
Abstract
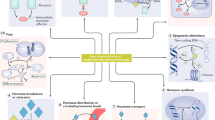
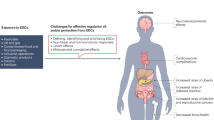
The emerging field of omics — large-scale data-rich biological measurements of the genome — provides new opportunities to advance and strengthen research into endocrine-disrupting chemicals (EDCs). Although some EDCs have been associated with adverse health effects in humans, our understanding of their impact remains incomplete. Progress in the field has been primarily limited by our inability to adequately estimate and characterize exposure and identify sensitive and measurable outcomes during windows of vulnerability. Evolving omics technologies in genomics, epigenomics and mitochondriomics have the potential to generate data that enhance exposure assessment to include the exposome — the totality of the lifetime exposure burden — and provide biology-based estimates of individual risks. Applying omics technologies to expand our knowledge of individual risk and susceptibility will augment biological data in the prediction of variability and response to disease, thereby further advancing EDC research. Together, refined exposure characterization and enhanced disease-risk prediction will help to bridge crucial gaps in EDC research and create opportunities to move the field towards a new vision — precision public health.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others
References
Nature Publishing Group. OmicsGateway. Nature http://www.nature.com/omics/about/index.html (2016).
Attene-Ramos, M. S. et al. Profiling of the Tox21 chemical collection for mitochondrial function to identify compounds that acutely decrease mitochondrial membrane potential. Environ. Health Perspect. 123, 49–56 (2015).
Casati, L., Sendra, R., Sibilia, V. & Celotti, F. Endocrine disrupters: the new players able to affect the epigenome. Front. Cell Dev. Biol. 3, 37 (2015).
Baccarelli, A., Pesatori, A. C. & Bertazzi, P. A. Occupational and environmental agents as endocrine disruptors: experimental and human evidence. J. Endocrinol. Invest. 23, 771–781 (2000).
Diamanti-Kandarakis, E. et al. Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr. Rev. 30, 293–342 (2009).
National Institute of Environmental Health Sciences. Endocrine disruptors. NIH https://www.niehs.nih.gov/health/topics/agents/endocrine/ (2017).
US Department of Health and Human Services. National Toxicology Program. NIH https://ntp.niehs.nih.gov/go/about (2016).
Gore, A. C. et al. EDC-2: the Endocrine Society's second scientific statement on endocrine-disrupting chemicals. Endocr. Rev. 36, E1–E150 (2015).
Attina, T. M. et al. Exposure to endocrine-disrupting chemicals in the USA: a population-based disease burden and cost analysis. Lancet Diabetes Endocrinol. 4, 996–1003 (2016).
The White House. Fact sheet: President Obama's precision medicine initiative. Obama White House https://obamawhitehouse.archives.gov/the-press-office/2015/01/30/fact-sheet-president-obama-s-precision-medicine-initiative (2015).
Centers for Disease Control and Prevention. Exposome and exposomics. CDC https://www.cdc.gov/niosh/topics/exposome/ (2014).
National Institutes of Health. About the All of Us Research Program. NIH https://allofus.nih.gov/about/about-all-us-research-program (2016).
Schug, T. T., Janesick, A., Blumberg, B. & Heindel, J. J. Endocrine disrupting chemicals and disease susceptibility. J. Steroid Biochem. Mol. Biol. 127, 204–215 (2011).
Kumar, V. et al. CYP 1A1 polymorphism and organochlorine pesticides levels in the etiology of prostate cancer. Chemosphere 81, 464–468 (2010).
Yoshida, R. et al. Association of cryptorchidism with a specific haplotype of the estrogen receptor alpha gene: implication for the susceptibility to estrogenic environmental endocrine disruptors. J. Clin. Endocrinol. Metab. 90, 4716–4721 (2005).
Bi, Y. et al. Diabetes genetic risk score modifies effect of bisphenol A exposure on deterioration in glucose metabolism. J. Clin. Endocrinol. Metab. 101, 143–150 (2016).
Martinez-Nava, G. A. et al. PPARγ and PPARGC1B polymorphisms modify the association between phthalate metabolites and breast cancer risk. Biomarkers 18, 493–501 (2013).
Hung, W. T., Lambert, G. H., Huang, P. W., Patterson, D. G. Jr & Guo, Y. L. Genetic susceptibility to dioxin-like chemicals' induction of cytochrome P4501A2 in the human adult linked to specific AhRR polymorphism. Chemosphere 90, 2358–2364 (2013).
Belinsky, S. A. et al. Aberrant promoter methylation in bronchial epithelium and sputum from current and former smokers. Cancer Res. 62, 2370–2377 (2002).
Yauk, C. et al. Germ-line mutations, DNA damage, and global hypermethylation in mice exposed to particulate air pollution in an urban/industrial location. Proc. Natl Acad. Sci. USA 105, 605–610 (2008).
Christensen, B. C. et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 5, e1000602 (2009).
Prins, G. S., Birch, L., Tang, W.-Y. & Ho, S.-M. Developmental estrogen exposures predispose to prostate carcinogenesis with aging. Reprod. Toxicol. 23, 374–382 (2007).
Anway, M. D., Cupp, A. S., Uzumcu, M. & Skinner, M. K. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308, 1466–1469 (2005).
Richards, E. Inherited epigenetic variation — revisiting soft inheritance. Nat. Rev. Genet. 7, 395–401 (2006).
Dolinoy, D. C., Weidman, J. R. & Jirtle, R. L. Epigenetic gene regulation: linking early developmental environment to adult disease. Reprod. Toxicol. 23, 297–307 (2007).
US National Library of Medicine. Help me understand genetics. NIH https://ghr.nlm.nih.gov/primer (2017).
Wu, M. C. et al. Powerful SNP-set analysis for case–control genome-wide association studies. Am. J. Hum. Genet. 86, 929–942 (2010).
Baccarelli, A. & Bollati, V. Epigenetics and environmental chemicals. Curr. Opin. Pediatr. 21, 243–251 (2009).
Brokken, L. J. & Giwercman, Y. L. Gene–environment interactions in male reproductive health: special reference to the aryl hydrocarbon receptor signaling pathway. Asian J. Androl. 16, 89–96 (2014).
Ottman, R. Gene–environment interaction: definitions and study designs. Prev. Med. 25, 764–770 (1996).
Dunaway, K. W. et al. Cumulative impact of polychlorinated biphenyl and large chromosomal duplications on DNA methylation, chromatin, and expression of autism candidate genes. Cell Rep. 17, 3035–3048 (2016).
Olden, K. & Wilson, S. Environmental health and genomics: visions and implications. Nat. Rev. Genet. 1, 149–153 (2000).
Alam, G. & Jones, B. C. Toxicogenetics: in search of host susceptibility to environmental toxicants. Front. Genet. 5, 327 (2014).
Lundberg Giwercman, Y. Androgen receptor genotype in humans and susceptibility to endocrine disruptors. Horm. Res. Paediatr. 86, 264–270 (2016).
Schwartz, D. A. Environmental genomics and human health. G. Ital. Med. Lav. Ergon. 33, 31–34 (2011).
Baccarelli, A. Epigenetics glossary. Columbia.edu https://www.mailman.columbia.edu/research/laboratory-precision-environmental-biosciences/epigenetics-glossary (2016).
Rivera, C. M. & Ren, B. Mapping human epigenomes. Cell 155, 39–55 (2013).
Stirzaker, C., Taberlay, P. C., Statham, A. L. & Clark, S. J. Mining cancer methylomes: prospects and challenges. Trends Genet. 30, 75–84 (2014).
Illumina. Introduction to methylation array analysis. Illumina https://www.illumina.com/techniques/microarrays/methylation-arrays.html (2016).
Dao, T., Hong, X., Wang, X. & Tang, W. Y. Aberrant 5′-CpG methylation of cord blood TNFα associated with maternal exposure to polybrominated diphenyl ethers. PLoS ONE 10, e0138815 (2015).
Lövkvist, C., Dodd, I. B., Sneppen, K. & Haerter, J. O. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 44, 5124–5132 (2016).
Gorber, S. C., Schofield-Hurwitz, S., Hardt, J., Levasseur, G. & Tremblay, M. The accuracy of self-reported smoking: a systematic review of the relationship between self-reported and cotinine-assessed smoking status. Nicotine Tob. Res. 11, 12–24 (2009).
Joubert, B. R. et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ. Health Perspect. 120, 1425–1431 (2012).
Joubert, B. R. et al. DNA methylation in newborns and maternal smoking in pregnancy: genome-wide consortium meta-analysis. Am. J. Hum. Genet. 98, 680–696 (2016).
Joehanes, R. et al. Epigenetic signatures of cigarette smoking. Circ. Cardiovasc. Genet. 9, 436–447 (2016).
Philibert, R. A., Beach, S. R. & Brody, G. H. Demethylation of the aryl hydrocarbon receptor repressor as a biomarker for nascent smokers. Epigenetics 7, 1331–1338 (2012).
Skinner, M. K., Bhandari, R. K., Haque, M. M. & Nilsson, E. E. Environmentally induced epigenetic transgenerational inheritance of altered SRY genomic binding during gonadal sex determination. Environ. Epigenet. 1, dvv004 (2015).
Skinner, M. K. Endocrine disruptors in 2015: epigenetic transgenerational inheritance. Nat. Rev. Endocrinol. 12, 68–70 (2016).
Heard, E. & Martienssen, R. A. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157, 95–109 (2014).
Blake, G. E. & Watson, E. D. Unravelling the complex mechanisms of transgenerational epigenetic inheritance. Curr. Opin. Chem. Biol. 33, 101–107 (2016).
Guerrero-Bosagna, C. in The Epigenome and Developmental Origins of Health and Disease (ed. Rosenfeld, C. S.) 425–437 (Academic Press, 2016).
Chen, J. et al. The mechanism of environmental endocrine disruptors (DEHP) induces epigenetic transgenerational inheritance of cryptorchidism. PLoS ONE 10, e0126403 (2015).
Manikkam, M., Haque, M. M., Guerrero-Bosagna, C., Nilsson, E. E. & Skinner, M. K. Pesticide methoxychlor promotes the epigenetic transgenerational inheritance of adult-onset disease through the female germline. PLoS ONE 9, e102091 (2014).
Guerrero-Bosagna, C. et al. Epigenetic transgenerational inheritance of vinclozolin induced mouse adult onset disease and associated sperm epigenome biomarkers. Reprod. Toxicol. 34, 694–707 (2012).
Adams, J. U. Essentials of Cell Biology (ed. O'Connor, C. M.) (NPG Education, 2010).
Yakes, F. M. & Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl Acad. Sci. USA 94, 514–519 (1997).
Guarnieri, M. & Balmes, J. R. Outdoor air pollution and asthma. Lancet 383, 1581–1592 (2014).
Pant, N. et al. Correlation of phthalate exposures with semen quality. Toxicol. Appl. Pharmacol. 231, 112–116 (2008).
Meyer, J. N. et al. Mitochondria as a target of environmental toxicants. Toxicol. Sci. 134, 1–17 (2013).
Gopalkrishnan, K., Padwal, V., D'Souza, S. & Shah, R. Severe asthenozoospermia: a structural and functional study. Int. J. Androl. 18 (Suppl. 1), 67–74 (1995).
Piasecka, M. & Kawiak, J. Sperm mitochondria of patients with normal sperm motility and with asthenozoospermia: morphological and functional study. Folia Histochem. Cytobiol. 41, 125–139 (2003).
Byun, H. M. & Baccarelli, A. A. Environmental exposure and mitochondrial epigenetics: study design and analytical challenges. Hum. Genet. 133, 247–257 (2014).
Chen, S. C., Liao, T. L., Wei, Y. H., Tzeng, C. R. & Kao, S. H. Endocrine disruptor, dioxin (TCDD)-induced mitochondrial dysfunction and apoptosis in human trophoblast-like JAR cells. Mol. Hum. Reprod. 16, 361–372 (2010).
Kaur, K., Chauhan, V., Gu, F. & Chauhan, A. Bisphenol A induces oxidative stress and mitochondrial dysfunction in lymphoblasts from children with autism and unaffected siblings. Free Radic. Biol. Med. 76, 25–33 (2014).
Liu, B. et al. CpG methylation patterns of human mitochondrial DNA. Sci. Rep. 6, 23421 (2016).
Dawid, I. B. 5-Methylcytidylic acid: absence from mitochondrial DNA of frogs and HeLa cells. Science 184, 80–81 (1974).
Shock, L. S., Thakkar, P. V., Peterson, E. J., Moran, R. G. & Taylor, S. M. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc. Natl Acad. Sci. USA 108, 3630–3635 (2011).
Byun, H. M. et al. Epigenetic effects of low perinatal doses of flame retardant BDE-47 on mitochondrial and nuclear genes in rat offspring. Toxicology 328, 152–159 (2015).
Crick, F. H. On protein synthesis. Symp. Soc. Exp. Biol. 12, 138–163 (1958).
Sheehan, D. The potential of proteomics for providing new insights into environmental impacts on human health. Rev. Environ. Health 22, 175–194 (2007).
Vidyasagar, M. Identifying predictive features in drug response using machine learning: opportunities and challenges. Annu. Rev. Pharmacol. Toxicol. 55, 15–34 (2015).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115–R115 (2013).
Chen, B. H. et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY) 8, 1844–1865 (2016).
Kourou, K., Exarchos, T. P., Exarchos, K. P., Karamouzis, M. V. & Fotiadis, D. I. Machine learning applications in cancer prognosis and prediction. Comput. Struct. Biotechnol. J. 13, 8–17 (2015).
Ornostay, A., Cowie, A. M., Hindle, M., Baker, C. J. & Martyniuk, C. J. Classifying chemical mode of action using gene networks and machine learning: a case study with the herbicide linuron. Comp. Biochem. Physiol. Part D Genomics Proteomics 8, 263–274 (2013).
Zhang, J. et al. In silico approach to identify potential thyroid hormone disruptors among currently known dust contaminants and their metabolites. Environ. Sci. Technol. 49, 10099–10107 (2015).
Kaye, J., Boddington, P., de Vries, J., Hawkins, N. & Melham, K. Ethical implications of the use of whole genome methods in medical research. Eur. J. Hum. Genet. 18, 398–403 (2010).
Strong, A. L. et al. Effects of the endocrine-disrupting chemical DDT on self-renewal and differentiation of human mesenchymal stem cells. Environ. Health Perspect. 123, 42–48 (2015).
Guyot, R., Chatonnet, F., Gillet, B., Hughes, S. & Flamant, F. Toxicogenomic analysis of the ability of brominated flame retardants TBBPA and BDE-209 to disrupt thyroid hormone signaling in neural cells. Toxicology 325, 125–132 (2014).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63 (2009).
Pidsley, R. et al. Critical evaluation of the Illumina MethylationEPIC BeadChip microarray for whole-genome DNA methylation profiling. Genome Biol. 17, 208 (2016).
Kurdyukov, S. & Bullock, M. DNA methylation analysis: choosing the right method. Biology (Basel) 5, E3 (2016).
Bannister, A. J. & Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 21, 381–395 (2011).
Wang, L., Jin, Q., Lee, J. E., Su, I. H. & Ge, K. Histone H3K27 methyltransferase Ezh2 represses Wnt genes to facilitate adipogenesis. Proc. Natl Acad. Sci. USA 107, 7317–7322 (2010).
Doherty, L. F., Bromer, J. G., Zhou, Y., Aldad, T. S. & Taylor, H. S. In utero exposure to diethylstilbestrol (DES) or bisphenol-A (BPA) increases EZH2 expression in the mammary gland: an epigenetic mechanism linking endocrine disruptors to breast cancer. Horm. Cancer 1, 146–155 (2010).
Bhan, A. et al. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-A and diethylstilbestrol. J. Mol. Biol. 426, 3426–3441 (2014).
Stel, J. & Legler, J. The role of epigenetics in the latent effects of early life exposure to obesogenic endocrine disrupting chemicals. Endocrinology 156, 3466–3472 (2015).
Wang, G. G., Allis, C. D. & Chi, P. Chromatin remodeling and cancer, part I: covalent histone modifications. Trends Mol. Med. 13, 363–372 (2007).
Rooney, J. P. et al. PCR based determination of mitochondrial DNA copy number in multiple species. Methods Mol. Biol. 1241, 23–38 (2015).
Furda, A., Santos, J. H., Meyer, J. N. & Van Houten, B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 1105, 419–437 (2014).
Kapoor, V., DeBry, R. W., Boccelli, D. L. & Wendell, D. Sequencing human mitochondrial hypervariable region II as a molecular fingerprint for environmental waters. Environ. Sci. Technol. 48, 10648–10655 (2014).
Cui, H. et al. Comprehensive next-generation sequence analyses of the entire mitochondrial genome reveal new insights into the molecular diagnosis of mitochondrial DNA disorders. Genet. Med. 15, 388–394 (2013).
Acknowledgements
The authors acknowledge the National Institute of Environmental Health Sciences (NIEHS) (NIEHS Center Grants ES000002 to C.M. and R.H., and P30ES009089 to A.A.B.; NIEHS Grants R01ES021733, R01ES021357, R21ES024841 and R21ES027087 to A.A.B., and R01ES009718 to R.H.), the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK; Grant R01DK100790 to A.A.B.) and the Centers for Disease Control and Prevention (CDC)/National Institute for Occupational Safety and Health (NIOSH) (CDC/NIOSH Training Grant T42OH008416 to R.M.M.).
Author information
Authors and Affiliations
Contributions
C.M. and R.M.M. researched data for the article and made substantial contributions to discussions of the content. C.M., R.M.M. and A.A.B. wrote the article. C.M., R.M.M., R.H. and A.A.B reviewed and/or edited the manuscript before submission. C.M. and R.M.M. contributed equally to all aspects of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Glossary
- DNA methylome
-
The set of methylation modifications in an organism's genome in a particular cell.
- DNA methyltransferases
-
A family of enzymes that catalyse the transfer of a methyl group to DNA.
- Epigenomics
-
The study of heritable changes in gene expression that do not result from changes in actual gene sequences.
- Exposome
-
An individual's lifetime exposure burden.
- Gene–environment interactions
-
(GxEs). The biological interactions between the environment and the human genome.
- Genomics
-
The study of an organism's genome or complete set of DNA, including all its genes.
- Histone modifications
-
Post-translational modifications to histones — referred to as marks — that regulate gene expression.
- Metabolomics
-
The study of the set of metabolites present within an organism, cell or tissue.
- Mitochondrial membrane potential
-
(MMP). The total force driving protons into the mitochondria.
- Mitochondriomics
-
The study of the properties of mitochondrial DNA.
- Proteomics
-
The large-scale study of proteins.
- Transcriptomics
-
The study of transcriptomes and their functions.
Rights and permissions
About this article

Cite this article
Messerlian, C., Martinez, R., Hauser, R. et al. 'Omics' and endocrine-disrupting chemicals — new paths forward. Nat Rev Endocrinol 13, 740–748 (2017). https://doi.org/10.1038/nrendo.2017.81
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrendo.2017.81
This article is cited by
-
Transcriptomic and metabolomic analyses revealed epiboly delayed mechanisms of 2,5-dichloro-1, 4-benuinone on zebrafish embryos
Environmental Science and Pollution Research (2023)
-
Advancing precision public health using human genomics: examples from the field and future research opportunities
Genome Medicine (2021)
-
The antiandrogenic vinclozolin induces differentiation delay of germ cells and changes in energy metabolism in 3D cultures of fetal ovaries
Scientific Reports (2020)
-
Endocrine disrupting chemicals: exposure, effects on human health, mechanism of action, models for testing and strategies for prevention
Reviews in Endocrine and Metabolic Disorders (2020)
