across 11 common diseases. Am. J. Hum. Genet. 95, 535–552 (2014). This study uses elegant whole-genome methods to estimate the relative contribution of functional genomic annotations to the heritability of several human traits and shows that by far the largest fraction is due to variants in putative regulatory elements.
Thurman, R. E. et al. The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012).
Nica, A. C. et al. Candidate causal regulatory effects by integration of expression QTLs with complex trait genetic associations. PLoS Genet. 6, e1000895 (2010).
Nicolae, D. L. et al. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 6, e1000888 (2010).
Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014).
Torres, J. M. et al. Cross-tissue and tissue-specific eQTLs: partitioning the heritability of a complex trait. Am. J. Hum. Genet. 95, 521–534 (2014).
Kapoor, A. et al. An enhancer polymorphism at the cardiomyocyte intercalated disc protein NOS1AP locus is a major regulator of the QT interval. Am. J. Hum. Genet. 94, 854–869 (2014).
Lonsdale, J. et al. The Genotype–Tissue Expression (GTEx) project. Nature Genet. 45, 580–585 (2013).
Almlöf, J. C. et al. Powerful identification of cis-regulatory SNPs in human primary monocytes using allele-specific gene expression. PLoS ONE 7, e52260 (2012).
Zeller, T. et al. Genetics and beyond — the transcriptome of human monocytes and disease susceptibility. PLoS ONE 5, e10693 (2010).
Adoue, V. et al. Allelic expression mapping across cellular lineages to establish impact of non-coding SNPs. Mol. Syst. Biol. 10, 754 (2014).
Ye, C. J. et al. Intersection of population variation and autoimmunity genetics in human T cell activation. Science 345, 1254665 (2014). References 16, 67, 74 and 150 detect previously hidden eQTLs in purified populations of primary immune cells once they are stimulated with triggers of the immune response.
Ferraro, A. et al. Interindividual variation in human T regulatory cells. Proc. Natl Acad. Sci. 111, E1111–E1120 (2014).
Ackermann, M., Sikora-Wohlfeld, W. & Beyer, A. Impact of natural genetic variation on gene expression dynamics. PLoS Genet. 9, e1003514 (2013).
Gerrits, A. et al. Expression quantitative trait loci are highly sensitive to cellular differentiation state. PLoS Genet. 5, e1000692 (2009).
Robinton, D. A. & Daley, G. Q. The promise of induced pluripotent stem cells in research and therapy. Nature 481, 295–305 (2012).
Rouhani, F. et al. Genetic background drives transcriptional variation in human induced pluripotent stem cells. PLoS Genet. 10, e1004432 (2014).
Kim, S. et al. Characterizing the genetic basis of innate immune response in TLR4-activated human monocytes. Nature Commun. 5, 5236 (2014).
Barreiro, L. B. et al. Deciphering the genetic architecture of variation in the immune response to Mycobacterium tuberculosis infection. Proc. Natl Acad. Sci. 109, 1204–1209 (2012).
Fairfax, B. P. & Knight, J. C. Genetics of gene expression in immunity to infection. Curr. Opin. Immunol. 30, 63–71 (2014).
Franco, L. M. et al. Integrative genomic analysis of the human immune response to influenza vaccination. eLife 2, e00299 (2013).
Schaub, M. A., Boyle, A. P., Kundaje, A., Batzoglou, S. & Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 22, 1748–1759 (2012).
Farh, K. K.-H. et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature http://dx.doi.org/10.1038/nature13835 (2014).
Schork, A. J. et al. All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 9, e1003449 (2013).
Trynka, G. et al. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nature Genet. 45, 124–130 (2012).
Pickrell, J. K. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 94, 559–573 (2014).
Kichaev, G. et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 10, e1004722 (2014).
Gagliano, S. A., Barnes, M. R., Weale, M. E. & Knight, J. A. Bayesian method to incorporate hundreds of functional characteristics with association evidence to improve variant prioritization. PLoS ONE 9, e98122 (2014). References 164–166 are examples of the formal integration of functional genomic information and genetic variation.
Chung, D., Yang, C., Li, C., Gelernter, J. & Zhao, H. GPA: a statistical approach to prioritizing GWAS results by integrating pleiotropy and annotation. PLoS Genet. 10, e1004787 (2014).
Knight, J., Barnes, M. R., Breen, G. & Weale, M. E. Using functional annotation for the empirical determination of Bayes factors for genome-wide association study analysis. PLoS ONE 6, e14808 (2011).
Gaffney, D. J. et al. Dissecting the regulatory architecture of gene expression QTLs. Genome Biol. 13, R7 (2012).
Brown, C. D., Mangravite, L. M. & Engelhardt, B. E. Integrative modeling of eQTLs and cis-regulatory elements suggests mechanisms underlying cell type specificity of eQTLs. PLoS Genet. 9, e1003649 (2013).
Flutre, T., Wen, X., Pritchard, J. & Stephens, M. A. Statistical framework for joint eQTL analysis in multiple tissues. PLoS Genet. 9, e1003486 (2013).
Manor, O. & Segal, E. Robust prediction of expression differences among human individuals using only genotype information. PLoS Genet. 9, e1003396 (2013).
Johansson, M. et al. Using prior information from the medical literature in GWAS of oral cancer identifies novel susceptibility variant on chromosome 4 — the AdAPT method. PLoS ONE 7, e36888 (2012).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Moffatt, M. F. et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature 448, 470–473 (2007).
Small, K. S. et al. Identification of an imprinted master trans regulator at the KLF14 locus related to multiple metabolic phenotypes. Nature Genet. 43, 561–564 (2011).
Cookson, W., Liang, L., Abecasis, G., Moffatt, M. & Lathrop, M. Mapping complex disease traits with global gene expression. Nature Rev. Genet. 10, 184–194 (2009).
Frayling, T. M. et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316, 889–894 (2007).
Fischer, J. et al. Inactivation of the Fto gene protects from obesity. Nature 458, 894–898 (2009).
Smemo, S. et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature 507, 371–375 (2014).
Zhao, Z. et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nature Genet. 38, 1341–1347 (2006).
Simonis, M. et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture–on-chip (4C). Nature Genet. 38, 1348–1354 (2006).
Dostie, J. et al. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 16, 1299–1309 (2006).
Dryden, N. H. et al. Unbiased analysis of potential targets of breast cancer susceptibility loci by capture Hi-C. Genome Res. 24, 1854–1868 (2014).
Dekker, J., Rippe, K., Dekker, M. & Kleckner, N. Capturing chromosome conformation. Science 295, 1306–1311 (2002).
Lieberman-Aiden, E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009).
Sterneckert, J. L., Reinhardt, P. & Schöler, H. R. Investigating human disease using stem cell models. Nature Rev. Genet. 15, 625–639 (2014).
Treutlein, B. et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 509, 371–375 (2014).
Jaitin, D. A. et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 343, 776–779 (2014).
Brem, R. B. & Kruglyak, L. The landscape of genetic complexity across 5,700 gene expression traits in yeast. Proc. Natl Acad. Sci. 102, 1572–1577 (2005).
Hemani, G. et al. Detection and replication of epistasis influencing transcription in humans. Nature 508, 249–253 (2014).
Brown, A. A. et al. Genetic interactions affecting human gene expression identified by variance association mapping. eLife 3, e01381 (2014).
Wood, A. R. et al. Another explanation for apparent epistasis. Nature 514, E3–E5 (2014).
Buil, A. et al. Gene–gene and gene–environment interactions detected by transcriptome sequence analysis in twins. Nature Genet. 47, 88–91 (2015).
Hill, W. G., Goddard, M. E. & Visscher, P. M. Data and theory point to mainly additive genetic variance for complex traits. PLoS Genet. 4, e1000008 (2008).
Maki-Tanila, A. & Hill, W. G. Influence of gene interaction on complex trait variation with multilocus models. Genetics 198, 355–367 (2014).
Mangravite, L. M. et al. A statin-dependent QTL for GATM expression is associated with statin-induced myopathy. Nature 502, 377–380 (2013).
Hsu, P. D., Lander, E. S. & Zhang, F. Development and applications of CRISPR–Cas9 for genome engineering. Cell 157, 1262–1278 (2014).
Sander, J. D. & Joung, J. K. CRISPR–Cas systems for editing, regulating and targeting genomes. Nature Biotech. 32, 347–355 (2014).
Leek, J. T. et al. Tackling the widespread and critical impact of batch effects in high-throughput data. Nature Rev. Genet. 11, 733–739 (2010).
Akey, J. M., Biswas, S., Leek, J. T. & Storey, J. D. On the design and analysis of gene expression studies in human populations. Nature Genet. 39, 807–808 (2007).
Leek, J. T. & Storey, J. D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 3, e161 (2007).
Stegle, O., Parts, L., Durbin, R. & Winn, J. A. Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput. Biol. 6, e1000770 (2010).
Goldinger, A. et al. Genetic and nongenetic variation revealed for the principal components of human gene expression. Genetics 195, 1117–1128 (2013).
Fusi, N., Stegle, O. & Lawrence, N. D. Joint modelling of confounding factors and prominent genetic regulators provides increased accuracy in genetical genomics studies. PLoS Comput. Biol. 8, e1002330 (2012).
Stegle, O., Parts, L., Piipari, M., Winn, J. & Durbin, R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nature Protoc. 7, 500–507 (2012).
Listgarten, J., Kadie, C., Schadt, E. E. & Heckerman, D. Correction for hidden confounders in the genetic analysis of gene expression. Proc. Natl Acad. Sci. 107, 16465–16470 (2010).
Kang, H. M., Ye, C. & Eskin, E. Accurate discovery of expression quantitative trait loci under confounding from spurious and genuine regulatory hotspots. Genetics 180, 1909–1925 (2008).
Gao, C. et al. HEFT: eQTL analysis of many thousands of expressed genes while simultaneously controlling for hidden factors. Bioinformatics 30, 369–376 (2014).
Yang, C., Wang, L., Zhang, S. & Zhao, H. Accounting for non-genetic factors by low-rank representation and sparse regression for eQTL mapping. Bioinformatics 29, 1026–1034 (2013).
Mostafavi, S. et al. Normalizing RNA-sequencing data by modeling hidden covariates with prior knowledge. PLoS ONE 8, e68141 (2013).
Parts, L., Stegle, O., Winn, J. & Durbin, R. Joint genetic analysis of gene expression data with inferred cellular phenotypes. PLoS Genet. 7, e1001276 (2011).
Lee, E. & Bussemaker, H. J. Identifying the genetic determinants of transcription factor activity. Mol. Syst. Biol. 6, 412 (2010).
Fazlollahi, M. et al. Harnessing natural sequence variation to dissect posttranscriptional regulatory networks in yeast. G3 (Bethesda) 4, 1539–1553 (2014).
Francesconi, M. & Lehner, B. The effects of genetic variation on gene expression dynamics during development. Nature 505, 208–211 (2013).
Powell, J. E. et al. The Brisbane Systems Genetics Study: genetical genomics meets complex trait genetics. PLoS ONE 7, e35430 (2012).
Grundberg, E. et al. Global analysis of the impact of environmental perturbation on cis-regulation of gene expression. PLoS Genet. 7, e1001279 (2011).
Ramasamy, A. et al. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nature Neurosci. 17, 1418–1428 (2014).
Zou, F. et al. Brain expression genome-wide association study (eGWAS) identifies human disease-associated variants. PLoS Genet. 8, e1002707 (2012).
Colantuoni, C. et al. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 478, 519–523 (2011).
Gibbs, J. R. et al. Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet. 6, e1000952 (2010).
Koopmann, T. T. et al. Genome-wide identification of expression quantitative trait loci (eQTLs) in human heart. PLoS ONE 9, e97380 (2014).
Innocenti, F. et al. Identification, replication, and functional fine-mapping of expression quantitative trait loci in primary human liver tissue. PLoS Genet. 7, e1002078 (2011).
Schadt, E. E. et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 6, e107 (2008).
Hao, K. et al. Lung eQTLs to help reveal the molecular underpinnings of asthma. PLoS Genet. 8, e1003029 (2012).
Ongen, H. et al. Putative cis-regulatory drivers in colorectal cancer. Nature 512, 87–90 (2014).
Li, Q. et al. Expression QTL-based analyses reveal candidate causal genes and loci across five tumor types. Hum. Mol. Genet. 23, 5294–5302 (2014).
Li, Q. et al. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 152, 633–641 (2013).
Acknowledgements
The authors thank members of L.K.'s laboratory and many colleagues for discussions. L.K. is supported by funds from the Howard Hughes Medical Institute, the US National Institutes of Health and the James S. McDonnell Foundation. F.W.A. was supported by the German Science Foundation (DFG).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- Expression quantitative trait loci
-
(eQTLs). Genomic regions that carry one or more DNA sequence variants that influence the expression level (typically mRNA abundance) of a given gene.
- Recombinant offspring
-
Offspring of sexually reproducing organisms that carry a random combination of the alleles that they have inherited from their parents.
- Protein QTLs
-
(pQTLs). Genomic regions that carry one or more DNA sequence variants that influence the protein abundance of a given gene.
- eQTL hot spots
-
Regions of the genome that contain more expression quantitative trait loci (eQTLs) than expected by chance.
- Organismal phenotypes
-
Traits that are detectable at the level of the whole organism, such as shape, size, colour, growth rate or the risk of developing a certain disease.
- Linkage disequilibrium
-
The phenomenon whereby specific allele combinations occur more frequently than expected by chance, typically because they are physically close to each other on the same chromosome.
- QTLs
-
Genomic regions that carry one or several DNA sequence variants which influence a continuously variable trait of interest.
- Homologous
-
Pertaining to the two copies of the same chromosome that were inherited from the mother and the father in diploid organisms (such as humans).
- RNA sequencing
-
A method to determine the sequence of RNA molecules in a biological sample. By counting the RNA molecules that were transcribed from each gene, RNA sequencing can be used to quantify mRNA expression levels.
- Genetic distance
-
A measure of how often two sites in a genome are separated during meiosis. Genetic distance is correlated with physical distance but can differ quantitatively because of variation in recombination rate along a chromosome.
- Linkage blocks
-
Continuous haplotypes that are not broken up in the population under study such that sequence variants within them all show the same patterns of association with a certain trait of interest.
- Haplotypes
-
Stretches of DNA that carry certain combinations of alleles at two or more DNA variants.
- Variance
-
A statistical measure of the variability of a trait in a population.
- Heritability
-
The fraction of variance in a trait that is due to genetic differences among the individuals in a population.
Rights and permissions
About this article

Cite this article
Albert, F., Kruglyak, L. The role of regulatory variation in complex traits and disease. Nat Rev Genet 16, 197–212 (2015). https://doi.org/10.1038/nrg3891
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrg3891
This article is cited by
-
Genomic dissection of the correlation between milk yield and various health traits using functional and evolutionary information about imputed sequence variants of 34,497 German Holstein cows
BMC Genomics (2024)
-
Diallel panel reveals a significant impact of low-frequency genetic variants on gene expression variation in yeast
Molecular Systems Biology (2024)
-
Haplotype-aware modeling of cis-regulatory effects highlights the gaps remaining in eQTL data
Nature Communications (2024)
-
Multi-omics and pathway analyses of genome-wide associations implicate regulation and immunity in verbal declarative memory performance
Alzheimer's Research & Therapy (2024)
-
Single-cell multiomics of the human retina reveals hierarchical transcription factor collaboration in mediating cell type-specific effects of genetic variants on gene regulation
Genome Biology (2023)
