
Abstract
Congenital hypogonadotropic hypogonadism (CHH) is a rare disorder caused by the deficient production, secretion or action of gonadotropin-releasing hormone (GnRH), which is the master hormone regulating the reproductive axis. CHH is clinically and genetically heterogeneous, with >25 different causal genes identified to date. Clinically, the disorder is characterized by an absence of puberty and infertility. The association of CHH with a defective sense of smell (anosmia or hyposmia), which is found in ∼50% of patients with CHH is termed Kallmann syndrome and results from incomplete embryonic migration of GnRH-synthesizing neurons. CHH can be challenging to diagnose, particularly when attempting to differentiate it from constitutional delay of puberty. A timely diagnosis and treatment to induce puberty can be beneficial for sexual, bone and metabolic health, and might help minimize some of the psychological effects of CHH. In most cases, fertility can be induced using specialized treatment regimens and several predictors of outcome have been identified. Patients typically require lifelong treatment, yet ∼10–20% of patients exhibit a spontaneous recovery of reproductive function. This Consensus Statement summarizes approaches for the diagnosis and treatment of CHH and discusses important unanswered questions in the field.
Similar content being viewed by others

Introduction
Congenital hypogonadotropic hypogonadism (CHH) is caused by deficient production, secretion or action of gonadotropin-releasing hormone (GnRH), a key neuropeptide that orchestrates mammalian reproduction.1 CHH is characterized by incomplete or absent puberty and infertility in the setting of isolated hypogonadotropic hypogonadism (that is, otherwise normal anterior pituitary function). CHH can present solely as congenital GnRH deficiency or be associated with other developmental anomalies such as cleft lip or palate, dental agenesis, ear anomalies, congenital hearing impairment, renal agenesis, bimanual synkinesis or skeletal anomalies. When associated with anosmia or hyposmia, CHH is termed Kallmann syndrome, which results from abnormal embryonic migration of GnRH neurons from their origin in the olfactory placode to the forebrain.2,3 CHH has a male predominance of 3–5 to 1.4,5 Most patients with CHH are diagnosed late in adolescence or early in adulthood as CHH is challenging to differentiate from other causes of delayed puberty. Effective therapies in both men and women are available for the development of secondary sexual characteristics (virilization and/or estrogenization) and induction of fertility.
A number of important developments in the field have emerged over the past decade. Reversal of CHH occurs in ∼10–20% of patients,6,7 which challenges the dogma that the condition is lifelong. These data have implications for the management of CHH and suggest a plasticity of the GnRH neuronal system. Furthermore, inhibin B, insulin-like 3 (INSL3), anti-Müllerian hormone (AMH) and kisspeptin have emerged as biomarkers for use in the diagnosis and treatment of CHH.8,9 In addition, hormonal therapies are being personalized to maximize fertility outcomes.10 Over the past few years, the traditional Mendelian view of CHH as a monogenic disorder has been revised following the identification of oligogenic forms of CHH,11 which has altered the approach adopted for genetic testing and genetic counselling of patients with CHH. Finally, the availability of next-generation sequencing has started to unravel the complex molecular basis of CHH. This Consensus Statement focuses on the pathogenesis, diagnosis and treatment of CHH in light of recent discoveries and differs from existing guidelines for the treatment of hypogonadism12,13,14 as it focuses exclusively on CHH. Definitions of several key terms used in this article are presented in the glossary (Box 1).
Methods
This Consensus Statement is the work of the European consortium studying GnRH biology (COST Action BM1105, http://www.gnrhnetwork.eu/). The COST network includes clinician investigators, geneticists, bioinformaticians, basic scientists and patient advocates from 28 countries. Our recommendations are based on a review of the literature published in the English language between 1990 and 2015 using the following search terms: “congenital hypogonadotropic hypogonadism”, “idiopathic GnRH deficiency”, “central hypogonadism” and “Kallmann syndrome”. The reference lists of identified papers were searched for additional relevant articles. A series of meetings and focused discussions with expert clinicians (specialized in paediatric and adult CHH) were conducted, with a final vetting process involving experts participating in the network from the fields of endocrinology, andrology, genetics and reproductive medicine.
Biology of the GnRH neuronal system
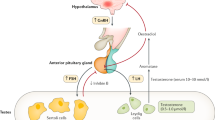
GnRH neurons are unusual neuroendocrine cells as they originate outside the central nervous system in the olfactory placode and then migrate into the brain during embryonic development.2,3 This route provides a developmental link between the central control of reproduction and the sense of smell, which are both affected in Kallmann syndrome. Data from the past few years suggest that GnRH neurons originate from both the neural crest and ectodermal progenitors, and migrate in close association with growing axons of olfactory and/or terminal nerves.15,16 Once in the hypothalamus, GnRH neurons detach from their axonal guides, disperse further into the brain parenchyma and stop migrating. At birth, GnRH neurons have reached their final destination in the brain and many of them project into the median eminence, where they release the GnRH decapeptide from their axon terminals into the hypophyseal portal vasculature.1 GnRH acts via the GnRH receptor, which is expressed on gonadotropic cells in the anterior pituitary gland. This action regulates both synthesis and release of gonadotropins such as luteinizing hormone (LH) and follicle-stimulating hormone (FSH), which control gonadal maturation and adult reproductive physiology via the hypothalamic–pituitary–gonadal (HPG) axis.
Several genes mutated in Kallmann syndrome affect the fate and migration of GnRH neurons. Genes encoding fibroblast growth factor 8 (FGF8) signalling pathway proteins,17,18,19,20,21,22 chromodomain helicase DNA-binding protein 7 (CHD7)23,24,25,26,27 and sex determining region Y-Box 10 (SOX10)28,29 affect the neurogenic niche in the nasal area and craniofacial development. Conversely, Kallmann syndrome protein, which is now officially known as anosmin 1 (encoded by KAL1; following nomenclature change, the gene is now denoted as ANOS1),2 prokineticin-2 and prokineticin receptor 2 (encoded by PROK2 and PROKR2, respectively),30,31,32,33 WD repeat domain 11 (encoded by WDR11),34,35 semaphorin 3A (encoded by SEMA3A)36,37,38 and FEZ family zinc finger 1 (encoded by FEZF1)39 influence migration of GnRH neurons.
Postmigratory GnRH neurons are embedded in a complex neuronal network of afferents that send information about permissive reproductive cues such as steroid and metabolic hormones to these cells. Individual components of the underlying neural circuits are beginning to emerge. Some key molecules have been discovered through the study of the genetics of CHH.1 Inactivating mutations in genes encoding kisspeptin-1 (KISS1)40 and its receptor (KISS1R)41,42 halt pubertal development in humans. Extensive experimental studies in various species have demonstrated that kisspeptin-producing neurons are major afferents to GnRH neurons and are essential for different aspects of GnRH function, ranging from the tonic feedback control of GnRH and/or gonadotropin secretion to generation of the pre-ovulatory surge responsible for ovulation.43 Interestingly, although kisspeptins do not seem to be mandatory for proper GnRH neuron migration, compelling experimental work has documented that populations of kisspeptin neurons undergo a dynamic process of prenatal and postnatal maturation that enables them to establish connections with GnRH neurons early in development44 (under the control of steroid hormones45,46,47). Similarly, identification of mutations in TAC3 (encoding tachykinin-3, which is cleaved to form neurokinin-B) and TACR3 (encoding tachykinin receptor 3; also known as neuromedin-K receptor [NKR])48,49,50 in patients with CHH highlights the important role of members of the tachykinin family in the control of GnRH neurons. Furthermore, these findings led to the identification of a subpopulation of afferent neurons in the arcuate and/or infundibular hypothalamic region, which co-express kisspeptins and NKB.51 Interestingly, data from the past few years suggest that the actual number of kisspeptin and/or NKB neurons might change during development.52,53 Mutations in proteins that regulate ubiquitination such as OTU domain-containing protein 4 (encoded by OTUD4) and E3 ubiquitin-protein ligase RNF216 (also known as ring finger protein 216; encoded by RNF216),54 as well as in proteins involved in lipid metabolism such as neuropathy target esterase (also known as patatin-like phospholipase domain-containing protein 6; encoded by PNPLA6)55,56 have been identified in patients with Gordon Holmes syndrome (with associated CHH and ataxia). Thus, mutations in these three genes give rise to a broad and progressive neurodegenerative syndrome that includes CHH. In 2014, haploinsufficiency of DMXL2, which encodes synaptic protein DmX-like protein 2, was shown to cause a complex new syndrome associating CHH with polyendocrine deficiencies and polyneuropathies.57
Peripheral signals that convey information about metabolic status indirectly modulate GnRH neurosecretion. This modulation is well illustrated by the reproductive phenotype of absent pubertal development and hypogonadotropic hypogonadism in patients with inactivating mutations in the genes encoding leptin (LEP) or its receptor (LEPR).58 Exactly how the effects of leptin are transmitted to GnRH neurons is unknown, as this neuronal population does not express the leptin receptor.59 Experimental data suggest that kisspeptin neurons are sensitive to changes in leptin concentrations and metabolic conditions,43 yet they apparently do not express functional leptin receptor, which indicates the mode of action is indirect.60 Primary leptin targets for conduction of its reproductive effects probably include nitric oxide-producing neurons in the pre-optic hypothalamus61 and neuronal circuits in the ventral premammillary nucleus,60 which might transmit metabolic information to GnRH neurons through kisspeptin dependent and/or independent pathways. Taken together, the identification of genes mutated in the different forms of CHH has facilitated an improved understanding of the neuroendocrine control of reproduction.
Puberty and reproduction
Puberty represents a period of transition from childhood into adulthood during which complete reproductive capacity is attained. Puberty is regulated by the HPG axis. The axis is active in utero and shortly after birth62,63 (a phenomenon referred to as mini-puberty), is subsequently silenced and remains quiescent for years until reawakening at the onset of puberty. Multiple central and peripheral inputs are integrated into pubertal reactivation of the GnRH pulse generator.64 At the onset of puberty, GnRH-induced pulses of LH are initially nocturnal and, gradually, the pulsatility extends to the daytime as puberty progresses.65,66,67
In boys, FSH stimulates proliferation of immature Sertoli cells and spermatogonia, whereas LH stimulates Leydig cells to produce testosterone. The concerted stimulation of Sertoli cells by FSH and intragonadal testosterone levels 50–100-fold higher than in the systemic circulation leads to the initiation of spermatogenesis. In girls, the early stages of follicular growth are primarily driven by intra-ovarian factors. FSH and LH are needed for follicular maturation, which leads to ovulation. More specifically, LH stimulates theca cells to produce androgens and FSH stimulates recruitment of secondary ovarian follicles and the secretion of estradiol from granulosa cells. Inhibin B and AMH are produced by prepubertal Sertoli cells and granulosa cells.68 Circulating levels of inhibin B increase during puberty in girls and boys.69,70 AMH concentrations show only minor fluctuations during female puberty.71 In boys, AMH concentrations fall as Sertoli cells differentiate and testosterone production increases.72 In male individuals, INSL3 is secreted by Leydig cells during fetal and immediate postnatal life.8,73 Testicular INSL3 secretion declines during childhood before increasing at the time of puberty and peaking during adulthood.74
Early signs of puberty manifest around the age of 10 years in girls (breast development) and 11.5 years in boys (testicular enlargement and spermarche—appearance of spermatozoa in first morning void).75,76,77 Considerable interindividual variation in the timing of pubertal onset exists, ranging from 8 years to 13 years in girls and 9 years to 14 years in boys.78,79 Subsequent hallmarks of puberty in boys include deepening of the voice and a growth spurt. In girls, menarche occurs at ∼12.5 years of age.78
Clinical presentation of CHH
Neonatal and childhood
Lack of neonatal activation of the HPG axis can provide early diagnostic cues for identifying CHH at birth or during mini-puberty.80,81,82,83 In male infants, cryptorchidism (that is, maldescended testes) and micropenis can be signs of GnRH deficiency (Figure 1), yet these features are by no means invariable.84,85,86 As penile growth occurs during infancy and childhood, micropenis can be assessed using available cross-sectional normative data.87,88 These two clinical clues warrant hormonal monitoring during mini-puberty. However, more severe genital anomalies, such as hypospadias, point away from a diagnosis of CHH and, instead, indicate a defect of human chorionic gonadotropin (hCG)-driven androgen secretion and/or action in early fetal life, before the initiation of endogenous GnRH activity. No specific clinical signs of CHH present in female neonates. Regardless of sex, when infants are born to parents with CHH, we recommend monitoring reproductive hormones during mini-puberty and performing genetic testing if mutations have been identified in the parents.

Normal GnRH and LH secretion from fetal life to adulthood (blue line) compared with non-reversible CHH (red line). Reproductive phenotypes observed in CHH are listed under the respective stage of development. Abbreviations: CHH, congenital hypogonadotropic hypogonadism; F, female; GnRH, gonadotropin-releasing hormone; HPG, hypothalamic–pituitary–gonadal; LH, luteinizing hormone; M, male.
Adolescent
The timing and onset of puberty varies widely in the general population.89 Delayed puberty is classically defined as absence of testicular enlargement (volume <4 ml) in boys by the age of 14 years and absent breast development in girls by the age of 13 years.90 In addition, use of the stage-line diagram enables the identification of patients with delayed puberty, which is based not only on pubertal onset but also on the tempo of pubertal development.91 In most of these individuals, puberty is initiated and eventually completed (that is, constitutional delay of growth and puberty [CDGP]). By contrast, adolescents with CHH lack pubertal activation of the HPG axis, which leads to absent puberty and infertility (Figure 1). In the majority of patients, puberty never occurs (absent puberty); less commonly, puberty is initiated then arrested (partial puberty).92,93,94 Adolescents with CHH exhibit steady linear growth and thus lack the so-called growth spurt.91,95,96 Given the delayed epiphyseal closure, these individuals often have eunuchoidal proportions. The most common complaints in male adolescents with CHH include absent and/or minimal virilization, low libido and lack of sexual function. Among female adolescents, lack of breast development and/or primary amenorrhoea by the age of 15 years are frequent complaints. In addition, patients with CHH often have low self-esteem, distorted body image, impaired psychosexual development and, in some cases, problems with sexual identity.97,98 Furthermore, small studies have shown increased anxiety and depression among adolescents with CHH and, as such, these symptoms merit attention.99,100
The clinical heterogeneity of CHH makes differentiation from CDGP, which is usually associated with short stature, poor growth velocity and delayed skeletal maturation, difficult.90,95 The presence of micropenis and/or cryptorchidism argues firmly in favour of CHH, as these features are rarely seen in CDPG. Associated congenital phenotypes are also very useful as they indicate a syndromic form of CHH. The classic, and by far most frequent, form being Kallmann syndrome, which includes anosmia. Other phenotypes like cleft palate or sensorineural deafness also suggest a syndromic form of CHH,94 most often associated with Kallmann syndrome, but not exclusively so.18 The clinical sign that points away from CDGP is a poor sense of smell, which suggests Kallmann syndrome.101,102,103 Other 'red flags' include congenital hearing impairment with or without pigmentation defects, bimanual synkinesia (mirror movements), dental agenesis, cleft lip and/or palate and cryptorchidism with or without micropenis.17,28,104,105,106,107,108 Family history indicative of autosomal inheritance cannot be used as a diagnostic tool, as this feature can be observed in both CHH and CDGP.109
Adulthood
CHH might be diagnosed after adolescence, when patients present for evaluation of infertility or even for osteoporotic fractures. This delay in diagnosis might be related to missed opportunities to diagnose CHH in adolescence or a reluctance of patients to seek medical evaluation.
Genetics of CHH
CHH is genetically heterogeneous, with both sporadic and familial cases. Several modes of inheritance have been identified, including X chromosome-linked recessive, autosomal recessive and dominant.1 By examining milder phenotypes (such as delayed puberty or isolated anosmia), the frequency of familial cases increases.110,111 Understanding of the molecular genetics of CHH and Kallmann syndrome has advanced tremendously in the past 20 years, since the first gene associated with Kallmann syndrome (KAL1 [ANOS1]) was identified by a positional cloning strategy in 1991 (Table 1).112,113,114,115KAL1 (ANOS1) encodes anosmin-1, a transiently expressed, locally restricted glycoprotein of embryonic extracellular matrices116 that is involved in fibroblast growth factor (FGF) signalling.117,118 To date, >25 different genes have been implicated in Kallmann syndrome and/or CHH, which accounts for ∼50% of cases.21 Causative genes for Kallmann syndrome include: KAL1 (ANOS1) in the X-linked form; FGFR1 (encoding fibroblast growth factor receptor 1),17,18FGF8,19,119CHD7,23,24,25,26,27HS6ST1 (encoding heparan-sulphate 6-O-sulphotransferase 1),20SOX10,28,29SEMA3A (encoding semaphorin-3A),36,37,38WDR11 (encoding WD repeat-containing protein 11)34,35 and IL17RD (encoding interleukin-17 receptor D)21 in the autosomal dominant form; and PROKR2 and/or PROK2,30,31,32,33 and FEZF139 in the autosomal recessive form, even though it should be noted that most patients carrying mutations in PROKR2 or PROK2 carry these mutations in the heterozygous state.120,121 Genes involved in CHH that are associated with a normal sense of smell include GNRHR (encoding gonadotropin-releasing hormone receptor),122,123GNRH1 (encoding gonadotropin-releasing hormone 1),124,125KISS1R,41,42KISS1,40,126TACR3 and TAC3.48,49,50 Other genes such as FGFR1 or PROKR2 can be mutated in patients with either Kallmann syndrome or CHH (Table 1). Remarkably, mutations in each Kallmann syndrome or CHH gene identified so far account for <10% of cases.11,21,127
CHH has classically been categorized as a monogenic disorder, which means that one defective gene is sufficient to account for the disease phenotype. As such, CHH and Kallmann syndrome caused by mutations in GNRHR (a recessive gene) and KAL1 (ANOS1; an X-linked gene), respectively, are highly penetrant,127 yet such a pattern is not observed for all genes involved in CHH and/or Kallmann syndrome. For instance, the p.Arg622X FGFR1 mutation has been reported in unrelated Kallmann syndrome and CHH probands (Figure 2). Examination of pedigrees reveals low penetrance for most CHH genes and variable expressivity among affected individuals carrying the same gene defect. Such an observation led to the hypothesis that multiple gene defects could synergize to produce a more severe CHH phenotype (that is, oligogenicity).30,128 Studies in large CHH cohorts indicate that at least 20% of cases are oligogenic11,21 (Table 1). This genetic model might be useful to predict genotype–phenotype correlations.5 Interestingly, genome-wide association studies have identified >100 loci affecting the age of menarche,129 which confirms a genetic contribution to pubertal timing. Surprisingly, little overlap exists between the genes implicated in age at menarche and CHH.129

In family 1, the CHH phenotype is fully penetrant.147 In the three other pedigrees, family members carrying the same FGFR1 mutation have variable reproductive and olfactory phenotypes and family 2 includes an asymptomatic carrier.17,148 + denotes wild-type allele. Abbreviation: CHH, congenital hypogonadotropic hypogonadism.
Diagnosis of CHH
Central to the evaluation process for diagnosing CHH is the exclusion of differential diagnoses such as pituitary tumour or functional causes (Box 2).
Clinical, biochemical & imaging investigations
The mini-puberty provides a brief opportunity to identify CHH. For male infants, cryptorchidism with or without micropenis can be suggestive of CHH (Figure 1). In such cases, hormone profiling at 4–8 weeks of life can be used to make a diagnosis83,85 based on comparisons with established reference ranges for gonadotropins, sex steroids and inhibin B levels.62,63,70,130 Female neonates born to parents with CHH should undergo biochemical evaluation; serum levels of FSH during the mini-puberty seem to be the most sensitive indicator of CHH in this setting.131 During childhood, diagnosis of CHH is very challenging as this interval is a physiologically hypogonadal period of development. Undetectable levels of FSH (but not of LH) or absence of response to a GnRH test in childhood might indicate CHH.
CHH diagnosis is typically made during adolescence or early adulthood (Table 2). Approximately half of patients with CHH have Kallmann syndrome.101 Self-report of anosmia is reliable, yet claims of having a 'good' sense of smell do not always align with formal smell testing. Most patients with Kallmann syndrome display olfactory bulb hypoplasia and/or aplasia on brain MRI; however, this finding is not fully concordant with sense of smell.102 An assessment of family history is important to identify familial cases. A family history of CDGP does not exclude CHH as pedigrees of patients with CHH seem to be enriched in delayed puberty, subfertility and anosmia or hyposmia.132 A detailed physical examination is needed, with particular attention paid to genitalia (that is, measurement of testicular volume using a Prader orchidometer) and virilization and/or estrogenization (that is, Tanner staging for breast development and pubic hair133,134). Eunuchoidal proportions are often present.135 A stage-line diagram is useful for providing developmental stages ±SD or as percentiles, as well as the tempo of pubertal development ±SD per year.91 Additionally, signs of associated developmental anomalies such as cryptorchidism with or without micropenis, midline defects or signs suggestive of combined pituitary hormone deficiency should be evaluated (Box 2).
Biochemical evaluation includes a variety of tests to exclude other causes of CHH such as pituitary tumour or functional causes (Table 3). Isolated hypogonadotropic hypogonadism is characterized by low serum levels of testosterone in male individuals (usually <2 nmol/l) in the setting of low or normal serum levels of gonadotropins (LH and FSH) with otherwise normal pituitary function (Figure 3). In female individuals, serum levels of estradiol are often low, sometimes undetectable in the setting of low-normal gonadotropin levels. Serum levels of estradiol seem to correlate with breast development, as most women with absent breast development have very low or undetectable levels, whereas women with breast development exceeding Tanner stage B2 usually have measurable serum levels of estradiol.

a | Serum testosterone levels. b | Serum gonadotropin (LH and FSH) levels. c | Serum inhibin B levels. d | Serum INSL3 levels. Healthy male individuals (controls; aged 17—27 years) versus untreated patients with CHH and/or KS (aged 17–29 years). Abbreviations: CHH, congenital hypogonadotropic hypogonadism; FSH, follicle-stimulating hormone; INSL3, insulin-like 3; LH, luteinizing hormone; KS, Kallmann syndrome. Adapted with permission from The Endocrine Society © Trabado, S. et al. J. Clin. Endocrinol. Metab. 99, E268–E275 (2014) and Young, J. J. Clin. Endocrinol. Metab. 97, 707–718 (2012).
In early adolescence (13–16 years), a diagnosis of CHH is challenging to make as isolated hypogonadotropic hypogonadism is common to both CHH and CDGP. In CHH, the GnRH stimulation test has a poor diagnostic value. In the absence of a gonadotropin response, this test can help to confirm severe GnRH deficiency, yet clinical signs are often already evident (that is, prepubertal testes or absence of breast development). By contrast, the GnRH stimulation test rarely enables differentiation of CDGP from partial forms of CHH—the most challenging differential diagnosis. A number of other tests have been proposed to differentiate between these two entities, including a combination of GnRH and hCG stimulation tests, as well as measurement of serum levels of inhibin B, AMH or INSL3.74,136,137,138,139,140,141,142 However to date, no gold-standard diagnostic test exists to fully differentiate CHH from CDGP.143
Inhibin B is a marker of Sertoli cell number and correlates with testicular volume.92 In men with CHH and severe GnRH deficiency, serum levels of inhibin B are typically very low (<30 pg/ml; Figure 3). However, for partial forms of CHH, inhibin B levels overlap with those in patients with CDGP and in healthy controls.138,139 INSL3 is a good marker of Leydig cell function144 and, as such, levels of INSL3 are typically low in male individuals with CHH (at diagnosis or on testosterone therapy) and increase with LH and/or hCG stimulation.74 However, further studies are needed to identify the potential clinical utility of INSL3 measurements, such as differentiating CHH from CDGP or as a predictor of reversible CHH. Kisspeptin is a potent stimulator of GnRH-induced LH secretion.9 Exogenous kisspeptin administration failed to induce a response in patients with CHH regardless of genotype. However, a male individual with reversible CHH exhibited a robust response to exogenous kisspeptin145 and continuous kisspeptin infusion was demonstrated to overcome genetic defects in the kisspeptin signalling pathway.146 In terms of clinical application, kisspeptin has been used with therapeutic effect in women with hypothalamic amenorrhoea,147 as well as for egg maturation during assisted fertility.148 Thus, although still investigational, kisspeptin seems to be emerging as useful in several therapeutic areas.
CHH work-up also includes cranial MRI to assess for tumours and/or space occupying lesions and to visualize inner ear and olfactory structures, bone age in adolescents to compare with chronological age and to assess epiphyseal closure, abdominal and/or pelvic ultrasonography to assess unilateral renal agenesis and to visualize the ovaries and/or uterus or testis, and bone densitometry (dual-energy X-ray absorptiometry) to assess bone density.
Genetic testing
Genetic testing is useful for diagnosis, prognosis149 and genetic counselling in CHH.127 The first step in prioritizing genes for genetic testing is to establish the inheritance pattern. Briefly, X-linked inheritance is observed in pedigrees when male-to-male transmission of the disease phenotype does not occur but the disease is observed in male members of the maternal line. This feature is typical for KAL1 (ANOS1) mutations underlying Kallmann syndrome.150 Autosomal dominant inheritance is observed in a family with vertical transmission of the disease phenotype across one or more generations. In this setting, male and female individuals are equally affected and male-to-male transmission can be observed. Importantly, incomplete penetrance and variable expressivity of the disease among people with identical mutations can be observed for most CHH genes (Figure 2).17,151,152 In autosomal recessive inheritance, vertical transmission of the disease phenotype does not occur and ∼25% of offspring are affected. Examples of autosomal recessive forms include those involving mutations in GNRHR, KISS1R, TACR3, PROKR2 or FEZF1 (Table 1).
Genetic testing can also be guided by the presence of additional phenotypic features. Cleft lip and/or palate and skeletal anomalies indicate mutations in genes encoding components of the FGF8 signalling pathway (for example, FGFR1, FGF8 and HS60ST1).17,20,21,104,153 Signs associated with CHARGE syndrome (CHARGE: coloboma, heart defects, atresia of choanae, retardation of growth and/or development, genital and/or urinary defects, and ear anomalies and/or deafness) favour screening for mutations in CHD7.24,25,26,104 Likewise, the association of CHH with congenital hearing impairment should direct screening towards CHD7, SOX10 and/or IL17RD.21,28,104 Bimanual synkinesia or renal agenesis are important clinical cues suggestive of KAL1 (ANOS1) mutations.101,104,114,154,155 Early onset of morbid obesity associated with CHH suggests mutations in LEP, LEPR or PCSK1.58,156 Indeed, combining CHH with specific associated phenotypes can increase the probability of finding causal mutations by targeted gene sequencing (Figure 4).21,28,104,157 CHH accompanied by other syndromic features such as congenital ichthyosis158 or spherocytosis159 is suggestive of a contiguous gene syndrome for which a karyotype or comparative genomic hybridization array analysis might be useful for identifying underlying chromosomal abnormalities.159,160,161

Genetic counselling
A legitimate question often raised by patients with CHH relates to the risk of passing on the disease to their offspring.26,127,162 When autosomal dominant transmission is suspected, the patient and partner should receive genetic counselling before fertility-inducing treatment. This counselling should include clear explanation of the 50% theoretical risk of transmitting the disease. In such cases, hormonal profiling during mini-puberty should be conducted. In proven autosomal recessive forms, which affect both male and female individuals, the risk of disease transmission to offspring is very low in the absence of consanguinity (that is, inter-related parents). This reduced risk is due to the very low frequency of heterozygous healthy carriers in the general population, which should be reassuring for intended parents. Male individuals with X-linked Kallmann syndrome should be informed of the automatic transmission of the mutation to their unborn daughters (obligate carriers) and the necessity for genetic counselling of these girls after the age of puberty. When patients carry several mutations in different CHH or Kallmann syndrome genes (that is, oligogenicity), genetic counselling is difficult and the transmission risk is variable.127 Finally, the advent of next-generation sequencing will probably improve the molecular genetic diagnosis of CHH.163 More specifically, this technology will help identify cases of oligogenicity and might promote personalized approaches to genetic counselling.
Treatment of CHH
No treatments exist for many of the CHH-associated phenotypes such as anosmia, bimanual synkinesia or renal agenesis. Others phenotypes such as cleft lip and/or palate, hearing loss or various skeletal defects require surgical and/or specialist intervention early in life. The approach to CHH treatment is largely determined by goals such as developing only virilization or estrogenization, or inducing fertility as well.
During infancy and childhood
In affected boys, the focus of most treatment is on appropriate testicular descent and penile growth. Cryptorchidism (particularly bilateral) can have far reaching negative effects on future fertility potential.164,165 As such, current recommendations advocate surgical correction at 6–12 months.166,167,168 Micropenis should be treated early using short-term, low-dose testosterone (dihydrotestosterone or testosterone esters) to induce penile growth (Table 4).86,169 Importantly, as the duration of treatment is brief, no major concerns regarding virilization or the development of secondary sexual characteristics exist; however, erections might be observed. Gonadotropins (LH and FSH) have been used to treat patients with micropenis and evidence of absent mini-puberty.170,171 Such an approach might be beneficial for the additional stimulatory effect on gonadal development. Indeed, treatment with FSH stimulates proliferation of immature Sertoli cells and spermatogonia in men with CHH before pubertal induction.10 Furthermore, neonatal gonadotropin therapy can increase intratesticular testosterone concentrations without fear of inducing spermatogenesis or negatively affecting the ultimate number of Sertoli cells (an important determinant of fertility), as Sertoli cells do not express the androgen receptor during the first 5 years of life.172,173 Although initial studies involving neonatal administration of gonadotropin have been promising, these studies are limited, both in number of studies and number of participants, and further investigations are needed to examine the effectiveness of gonadotropin and its effect on long-term outcomes such as fertility (Box 3).
During adolescence and adulthood
The aim of treatment is to induce virilization or estrogenization and normal sexual function, to stimulate statural growth, to promote bone health and to address concerns about future fertility as well as psychological and emotional wellbeing.174,175 Sex steroid therapy (that is, testosterone in male individuals and estradiol before estradiol + progesterone in female individuals) induces the characteristic changes of puberty, which includes psychosocial development (Table 4).
Induction of male sexual characteristics
Virilization is typically the primary objective of treating CHH174 in order to diminish the psychological suffering caused by sexual infantilism.97,98,176 Accordingly, prompt initiation of therapy after diagnosis is important and usually involves an injectable testosterone ester (aromatizable androgen such as enanthate, cypionate or undecanoate) or transdermal testosterone application with the precise protocol depending on the age at diagnosis and local practice.94,177,178 Paediatric endocrinologists treating young patients (that is, from 12 years of age) usually begin treatment with low-dose testosterone (for example, 50 mg of testosterone enanthate monthly, 10 mg transdermal testosterone every second day, or 40 mg oral testosterone undecanoate daily) and gradually increase to full adult dosing over the course of 18–24 months, depending on age at the start of treatment. Such regimens help mimic natural puberty and maximize statural growth while affording time for psychosexual development and minimizing the risk of precocious sexual activity.95,174
Adult endocrinologists often see patients with CHH in late adolescence or early adulthood when the main complaint is the lack of pubertal development.94 In such cases, the therapeutic approach is often more incisive than for younger patients, involving higher initial testosterone doses (200 mg testosterone enanthate monthly, then every 2–3 weeks) than those used by paediatric endocrinologists to induce more rapid virilization (Box 4). The frequency of injections should be guided by trough serum testosterone measurement targeting the lower end of the normal range to avoid periods of hypogonadism between injections. Of note, testosterone treatment will not induce gonadal maturation or fertility in these patients.94,177 Importantly, increased testicular volume following testosterone treatment is an indicator of reversal6,7,179,180,181,182,183,184,185 and requires re-assessment of the HPG axis in the absence of testosterone replacement therapy. An estimated 10–20% of patients with CHH undergo spontaneous recovery of reproductive function,6,7 and this includes patients with mutations in known CHH genes (Table 1). Moreover, reversal is not always lasting7,184 and, as yet, no predictors to identify those who will reverse or relapse exist (Box 3). Ongoing monitoring is thus justified.
Virilization in adolescents or young adults can also be achieved with pulsatile GnRH186,187,188 or gonadotropin therapy (hCG alone or combined with FSH).189,190 These fertility-inducing treatments stimulate endogenous testosterone (and estradiol) production by testicular Leydig cells and are associated with good physical and psychological outcomes.191 In clinical practice, testosterone enanthate is the most frequently used treatment for young adults, yet clinicians should avoid adherence to an overly dogmatic approach. Few published studies have examined the effects of treatment to induce virilization, as well as fertility treatment, on quality of life and sexuality in men with CHH,97,98,192 particularly in those with severe CHH (micropenis and cryptorchidism). Regardless of the therapy considered, patients and their entourage (that is, spouse, partner or family) should be clearly informed that treatment is likely to be lifelong and to require regular monitoring for optimal benefit.
Induction of male fertility
CHH manifests as a lack of gonadal development and infertility. Importantly, CHH is one of the rare treatable causes of male infertility. Spermatogenesis can be induced either by long-term pulsatile GnRH administration (pump)193 or more commonly by subcutaneous gonadotropin injections (2–3 times weekly).194,195,196,197,198 These therapies induce both testicular testosterone production by Leydig cells and spermatogenesis in the seminiferous tubules.199,200,201,202 The majority of patients with CHH develop sperm in their ejaculate with long-term therapy.149,193,196,197 Although these treatments seem to have similar fertility outcomes,199,200,201,202 comparing their efficacy is difficult owing to the small numbers of patients studied, heterogeneity in terms of degrees of GnRH deficiency (testicular volume before treatment), prior treatment and a lack of randomized studies performed to date.
A number of predictors of fertility outcome have emerged in CHH. First, cryptorchidism indicates a poor fertility prognosis.193,195 Men with CHH who have cryptorchidism often require extended courses of treatment (18–24 months).203,204 Prepubertal testicular volume (<4 ml) and/or low serum levels of inhibin B are also negative determinants of fertility outcome.193,194,195,196,197 Clinicians should be aware that although ∼75% of men will develop sperm, counts rarely reach the normal range with long-term therapy. A 2014 meta-analysis identified a mean sperm count of 5.9 × 106/ml (range: 4.7–7.1 × 106/ml) for gonadotropin therapy and 4.3 × 106/ml (range: 1.8–6.7 × 106/ml) for GnRH therapy.200 However, an important feature of patients with CHH is that low sperm concentrations do not preclude fertility.205
In clinical practice, when testicular volume is <4 ml, the classic treatment is either pulsatile GnRH (25 ng/kg every 2 h, titrated for trough serum testosterone level) or combined gonadotropin therapy (hCG: 1,000–1,500 IU + FSH: 75–150 IU 2–3 times weekly, based on available formulations). In patients with some gonadal development (testicular volume >4 ml) and no history of cryptorchidism, induction of spermatogenesis can be achieved with hCG monotherapy.194,206,207,208 If no sperm is present in the ejaculate after 3–6 months of treatment, FSH can be used to stimulate the seminiferous tubules.198 In the most severely affected men with CHH (testicular volume <4 ml), a sequential treatment has emerged in an attempt to maximize fertility potential.10,209,210 The unopposed FSH treatment before maturation by hCG (or alternatively, GnRH-induced LH) stimulates proliferation of immature Sertoli and germ cells. Outcomes of patients treated with this regimen demonstrate successful induction of testicular development and fertility in men with CHH who have prepubertal testes.10,210 To definitively determine the optimal approach to fertility treatment in men with CHH and severe GnRH deficiency (with or without cryptorchidism), an international, multicentre randomized trial is needed (Box 3).
Assessment of the fertility status of the female partner is crucial before induction of spermatogenesis and includes a detailed reproductive and/or menstrual history and evaluation of the integrity of the uterine cavity and tubal permeability (by use of sono-hysterosalpingography or hysterosalpingography). Anovulation and ovarian reserve can be assessed via a combination of history of menstrual cycle abnormalities, laboratory examination and pelvic ultrasonography. Couples should be counselled appropriately on the basis of the identified predictors of CHH fertility-inducing treatment,193,194,195,196,197,198,200 as well as the partner's reproductive status and age in order to develop realistic expectations regarding treatment outcome.
For men with CHH who have severely impaired sperm counts (or quality), assisted reproductive technology (ART) treatments such as in vitro fertilization can be used to improve fertility. More invasive procedures than ART include surgical testicular sperm extraction followed by in vitro intracytoplasmic sperm injection (ICSI). ICSI was initially used in men with CHH as a means to shorten the duration of treatment;211 however, outcomes are improved after maximal testicular volume is attained. Overall, fertilization is achieved in 50–60% of cases of ICSI, with pregnancy occurring in about a third of cycles.212,213,214,215 Although a study of sperm quality in men with CHH who received fertility-inducing treatment revealed neither altered DNA integrity nor an increased risk of chromosomal abnormalities,216 the possibility of an increased risk of birth defects with ART remains controversial. Once pregnancy is achieved, men with CHH can be transitioned to testosterone replacement therapy (injectable or transdermal preparations) for long-term treatment. Our consensus is that treatment be continued through the first trimester of pregnancy, in order to maintain male fertility capability in the event of miscarriage. Furthermore, although previous gonadotropin cycles seem to shorten the time to fertility in subsequent treatments,196 fertility in the future is not guaranteed. We, thus, recommend cryopreserving sperm once fertility has been attained.
Induction of female sexual characteristics
Puberty can be induced by oral or preferably transdermal estradiol administration in girls. The use of specific low-dose estradiol formulations (for example, 0.1 mg daily) are important.174 Transdermal estradiol administration is started (from age 10 years at the earliest) at low doses (typically 0.05–0.07 μg/kg nocturnally), with the goal of mimicking estradiol levels during gonadarche. In older girls, when breast development is a high priority, the starting dose of transdermal estradiol can be 0.08–0.12 μg/kg.217 The estradiol dose is slowly increased over 12–24 months, after which time cyclic gestagen is added (or after the first menstrual bleed) in order to maximize breast development. In adulthood, estradiol is typically given orally (at a dose of 1–2 mg) or transdermally (50 μg daily by patch or 1–2 pumps of 0.06% gel daily) as a maintenance dose with a cyclic progestin regimen (progesterone 200 mg for 14 days of the cycle, depending on formulation) to avoid endometrial hyperplasia. In the majority of women with CHH, estroprogestin therapy is effective in inducing harmonious development of the breasts and genitals, as well as an increased sense of femininity that contributes to a satisfactory emotional and sexual life. Estrogen treatment increases uterine size and combined estrogen and progestin therapy induces monthly withdrawal bleeding, but does not induce ovulation. For fertility, gonadotropins or GnRH therapy are necessary and effective.
Induction of female fertility
Infertility in women with CHH is related to insufficient follicular maturation, which leads to chronic anovulation.218 GnRH-induced pituitary gonadotropins induce absent or decreased physiological ovarian stimulation; however, no evidence of a decreased follicular reserve exists. This point must be emphasized as decreased circulating AMH concentrations observed in female patients with CHH and severe GnRH deficiency could wrongly suggest an alteration in ovarian reserve and therefore a poor fertility prognosis.219 By contrast, women with CHH should be informed that their infertility is treatable and that their reproductive prognosis is quite good in the absence of a male factor of infertility.
Before considering ovulation induction, sono-hysterosalpingography or hysterosalpingography should be performed to evaluate the integrity of the uterine cavity and fallopian tubes. One must also ensure regular sexual intercourse takes place and rule-out an associated male factor in infertility by semen analysis.220 Couples should be advised on the optimal timing of sexual intercourse during the ovulation induction as this first-line therapy does not require in vitro fertilization.221 The goal of ovulation induction therapy in female patients with CHH is to obtain a single ovulation and to avoid multiple pregnancies. Ovulation can be achieved either with pulsatile GnRH therapy222,223,224 or, alternatively, with FSH treatment followed by hCG or LH to trigger ovulation.225 One caveat is that most women with CHH do not secrete endogenous LH and, thus, require a complement of LH to stimulate local production of androgen substrates by theca cells, which facilitates sufficient secretion of estradiol by the dominant follicle.226,227,228 Physiologic estradiol secretion promotes optimal endometrial development and the cervical mucus production needed for sperm transit and embryo implantation.229 Typically, subcutaneous FSH doses of 75–150 IU per day are sufficient.223,224,225,229 The usual time required to obtain a dominant mature follicle (>18 mm) is ∼12 days.223,224,225,229 The starting dose of FSH is often increased or decreased depending on the ovarian response, as assessed by repeated serum estradiol measurements or by counting maturing follicles (using ultrasonography) performed approximately every 3–4 days. This regimen helps to minimize multiple pregnancies and the risk of ovarian hyperstimulation syndrome.230 After ovulation, progesterone production can be stimulated by repeated hCG injections or direct administration of progesterone during the postovulatory phase, until embryonic endogenous hCG secretion takes over.
Ovulation induction with a pulsatile GnRH pump can be effective even in the presence of GnRH resistance, such as in women with CHH who have mutations in GNRHR.231 GnRH can be administered at different pulse frequencies across the cycle, mimicking physiological ovarian stimulation.232 Alternatively, pulsatile GnRH at a constant frequency also induces maturation of ovarian follicles and triggers an LH peak. The usual doses comprise 3–10 μg per pulse, injected subcutaneously every 90 min. Monitoring ovulation induction with GnRH is useful, as the risk of multiple pregnancy and ovarian hyperstimulation syndrome is much lower than that with gonadotropin therapy.233 Once ovulation is obtained, the corpus luteum is stimulated to produce progesterone, which is necessary for embryo implantation. Short-term GnRH treatment is necessary to induce progesterone release until the endogenous secretion of hCG from the embryo begins.
Reducing long-term health effects of CHH
With appropriate and long-term treatment, many of the long-term effects of hypogonadism can be minimized. Adherence to treatment is key to supporting pubertal development, sexual function and psychological health.234 Patients with psychosexual problems, low self-esteem and distorted body image might require psychological counselling and often benefit from peer–peer support.98,192 Bone-health concerns in patients with CHH are well-recognized.235 Lack of sex steroids is the presumed cause of osteoporosis in patients with CHH236,237,238 and although sex steroid treatment typically improves bone density, it does not fully reverse the phenotype. Notably, mutations in FGF8, FGFR1 or SEMA3A might not only cause GnRH deficiency but also directly affect bone.153,239 We recommend a baseline bone density measurement in all patients with CHH at final height and after 2 years of treatment. To date, insufficient evidence exists to use the WHO–FRAX risk calculator240 in this population of patients. Long-term effects of hypogonadism also include an increased risk of developing metabolic problems. Several meta-analyses have demonstrated an association between low testosterone levels and the metabolic syndrome.241,242,243,244 In male individuals, low testosterone levels are also strongly associated with an increased risk of developing type 2 diabetes mellitus.245 However, few studies have examined metabolic risk specifically in patients with CHH. Interestingly, even short-term hypogonadism (such as that resulting from discontinuation of sex steroids for 2 weeks) induces increased fasting insulin concentrations in young men with CHH,246 which suggests that adherence to treatment is also important for promoting metabolic health. Accordingly, therapeutic education is important for promoting adherence in the ongoing management of CHH.
Transitional care for CHH
Transition of young adults from paediatric care to adult care is a well-recognized challenge for patients with chronic endocrine conditions247,248 including CHH.175 For patients with CHH, discontinuation of care and resulting gaps in treatment can have considerable health consequences. Several types of model for transition exist from a simple transfer of care to more structured programmes.249 Consensus among paediatric and adult endocrinologists in the European consortium studying GnRH biology (COST Action BM1105, http://www.gnrhnetwork.eu/) is that a structured transition is preferable and the process should include an evaluation of patient readiness, communication between providers and follow-up to ensure that the initial consultation has been completed. The timing of transition not only depends on structural aspects of a given health-care system but also on the individual patient's psychological and emotional development and capacity for autonomy and self-care.
Conclusions
CHH is a condition that is clinically and genetically heterogeneous. It is often challenging to diagnose, particularly during adolescence. In male individuals, cryptorchidism with or without micropenis might suggest neonatal CHH; however, similarly useful signs are lacking for female individuals. During adolescence, the diagnosis of CHH is difficult to discern from constitutional delay of growth and puberty. The presence of anosmia and/or olfactory bulb hypoplasia and/or aplasia (visualized by MRI) points to Kallmann syndrome, yet measurement of levels of serum gonadotropins, sex steroids, AMH and inhibin B are not always fully informative in confirming the diagnosis. At this stage, pubertal induction is commenced and a clinical and/or biochemical reassessment is conducted following therapeutic pause at final height, thus enabling a definitive diagnosis of CHH. Effective treatment is available not only for inducing virilization or estrogenization, but also for successful development of fertility. Advances in our understanding of the molecular basis of CHH have helped explain the genetic cause in 50% of cases and uncovered a complex model of genetics (oligogenicity) that might apply to a large proportion of patients. Furthermore, cases of reversal in adulthood point to gene–environment interactions.6,7 To advance the field, identification of novel biomarkers to aid early diagnosis of CHH, assembly of a large CHH population to study the complex molecular basis of the disease, and development of targeted treatments to optimize fertility outcomes are important. The COST network was created to address these issues as well as to promote translational research into human reproduction and improve the management of CHH.
References
Bianco, S. D. & Kaiser, U. B. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat. Rev. Endocrinol. 5, 569–576 (2009).
Schwanzel-Fukuda, M., Bick, D. & Pfaff, D. W. Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res. Mol. Brain Res. 6, 311–326 (1989).
Teixeira, L. et al. Defective migration of neuroendocrine GnRH cells in human arrhinencephalic conditions. J. Clin. Invest. 120, 3668–3672 (2010).
Seminara, S. B., Hayes, F. J. & Crowley, W. F. Jr. Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations. Endocr. Rev. 19, 521–539 (1998).
Mitchell, A. L., Dwyer, A., Pitteloud, N. & Quinton, R. Genetic basis and variable phenotypic expression of Kallmann syndrome: towards a unifying theory. Trends Endocrinol. Metab. 22, 249–258 (2011).
Raivio, T. et al. Reversal of idiopathic hypogonadotropic hypogonadism. N. Engl. J. Med. 357, 863–873 (2007).
Sidhoum, V. F. et al. Reversal and relapse of hypogonadotropic hypogonadism: resilience and fragility of the reproductive neuroendocrine system. J. Clin. Endocrinol. Metab. 99, 861–870 (2014).
Rey, R. A. et al. Male hypogonadism: an extended classification based on a developmental, endocrine physiology-based approach. Andrology 1, 3–16 (2013).
Chan, Y. M. Effects of kisspeptin on hormone secretion in humans. Adv. Exp. Med. Biol. 784, 89–112 (2013).
Dwyer, A. A. et al. Trial of recombinant follicle-stimulating hormone pretreatment for GnRH-induced fertility in patients with congenital hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 98, E1790–E1795 (2013).
Sykiotis, G. P. et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl Acad. Sci. USA 107, 15140–15144 (2010).
Petak, S. M. et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the evaluation and treatment of hypogonadism in adult male patients—2002 update. Endocr. Pract. 8, 440–456 (2002).
Wang, C. et al. Investigation, treatment and monitoring of late-onset hypogonadism in males: ISA, ISSAM, EAU, EAA and ASA recommendations. Eur. J. Endocrinol. 159, 507–514 (2008).
Bhasin, S. et al. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J. Clin. Endocrinol. Metab. 95, 2536–2559 (2010).
Forni, P. E., Taylor-Burds, C., Melvin, V. S., Williams, T. & Wray, S. Neural crest and ectodermal cells intermix in the nasal placode to give rise to GnRH-1 neurons, sensory neurons, and olfactory ensheathing cells. J. Neurosci. 31, 6915–6927 (2011).
Forni, P. E. & Wray, S. GnRH, anosmia and hypogonadotropic hypogonadism—where are we? Front. Neuroendocrinol. 36, 167–177 (2014).
Dode, C. et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 33, 463–465 (2003).
Pitteloud, N. et al. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl Acad. Sci. USA 103, 6281–6286 (2006).
Falardeau, J. et al. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Invest. 118, 2822–2831 (2008).
Tornberg, J. et al. Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc. Natl Acad. Sci. USA 108, 11524–11529 (2011).
Miraoui, H. et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am. J. Hum. Genet. 92, 725–743 (2013).
Gill, J. C., Moenter, S. M. & Tsai, P. S. Developmental regulation of gonadotropin-releasing hormone neurons by fibroblast growth factor signaling. Endocrinology 145, 3830–3839 (2004).
Ogata, T. et al. Kallmann syndrome phenotype in a female patient with CHARGE syndrome and CHD7 mutation. Endocr. J. 53, 741–743 (2006).
Jongmans, M. C. et al. CHD7 mutations in patients initially diagnosed with Kallmann syndrome—the clinical overlap with CHARGE syndrome. Clin. Genet. 75, 65–71 (2009).
Kim, H. G. et al. Mutations in CHD7, encoding a chromatin-remodeling protein, cause idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 83, 511–519 (2008).
Marcos, S. et al. The prevalence of CHD7 missense versus truncating mutations is higher in patients with Kallmann syndrome than in typical CHARGE patients. J. Clin. Endocrinol. Metab. 99, E2138–E2143 (2014).
Balasubramanian, R. et al. Functionally compromised CHD7 alleles in patients with isolated GnRH deficiency. Proc. Natl Acad. Sci. USA 111, 17953–17958 (2014).
Pingault, V. et al. Loss-of-function mutations in SOX10 cause Kallmann syndrome with deafness. Am. J. Hum. Genet. 92, 707–724 (2013).
Vaaralahti, K. et al. De novo SOX10 nonsense mutation in a patient with Kallmann syndrome and hearing loss. Pediatr. Res. 76, 115–116 (2014).
Dode, C. et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2, e175 (2006).
Leroy, C. et al. Biallelic mutations in the prokineticin-2 gene in two sporadic cases of Kallmann syndrome. Eur. J. Hum. Genet. 16, 865–868 (2008).
Cole, L. W. et al. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: molecular genetics and clinical spectrum. J. Clin. Endocrinol. Metab. 93, 3551–3559 (2008).
Abreu, A. P. et al. Loss-of-function mutations in the genes encoding prokineticin-2 or prokineticin receptor-2 cause autosomal recessive Kallmann syndrome. J. Clin. Endocrinol. Metab. 93, 4113–4118 (2008).
Kim, H. G. et al. WDR11, a WD protein that interacts with transcription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Am. J. Hum. Genet. 87, 465–479 (2010).
Quaynor, S. D. et al. The prevalence of digenic mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil. Steril. 96, 1424–1430 (2011).
Hanchate, N. K. et al. SEMA3A, a gene involved in axonal pathfinding, is mutated in patients with Kallmann syndrome. PLoS Genet. 8, e1002896 (2012).
Young, J. et al. SEMA3A deletion in a family with Kallmann syndrome validates the role of semaphorin 3A in human puberty and olfactory system development. Hum. Reprod. 27, 1460–1465 (2012).
Kansakoski, J. et al. Mutation screening of SEMA3A and SEMA7A in patients with congenital hypogonadotropic hypogonadism. Pediatr. Res. 75, 641–644 (2014).
Kotan, L. D. et al. Mutations in FEZF1 cause Kallmann syndrome. Am. J. Hum. Genet. 95, 326–331 (2014).
Topaloglu, A. K. et al. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N. Engl. J. Med. 366, 629–635 (2012).
de Roux, N. et al. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl Acad. Sci. USA 100, 10972–10976 (2003).
Seminara, S. B. et al. The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 349, 1614–1627 (2003).
Pinilla, L., Aguilar, E., Dieguez, C., Millar, R. P. & Tena-Sempere, M. Kisspeptins and reproduction: physiological roles and regulatory mechanisms. Physiol. Rev. 92, 1235–1316 (2012).
Kumar, D. et al. Murine arcuate nucleus kisspeptin neurons communicate with GnRH neurons in utero. J. Neurosci. 34, 3756–3766 (2014).
Clarkson, J. & Herbison, A. E. Oestrogen, kisspeptin, GPR54 and the pre-ovulatory luteinising hormone surge. J. Neuroendocrinol. 21, 305–311 (2009).
Kauffman, A. S. et al. Sexual differentiation of Kiss1 gene expression in the brain of the rat. Endocrinology 148, 1774–1783 (2007).
Mayer, C. et al. Timing and completion of puberty in female mice depend on estrogen receptor α-signaling in kisspeptin neurons. Proc. Natl Acad. Sci. USA 107, 22693–22698 (2010).
Topaloglu, A. K. et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin B in the central control of reproduction. Nat. Genet. 41, 354–358 (2009).
Young, J. et al. TAC3 and TACR3 defects cause hypothalamic congenital hypogonadotropic hypogonadism in humans. J. Clin. Endocrinol. Metab. 95, 2287–2295 (2010).
Gianetti, E. et al. TAC3/TACR3 mutations reveal preferential activation of gonadotropin-releasing hormone release by neurokinin B in neonatal life followed by reversal in adulthood. J. Clin. Endocrinol. Metab. 95, 2857–2867 (2010).
Navarro, V. M. & Tena-Sempere, M. Neuroendocrine control by kisspeptins: role in metabolic regulation of fertility. Nat. Rev. Endocrinol. 8, 40–53 (2012).
Hrabovszky, E. et al. Glutamatergic and GABAergic innervation of human gonadotropin-releasing hormone-I neurons. Endocrinology 153, 2766–2776 (2012).
Overgaard, A., Ruiz-Pino, F., Castellano, J. M., Tena-Sempere, M. & Mikkelsen, J. D. Disparate changes in kisspeptin and neurokinin B expression in the arcuate nucleus after sex steroid manipulation reveal differential regulation of the two KNDy peptides in rats. Endocrinology 155, 3945–3955 (2014).
Margolin, D. H. et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med. 368, 1992–2003 (2013).
Synofzik, M. et al. PNPLA6 mutations cause Boucher-Neuhauser and Gordon Holmes syndromes as part of a broad neurodegenerative spectrum. Brain 137, 69–77 (2014).
Topaloglu, A. K. et al. Loss-of-function mutations in PNPLA6 encoding neuropathy target esterase underlie pubertal failure and neurological deficits in Gordon Holmes syndrome. J. Clin. Endocrinol. Metab. 99, E2067–E2075 (2014).
Tata, B. et al. Haploinsufficiency of Dmxl2, encoding a synaptic protein, causes infertility associated with a loss of GnRH neurons in mouse. PLoS Biol. 12, e1001952 (2014).
Farooqi, I. S. & O'Rahilly, S. Mutations in ligands and receptors of the leptin-melanocortin pathway that lead to obesity. Nat. Clin. Pract Endocrinol. Metab. 4, 569–577 (2008).
Quennell, J. H. et al. Leptin indirectly regulates gonadotropin-releasing hormone neuronal function. Endocrinology 150, 2805–2812 (2009).
Donato, J. Jr et al. Leptin's effect on puberty in mice is relayed by the ventral premammillary nucleus and does not require signaling in Kiss1 neurons. J. Clin. Invest. 121, 355–368 (2011).
Bellefontaine, N. et al. Leptin-dependent neuronal NO signaling in the preoptic hypothalamus facilitates reproduction. J. Clin. Invest. 124, 2550–2559 (2014).
Kuiri-Hanninen, T. et al. Increased activity of the hypothalamic–pituitary–testicular axis in infancy results in increased androgen action in premature boys. J. Clin. Endocrinol. Metab. 96, 98–105 (2011).
Kuiri-Hanninen, T. et al. Postnatal developmental changes in the pituitary–ovarian axis in preterm and term infant girls. J. Clin. Endocrinol. Metab. 96, 3432–3439 (2011).
Sisk, C. L. & Foster, D. L. The neural basis of puberty and adolescence. Nat. Neurosci. 7, 1040–1047 (2004).
Boyar, R. M. et al. Human puberty. Simultaneous augmented secretion of luteinizing hormone and testosterone during sleep. J. Clin. Invest. 54, 609–618 (1974).
Wu, F. C., Butler, G. E., Kelnar, C. J. & Sellar, R. E. Patterns of pulsatile luteinizing hormone secretion before and during the onset of puberty in boys: a study using an immunoradiometric assay. J. Clin. Endocrinol. Metab. 70, 629–637 (1990).
Dunkel, L. et al. Developmental changes in 24-hour profiles of luteinizing hormone and follicle-stimulating hormone from prepuberty to midstages of puberty in boys. J. Clin. Endocrinol. Metab. 74, 890–897 (1992).
Valeri, C., Schteingart, H. F. & Rey, R. A. The prepubertal testis: biomarkers and functions. Curr. Opin. Endocrinol. Diabetes Obes. 20, 224–233 (2013).
Sehested, A. et al. Serum inhibin A and inhibin B in healthy prepubertal, pubertal, and adolescent girls and adult women: relation to age, stage of puberty, menstrual cycle, follicle-stimulating hormone, luteinizing hormone, and estradiol levels. J. Clin. Endocrinol. Metab. 85, 1634–1640 (2000).
Andersson, A. M. et al. Serum inhibin B in healthy pubertal and adolescent boys: relation to age, stage of puberty, and follicle-stimulating hormone, luteinizing hormone, testosterone, and estradiol levels. J. Clin. Endocrinol. Metab. 82, 3976–3981 (1997).
Hagen, C. P. et al. Serum levels of anti-Mullerian hormone as a marker of ovarian function in 926 healthy females from birth to adulthood and in 172 Turner syndrome patients. J. Clin. Endocrinol. Metab. 95, 5003–5010 (2010).
Aksglaede, L. et al. Changes in anti-Mullerian hormone (AMH) throughout the life span: a population-based study of 1027 healthy males from birth (cord blood) to the age of 69 years. J. Clin. Endocrinol. Metab. 95, 5357–5364 (2010).
Ivell, R., Heng, K. & Anand-Ivell, R. Insulin-like factor 3 and the HPG axis in the male. Front. Endocrinol. (Lausanne) 5, 6 (2014).
Trabado, S. et al. Insulin-like peptide 3 (INSL3) in men with congenital hypogonadotropic hypogonadism/Kallmann syndrome and effects of different modalities of hormonal treatment: a single-center study of 281 patients. J. Clin. Endocrinol. Metab. 99, E268–E275 (2014).
Nielsen, C. T. et al. Onset of the release of spermatozoa (spermarche) in boys in relation to age, testicular growth, pubic hair, and height. J. Clin. Endocrinol. Metab. 62, 532–535 (1986).
Biro, F. M. et al. Onset of breast development in a longitudinal cohort. Pediatrics 132, 1019–1027 (2013).
Herman-Giddens, M. E. et al. Secondary sexual characteristics in boys: data from the Pediatric Research in Office Settings Network. Pediatrics 130, e1058–e1068 (2012).
Aksglaede, L., Sorensen, K., Petersen, J. H., Skakkebaek, N. E. & Juul, A. Recent decline in age at breast development: the Copenhagen Puberty Study. Pediatrics 123, e932–e939 (2009).
Sorensen, K., Aksglaede, L., Petersen, J. H. & Juul, A. Recent changes in pubertal timing in healthy Danish boys: associations with body mass index. J. Clin. Endocrinol. Metab. 95, 263–270 (2010).
Evain-Brion, D., Gendrel, D., Bozzola, M., Chaussain, J. L. & Job, J. C. Diagnosis of Kallmann's syndrome in early infancy. Acta Paediatr. Scand. 71, 937–940 (1982).
Grumbach, M. M. A window of opportunity: the diagnosis of gonadotropin deficiency in the male infant. J. Clin. Endocrinol. Metab. 90, 3122–3127 (2005).
Kaplan, J. D., Bernstein, J. A., Kwan, A. & Hudgins, L. Clues to an early diagnosis of Kallmann syndrome. Am. J. Med. Genet. A 152A, 2796–2801 (2010).
Main, K. M., Schmidt, I. M. & Skakkebaek, N. E. A possible role for reproductive hormones in newborn boys: progressive hypogonadism without the postnatal testosterone peak. J. Clin. Endocrinol. Metab. 85, 4905–4907 (2000).
Wohlfahrt-Veje, C. et al. Acquired cryptorchidism is frequent in infancy and childhood. Int. J. Androl. 32, 423–428 (2009).
Baetens, D. et al. Extensive clinical, hormonal and genetic screening in a large consecutive series of 46,XY neonates and infants with atypical sexual development. Orphanet J. Rare Dis. 9, 209 (2014).
Hatipoglu, N. & Kurtoglu, S. Micropenis: etiology, diagnosis and treatment approaches. J. Clin. Res. Pediatr. Endocrinol. 5, 217–223 (2013).
Boas, M. et al. Postnatal penile length and growth rate correlate to serum testosterone levels: a longitudinal study of 1962 normal boys. Eur. J. Endocrinol. 154, 125–129 (2006).
Tomova, A. et al. Growth and development of male external genitalia: a cross-sectional study of 6200 males aged 0 to 19 years. Arch. Pediatr. Adolesc. Med. 164, 1152–1157 (2010).
Juul, A. et al. Pubertal development in Danish children: comparison of recent European and US data. Int. J. Androl. 29, 247–255 (2006).
Palmert, M. R. & Dunkel, L. Clinical practice. Delayed puberty. N. Engl. J. Med. 366, 443–453 (2012).
van Buuren, S. & Ooms, J. C. Stage line diagram: an age-conditional reference diagram for tracking development. Stat. Med. 28, 1569–1579 (2009).
Pitteloud, N. et al. The role of prior pubertal development, biochemical markers of testicular maturation, and genetics in elucidating the phenotypic heterogeneity of idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 87, 152–160 (2002).
Shaw, N. D. et al. Expanding the phenotype and genotype of female GnRH deficiency. J. Clin. Endocrinol. Metab. 96, E566–E576 (2011).
Young, J. Approach to the male patient with congenital hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 97, 707–718 (2012).
Lawaetz, J. G. et al. Evaluation of 451 Danish boys with delayed puberty: diagnostic use of a new puberty nomogram and effects of oral testosterone therapy. J. Clin. Endocrinol. Metab. 100, 1376–1385 (2015).
Hero, M. et al. Childhood growth of females with Kallmann syndrome and FGFR1 mutations. Clin. Endocrinol. (Oxf.) 82, 122–126 (2015).
Huffer, V., Scott, W. H., Connor, T. B. & Lovice, H. Psychological studies of adult male patients with sexual infantilism before and after androgen therapy. Ann. Intern. Med. 61, 255–268 (1964).
Dwyer, A. A., Quinton, R., Morin, D. & Pitteloud, N. Identifying the unmet health needs of patients with congenital hypogonadotropic hypogonadism using a web-based needs assessment: implications for online interventions and peer-to-peer support. Orphanet J. Rare Dis. 9, 83 (2014).
Aydogan, U. et al. Increased frequency of anxiety, depression, quality of life and sexual life in young hypogonadotropic hypogonadal males and impacts of testosterone replacement therapy on these conditions. Endocr. J. 59, 1099–1105 (2012).
Lasaite, L., Ceponis, J., Preiksa, R. T. & Zilaitiene, B. Impaired emotional state, quality of life and cognitive functions in young hypogonadal men. Andrologia 46, 1107–1112 (2014).
Quinton, R. et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin. Endocrinol. (Oxf.) 55, 163–174 (2001).
Lewkowitz-Shpuntoff, H. M. et al. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J. Clin. Endocrinol. Metab. 97, E136–E144 (2012).
Della Valle, E. et al. Prevalence of olfactory and other developmental anomalies in patients with central hypogonadotropic hypogonadism. Front. Endocrinol. (Lausanne) 4, 70 (2013).
Costa-Barbosa, F. A. et al. Prioritizing genetic testing in patients with Kallmann syndrome using clinical phenotypes. J. Clin. Endocrinol. Metab. 98, E943–E953 (2013).
Krams, M. et al. Kallmann's syndrome: mirror movements associated with bilateral corticospinal tract hypertrophy. Neurology 52, 816–822 (1999).
Laitinen, E. M. et al. Incidence, phenotypic features and molecular genetics of Kallmann syndrome in Finland. Orphanet J. Rare Dis. 6, 41 (2011).
Bailleul-Forestier, I. et al. Dental agenesis in Kallmann syndrome individuals with FGFR1 mutations. Int. J. Paediatr. Dent. 20, 305–312 (2010).
Molsted, K., Kjaer, I., Giwercman, A., Vesterhauge, S. & Skakkebaek, N. E. Craniofacial morphology in patients with Kallmann's syndrome with and without cleft lip and palate. Cleft Palate Craniofac. J. 34, 417–424 (1997).
Wehkalampi, K., Widen, E., Laine, T., Palotie, A. & Dunkel, L. Patterns of inheritance of constitutional delay of growth and puberty in families of adolescent girls and boys referred to specialist pediatric care. J. Clin. Endocrinol. Metab. 93, 723–728 (2008).
Waldstreicher, J. et al. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J. Clin. Endocrinol. Metab. 81, 4388–4395 (1996).
Zhu, J. et al. A shared genetic basis for self-limited delayed puberty and idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 100, E646–E654 (2015).
Franco, B. et al. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature 353, 529–536 (1991).
Legouis, R. et al. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell 67, 423–435 (1991).
Hardelin, J. P. et al. X chromosome-linked Kallmann syndrome: stop mutations validate the candidate gene. Proc. Natl Acad. Sci. USA 89, 8190–8194 (1992).
Bick, D. et al. Brief report: intragenic deletion of the KALIG-1 gene in Kallmann's syndrome. N. Engl. J. Med. 326, 1752–1755 (1992).
Hardelin, J. P. et al. Anosmin-1 is a regionally restricted component of basement membranes and interstitial matrices during organogenesis: implications for the developmental anomalies of X chromosome-linked Kallmann syndrome. Dev. Dyn. 215, 26–44 (1999).
Gonzalez-Martinez, D. et al. Anosmin-1 modulates fibroblast growth factor receptor 1 signaling in human gonadotropin-releasing hormone olfactory neuroblasts through a heparan sulfate-dependent mechanism. J. Neurosci. 24, 10384–10392 (2004).
Endo, Y., Ishiwata-Endo, H. & Yamada, K. M. Extracellular matrix protein anosmin promotes neural crest formation and regulates, FGF, BMP, and WNT activities. Dev. Cell 23, 305–316 (2012).
Trarbach, E. B. et al. Nonsense mutations in FGF8 gene causing different degrees of human gonadotropin-releasing deficiency. J. Clin. Endocrinol. Metab. 95, 3491–3496 (2010).
Sarfati, J. et al. A comparative phenotypic study of kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J. Clin. Endocrinol. Metab. 95, 659–669 (2010).
Martin, C. et al. The role of the prokineticin 2 pathway in human reproduction: evidence from the study of human and murine gene mutations. Endocr. Rev. 32, 225–246 (2011).
de Roux, N. et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N. Engl. J. Med. 337, 1597–1602 (1997).
Layman, L. C. et al. Mutations in gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nat. Genet. 18, 14–15 (1998).
Bouligand, J. et al. Isolated familial hypogonadotropic hypogonadism and a GNRH1 mutation. N. Engl. J. Med. 360, 2742–2748 (2009).
Chan, Y. M. et al. GNRH1 mutations in patients with idiopathic hypogonadotropic hypogonadism. Proc. Natl Acad. Sci. USA 106, 11703–11708 (2009).
Chan, Y. M. et al. GnRH-deficient phenotypes in humans and mice with heterozygous variants in KISS1/Kiss1. J. Clin. Endocrinol. Metab. 96, E1771–E1781 (2011).
Au, M. G., Crowley, W. F. Jr & Buck, C. L. Genetic counseling for isolated GnRH deficiency. Mol. Cell. Endocrinol. 346, 102–109 (2011).
Pitteloud, N. et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Invest. 117, 457–463 (2007).
Perry, J. R. et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature 514, 92–97 (2014).
Andersson, A. M. et al. Longitudinal reproductive hormone profiles in infants: peak of inhibin B levels in infant boys exceeds levels in adult men. J. Clin. Endocrinol. Metab. 83, 675–681 (1998).
Chellakooty, M. et al. Inhibin A, inhibin B, follicle-stimulating hormone, luteinizing hormone, estradiol, and sex hormone-binding globulin levels in 473 healthy infant girls. J. Clin. Endocrinol. Metab. 88, 3515–3520 (2003).
Waldstreicher, J. et al. The genetic and clinical heterogeneity of gonadotropin-releasing hormone deficiency in the human. J. Clin. Endocrinol. Metab. 81, 4388–4395 (1996).
Marshall, W. A. & Tanner, J. M. Variations in pattern of pubertal changes in girls. Arch. Dis. Child. 44, 291–303 (1969).
Marshall, W. A. & Tanner, J. M. Variations in the pattern of pubertal changes in boys. Arch. Dis. Child. 45, 13–23 (1970).
Kallmann, F. J., Schoenfeld, W. A. & Barrera, S. E. The genetic aspects of primary eunuchoidism. Am. J. Ment. Defic. 48, 203–236 (1944).
Segal, T. Y., Mehta, A., Anazodo, A., Hindmarsh, P. C. & Dattani, M. T. Role of gonadotropin-releasing hormone and human chorionic gonadotropin stimulation tests in differentiating patients with hypogonadotropic hypogonadism from those with constitutional delay of growth and puberty. J. Clin. Endocrinol. Metab. 94, 780–785 (2009).
Binder, G., Schweizer, R., Blumenstock, G. & Braun, R. Inhibin B plus LH vs GnRH agonist test for distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism in boys. Clin. Endocrinol. (Oxf.) 82, 100–105 (2015).
Coutant, R. et al. Baseline inhibin B and anti-Mullerian hormone measurements for diagnosis of hypogonadotropic hypogonadism (HH) in boys with delayed puberty. J. Clin. Endocrinol. Metab. 95, 5225–5232 (2010).
Adan, L. et al. Plasma inhibin B and antimullerian hormone concentrations in boys: discriminating between congenital hypogonadotropic hypogonadism and constitutional pubertal delay. Med. Sci. Monit. 16, CR511–CR517 (2010).
Young, J. et al. Antimullerian hormone in patients with hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 84, 2696–2699 (1999).
Vizeneux, A. et al. Congenital hypogonadotropic hypogonadism during childhood: presentation and genetic analyses in 46 boys. PLoS ONE 8, e77827 (2013).
Bay, K. et al. Insulin-like factor 3 serum levels in 135 normal men and 85 men with testicular disorders: relationship to the luteinizing hormone-testosterone axis. J. Clin. Endocrinol. Metab. 90, 3410–3418 (2005).
Harrington, J. & Palmert, M. R. Clinical review: Distinguishing constitutional delay of growth and puberty from isolated hypogonadotropic hypogonadism: critical appraisal of available diagnostic tests. J. Clin. Endocrinol. Metab. 97, 3056–3067 (2012).
Johansen, M. L. et al. Serum levels of insulin-like factor 3, anti-Mullerian hormone, inhibin B, and testosterone during pubertal transition in healthy boys: a longitudinal pilot study. Reproduction 147, 529–535 (2014).
Chan, Y. M. et al. Exogenous kisspeptin administration as a probe of GnRH neuronal function in patients with idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 99, E2762–E2771 (2014).
Young, J. et al. Kisspeptin restores pulsatile LH secretion in patients with neurokinin B signaling deficiencies: physiological, pathophysiological and therapeutic implications. Neuroendocrinology 97, 193–202 (2013).
Jayasena, C. N. et al. Twice-weekly administration of kisspeptin-54 for 8 weeks stimulates release of reproductive hormones in women with hypothalamic amenorrhea. Clin. Pharmacol. Ther. 88, 840–847 (2010).
Jayasena, C. N. et al. Kisspeptin-54 triggers egg maturation in women undergoing in vitro fertilization. J. Clin. Invest. 124, 3667–3677 (2014).
Sykiotis, G. P. et al. Congenital idiopathic hypogonadotropic hypogonadism: evidence of defects in the hypothalamus, pituitary, and testes. J. Clin. Endocrinol. Metab. 95, 3019–3027 (2010).
Hardelin, J. P. et al. Heterogeneity in the mutations responsible for X chromosome-linked Kallmann syndrome. Hum. Mol. Genet. 2, 373–377 (1993).
Xu, N. et al. A mutation in the fibroblast growth factor receptor 1 gene causes fully penetrant normosmic isolated hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 92, 1155–1158 (2007).
Pitteloud, N. et al. Reversible Kallmann syndrome, delayed puberty, and isolated anosmia occurring in a single family with a mutation in the fibroblast growth factor receptor 1 gene. J. Clin. Endocrinol. Metab. 90, 1317–1322 (2005).
Miraoui, H., Dwyer, A. & Pitteloud, N. Role of fibroblast growth factor (FGF) signaling in the neuroendocrine control of human reproduction. Mol. Cell. Endocrinol. 346, 37–43 (2011).
Kirk, J. M. et al. Unilateral renal aplasia in X-linked Kallmann's syndrome. Clin. Genet. 46, 260–262 (1994).
Georgopoulos, N. A. et al. Renal dysgenesis and KAL1 gene defects in patients with sporadic Kallmann syndrome. Fertil. Steril. 88, 1311–1317 (2007).
Jackson, R. S. et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat. Genet. 16, 303–306 (1997).
Villanueva, C. et al. Congenital hypogonadotropic hypogonadism with split hand/foot malformation: a clinical entity with a high frequency of FGFR1 mutations. Genet. Med. http://dx.doi.org/10.1038/gim.2014.166.
Ballabio, A. et al. Deletions of the steroid sulphatase gene in “classical” X-linked ichthyosis and in X-linked ichthyosis associated with Kallmann syndrome. Hum. Genet. 77, 338–341 (1987).
Vermeulen, S. et al. Kallmann syndrome in a patient with congenital spherocytosis and an interstitial 8p11.2 deletion. Am. J. Med. Genet. 108, 315–318 (2002).
Ballabio, A. & Andria, G. Deletions and translocations involving the distal short arm of the human X chromosome: review and hypotheses. Hum. Mol. Genet. 1, 221–227 (1992).
Vasson, A. et al. Custom oligonucleotide array-based CGH: a reliable diagnostic tool for detection of exonic copy-number changes in multiple targeted genes. Eur. J. Hum. Genet. 21, 977–987 (2013).
Dode, C. & Hardelin, J. P. Clinical genetics of Kallmann syndrome. Ann. Endocrinol. (Paris) 71, 149–157 (2010).
Lu, J. T., Campeau, P. M. & Lee, B. H. Genotype–phenotype correlation—promiscuity in the era of next-generation sequencing. N. Engl. J. Med. 371, 593–596 (2014).
Lee, P. A. et al. Paternity after unilateral cryptorchidism: a controlled study. Pediatrics 98, 676–679 (1996).
van Brakel, J. et al. Fertility potential in men with a history of congenital undescended testes: a long-term follow-up study. Andrology 1, 100–108 (2013).
Ritzen, E. M. et al. Nordic consensus on treatment of undescended testes. Acta Paediatr. 96, 638–643 (2007).
Penson, D., Krishnaswami, S., Jules, A. & McPheeters, M. L. Effectiveness of hormonal and surgical therapies for cryptorchidism: a systematic review. Pediatrics 131, e1897–e1907 (2013).
Chan, E., Wayne, C., Nasr, A. & FRCSC for Canadian Association of Pediatric Surgeon Evidence-Based Resource. Ideal timing of orchiopexy: a systematic review. Pediatr. Surg. Int. 30, 87–97 (2014).
Bin-Abbas, B., Conte, F. A., Grumbach, M. M. & Kaplan, S. L. Congenital hypogonadotropic hypogonadism and micropenis: effect of testosterone treatment on adult penile size why sex reversal is not indicated. J. Pediatr. 134, 579–583 (1999).
Main, K. M., Schmidt, I. M., Toppari, J. & Skakkebaek, N. E. Early postnatal treatment of hypogonadotropic hypogonadism with recombinant human FSH and LH. Eur. J. Endocrinol. 146, 75–79 (2002).
Bouvattier, C. et al. Neonatal gonadotropin therapy in male congenital hypogonadotropic hypogonadism. Nat. Rev. Endocrinol. 8, 172–182 (2012).
Chemes, H. E. et al. Physiological androgen insensitivity of the fetal, neonatal, and early infantile testis is explained by the ontogeny of the androgen receptor expression in Sertoli cells. J. Clin. Endocrinol. Metab. 93, 4408–4412 (2008).
Rey, R. A., Musse, M., Venara, M. & Chemes, H. E. Ontogeny of the androgen receptor expression in the fetal and postnatal testis: its relevance on Sertoli cell maturation and the onset of adult spermatogenesis. Microsc. Res. Tech. 72, 787–795 (2009).
Dunkel, L. & Quinton, R. Transition in endocrinology: induction of puberty. Eur. J. Endocrinol. 170, R229–R239 (2014).
Dwyer, A. A., Phan-Hug, F., Hauschild, M., Elowe-Gruau, E. & Pitteloud, N. Transition in endocrinology: hypogonadism in adolescence. Eur. J. Endocrinol. 173, R15–R24 (2015).
Bobrow, N. A., Money, J. & Lewis, V. G. Delayed puberty, eroticism, and sense of smell: a psychological study of hypogonadotropinism, osmatic and anosmatic (Kallmann's syndrome). Arch. Sex. Behav. 1, 329–344 (1971).
Han, T. S. & Bouloux, P. M. What is the optimal therapy for young males with hypogonadotropic hypogonadism? Clin. Endocrinol. 72, 731–737 (2010).
Santhakumar, A., Miller, M. & Quinton, R. Pubertal induction in adult males with isolated hypogonadotropic hypogonadism using long-acting intramuscular testosterone undecanoate 1-g depot (Nebido). Clin. Endocrinol. (Oxf.) 80, 155–157 (2014).
Quinton, R. et al. Kallmann's syndrome: is it always for life? Clin. Endocrinol. (Oxf.) 50, 481–485 (1999).
Sinisi, A. A. et al. Homozygous mutation in the prokineticin-receptor 2 gene (Val274Asp) presenting as reversible Kallmann syndrome and persistent oligozoospermia: case report. Hum. Reprod. 23, 2380–2384 (2008).
Tommiska, J., Jorgensen, N., Christiansen, P., Juul, A. & Raivio, T. A homozygous R262Q mutation in the gonadotropin-releasing hormone receptor presenting as reversal of hypogonadotropic hypogonadism and late-onset hypogonadism. Clin. Endocrinol. (Oxf.) 78, 316–317 (2013).
Laitinen, E. M. et al. Reversible congenital hypogonadotropic hypogonadism in patients with CHD7, FGFR1 or GNRHR mutations. PLoS ONE 7, e39450 (2012).
Kulshreshtha, B., Khadgawat, R., Gupta, N. & Ammini, A. Progression of puberty after initiation of androgen therapy in patients with idiopathic hypogonadotropic hypogonadism. Indian J. Endocrinol. Metab. 17, 851–854 (2013).
Santhakumar, A., Balasubramanian, R., Miller, M. & Quinton, R. Reversal of isolated hypogonadotropic hypogonadism: long-term integrity of hypothalamo–pituitary–testicular axis in two men is dependent on intermittent androgen exposure. Clin. Endocrinol. (Oxf.) 81, 473–476 (2014).
Mao, J. F. et al. Reversal of idiopathic hypogonadotropic hypogonadism: a cohort study in Chinese patients. Asian J. Androl. 17, 497–502 (2014).
Delemarre-Van de Waal, H. A. Induction of testicular growth and spermatogenesis by pulsatile, intravenous administration of gonadotrophin-releasing hormone in patients with hypogonadotrophic hypogonadism. Clin. Endocrinol. (Oxf.) 38, 473–480 (1993).
Hoffman, A. R. & Crowley, W. F. Jr. Induction of puberty in men by long-term pulsatile administration of low-dose gonadotropin-releasing hormone. N. Engl. J. Med. 307, 1237–1241 (1982).
Gong, C., Liu, Y., Qin, M., Wu, D. & Wang, X. Pulsatile GnRH is superior to hCG in therapeutic efficacy in adolescent boys with hypogonadotropic hypogonadodism. J. Clin. Endocrinol. Metab. http://dx.doi.org/10.1210/jc.2015-1343.
Bouvattier, C., Tauber, M., Jouret, B., Chaussain, J. L. & Rochiccioli, P. Gonadotropin treatment of hypogonadotropic hypogonadal adolescents. J. Pediatr. Endocrinol. Metab. 12 (Suppl. 1), 339–344 (1999).
Barrio, R., de Luis, D., Alonso, M., Lamas, A. & Moreno, J. C. Induction of puberty with human chorionic gonadotropin and follicle-stimulating hormone in adolescent males with hypogonadotropic hypogonadism. Fertil. Steril. 71, 244–248 (1999).
Shiraishi, K., Oka, S. & Matsuyama, H. Assessment of quality of life during gonadotrophin treatment for male hypogonadotrophic hypogonadism. Clin. Endocrinol. (Oxf.) 81, 259–265 (2014).
Varimo, T., Hero, M., Laitinen, E. M., Sintonen, H. & Raivio, T. Health-related quality of life in male patients with congenital hypogonadotropic hypogonadism. Clin. Endocrinol. (Oxf.) http://dx.doi.org/10.1111/cen.12701.
Pitteloud, N. et al. Predictors of outcome of long-term GnRH therapy in men with idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 87, 4128–4136 (2002).
Burris, A. S., Rodbard, H. W., Winters, S. J. & Sherins, R. J. Gonadotropin therapy in men with isolated hypogonadotropic hypogonadism: the response to human chorionic gonadotropin is predicted by initial testicular size. J. Clin. Endocrinol. Metab. 66, 1144–1151 (1988).
Miyagawa, Y. et al. Outcome of gonadotropin therapy for male hypogonadotropic hypogonadism at university affiliated male infertility centers: a 30-year retrospective study. J. Urol. 173, 2072–2075 (2005).
Liu, P. Y. et al. Induction of spermatogenesis and fertility during gonadotropin treatment of gonadotropin-deficient infertile men: predictors of fertility outcome. J. Clin. Endocrinol. Metab. 94, 801–808 (2009).
Warne, D. W. et al. A combined analysis of data to identify predictive factors for spermatogenesis in men with hypogonadotropic hypogonadism treated with recombinant human follicle-stimulating hormone and human chorionic gonadotropin. Fertil. Steril. 92, 594–604 (2009).
Dwyer, A. A., Raivio, T. & Pitteloud, N. Gonadotrophin replacement for induction of fertility in hypogonadal men. Best Pract. Res. Clin. Endocrinol. Metab. 29, 91–103 (2015).
Buchter, D., Behre, H. M., Kliesch, S. & Nieschlag, E. Pulsatile GnRH or human chorionic gonadotropin/human menopausal gonadotropin as effective treatment for men with hypogonadotropic hypogonadism: a review of 42 cases. Eur. J. Endocrinol. 139, 298–303 (1998).
Rastrelli, G., Corona, G., Mannucci, E. & Maggi, M. Factors affecting spermatogenesis upon gonadotropin-replacement therapy: a meta-analytic study. Andrology 2, 794–808 (2014).
Schopohl, J., Mehltretter, G., von Zumbusch, R., Eversmann, T. & von Werder, K. Comparison of gonadotropin-releasing hormone and gonadotropin therapy in male patients with idiopathic hypothalamic hypogonadism. Fertil. Steril. 56, 1143–1150 (1991).
Liu, L., Banks, S. M., Barnes, K. M. & Sherins, R. J. Two-year comparison of testicular responses to pulsatile gonadotropin-releasing hormone and exogenous gonadotropins from the inception of therapy in men with isolated hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 67, 1140–1145 (1988).
Kirk, J. M., Savage, M. O., Grant, D. B., Bouloux, P. M. & Besser, G. M. Gonadal function and response to human chorionic and menopausal gonadotrophin therapy in male patients with idiopathic hypogonadotrophic hypogonadism. Clin. Endocrinol. (Oxf.) 41, 57–63 (1994).
Bouloux, P. M. et al. Induction of spermatogenesis by recombinant follicle-stimulating hormone (puregon) in hypogonadotropic azoospermic men who failed to respond to human chorionic gonadotropin alone. J. Androl. 24, 604–611 (2003).
Burris, A. S., Clark, R. V., Vantman, D. J. & Sherins, R. J. A low sperm concentration does not preclude fertility in men with isolated hypogonadotropic hypogonadism after gonadotropin therapy. Fertil. Steril. 50, 343–347 (1988).
Finkel, D. M., Phillips, J. L. & Snyder, P. J. Stimulation of spermatogenesis by gonadotropins in men with hypogonadotropic hypogonadism. N. Engl. J. Med. 313, 651–655 (1985).
Vicari, E. et al. Therapy with human chorionic gonadotrophin alone induces spermatogenesis in men with isolated hypogonadotrophic hypogonadism—long-term follow-up. Int. J. Androl. 15, 320–329 (1992).
Kung, A. W., Zhong, Y. Y., Lam, K. S. & Wang, C. Induction of spermatogenesis with gonadotrophins in Chinese men with hypogonadotrophic hypogonadism. Int. J. Androl. 17, 241–247 (1994).
Raivio, T., Toppari, J., Perheentupa, A., McNeilly,A. S. & Dunkel, L. Treatment of prepubertal gonadotrophin-deficient boys with recombinant human follicle-stimulating hormone. Lancet 350, 263–264 (1997).
Raivio, T., Wikstrom, A. M. & Dunkel, L. Treatment of gonadotropin-deficient boys with recombinant human FSH: long-term observation and outcome. Eur. J. Endocrinol. 156, 105–111 (2007).
Yong, E. L., Lee, K. O., Ng, S. C. & Ratnam, S. S. Induction of spermatogenesis in isolated hypogonadotrophic hypogonadism with gonadotrophins and early intervention with intracytoplasmic sperm injection. Hum. Reprod. 12, 1230–1232 (1997).
Fahmy, I. et al. ICSI using testicular sperm in male hypogonadotrophic hypogonadism unresponsive to gonadotrophin therapy. Hum. Reprod. 19, 1558–1561 (2004).
Zorn, B., Pfeifer, M., Virant-Klun, I. & Meden-Vrtovec, H. Intracytoplasmic sperm injection as a complement to gonadotrophin treatment in infertile men with hypogonadotrophic hypogonadism. Int. J. Androl. 28, 202–207 (2005).
Bakircioglu, M. E., Erden, H. F., Ciray, H. N., Bayazit, N. & Bahceci, M. Gonadotrophin therapy in combination with ICSI in men with hypogonadotrophic hypogonadism. Reprod. Biomed. Online 15, 156–160 (2007).
Resorlu, B., Abdulmajed, M. I., Kara, C., Unsal, A. & Aydos, K. Is intracytoplasmic sperm injection essential for the treatment of hypogonadotrophic hypogonadism? A comparison between idiopathic and secondary hypogonadotrophic hypogonadism. Hum. Fertil. (Camb.) 12, 204–208 (2009).
Krabchi, K. et al. Quality assessment of induced spermatogenesis in hypogonadotrophic hypogonadic men treated with gonadotrophins. Reprod. Biomed. Online 22, 277–283 (2011).
Ankarberg-Lindgren, C., Kristrom, B. & Norjavaara, E. Physiological estrogen replacement therapy for puberty induction in girls: a clinical observational study. Horm. Res. Paediatr. 81, 239–244 (2014).
[No authors listed] Recombinant human luteinizing hormone (LH) to support recombinant human follicle-stimulating hormone (FSH)-induced follicular development in LH- and FSH-deficient anovulatory women: a dose-finding study. The European Recombinant Human LH Study Group. J. Clin. Endocrinol. Metab. 83, 1507–1514 (1998).
Deubzer, B., Weber, K., Lawrenz, B., Schweizer, R. & Binder, G. Anti-mullerian hormone deficiency in girls with congenital multiple pituitary hormone deficiency. J. Clin. Endocrinol. Metab. 99, E1045–E1049 (2014).
Anawalt, B. D. Approach to male infertility and induction of spermatogenesis. J. Clin. Endocrinol. Metab. 98, 3532–3542 (2013).
Kulkarni, A. D. et al. Fertility treatments and multiple births in the United States. N. Engl. J. Med. 369, 2218–2225 (2013).
Silveira, L. F. & Latronico, A. C. Approach to the patient with hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 98, 1781–1788 (2013).
Abel, B. S. et al. Responsiveness to a physiological regimen of GnRH therapy and relation to genotype in women with isolated hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 98, E206–E216 (2013).
Li, R. H. & Ng, E. H. Management of anovulatory infertility. Best Pract. Res. Clin. Obstet. Gynaecol. 26, 757–768 (2012).
Shoham, Z. et al. Recombinant LH (lutropin alfa) for the treatment of hypogonadotrophic women with profound LH deficiency: a randomized, double-blind, placebo-controlled, proof-of-efficacy study. Clin. Endocrinol. (Oxf.) 69, 471–478 (2008).
Couzinet, B., Lestrat, N., Brailly, S., Forest, M. & Schaison, G. Stimulation of ovarian follicular maturation with pure follicle-stimulating hormone in women with gonadotropin deficiency. J. Clin. Endocrinol. Metab. 66, 552–556 (1988).
Schoot, D. C. et al. Recombinant human follicle-stimulating hormone and ovarian response in gonadotrophin-deficient women. Hum. Reprod. 9, 1237–1242 (1994).
Kaufmann, R. et al. Recombinant human luteinizing hormone, lutropin alfa, for the induction of follicular development and pregnancy in profoundly gonadotrophin-deficient women. Clin. Endocrinol. (Oxf.) 67, 563–569 (2007).
Messinis, I. E. Ovulation induction: a mini review. Hum. Reprod. 20, 2688–2697 (2005).
Kwan, I., Bhattacharya, S., Kang, A. & Woolner, A. Monitoring of stimulated cycles in assisted reproduction (IVF and ICSI). Cochrane Database of Systematic Reviews, Issue 8, Art. No.: CD005289 http://dx.doi.org/10.1002/14651858.CD005289.pub3.
Seminara, S. B. et al. Successful use of pulsatile gonadotropin-releasing hormone (GnRH) for ovulation induction and pregnancy in a patient with GnRH receptor mutations. J. Clin. Endocrinol. Metab. 85, 556–562 (2000).
Crowley, W. F. Jr & McArthur, J. W. Simulation of the normal menstrual cycle in Kallman's syndrome by pulsatile administration of luteinizing hormone-releasing hormone (LHRH). J. Clin. Endocrinol. Metab. 51, 173–175 (1980).
Santoro, N., Filicori, M. & Crowley, W. F. Jr. Hypogonadotropic disorders in men and women: diagnosis and therapy with pulsatile gonadotropin-releasing hormone. Endocr. Rev. 7, 11–23 (1986).
Wang, C. et al. Long-term testosterone gel (AndroGel) treatment maintains beneficial effects on sexual function and mood, lean and fat mass, and bone mineral density in hypogonadal men. J. Clin. Endocrinol. Metab. 89, 2085–2098 (2004).
Finkelstein, J. S. et al. Osteoporosis in men with idiopathic hypogonadotropic hypogonadism. Ann. Intern. Med. 106, 354–361 (1987).
Finkelstein, J. S. et al. Increases in bone density during treatment of men with idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 69, 776–783 (1989).
Behre, H. M., Kliesch, S., Leifke, E., Link, T. M. & Nieschlag, E. Long-term effect of testosterone therapy on bone mineral density in hypogonadal men. J. Clin. Endocrinol. Metab. 82, 2386–2390 (1997).
Laitinen, E. M., Hero, M., Vaaralahti, K., Tommiska, J. & Raivio, T. Bone mineral density, body composition and bone turnover in patients with congenital hypogonadotropic hypogonadism. Int. J. Androl. 35, 534–540 (2012).
Hayashi, M. et al. Osteoprotection by semaphorin 3A. Nature 485, 69–74 (2012).
Cauley, J. A., El-Hajj Fuleihan, G., Luckey, M. M. & FRAX® Position Development Conference Members. FRAX® International Task Force of the 2010 Joint International Society for Clinical Densitometry & International Osteoporosis Foundation Position Development Conference. J. Clin. Densitom. 14, 237–239 (2011).
Ding, E. L., Song, Y., Malik, V. S. & Liu, S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA 295, 1288–1299 (2006).
Corona, G. et al. Type 2 diabetes mellitus and testosterone: a meta-analysis study. Int. J. Androl. 34, 528–540 (2011).
Brand, J. S., van der Tweel, I., Grobbee, D. E., Emmelot-Vonk, M. H. & van der Schouw, Y. T. Testosterone, sex hormone-binding globulin and the metabolic syndrome: a systematic review and meta-analysis of observational studies. Int. J. Epidemiol. 40, 189–207 (2011).
Brand, J. S. et al. Testosterone, sex hormone-binding globulin and the metabolic syndrome in men: an individual participant data meta-analysis of observational studies. PLoS ONE 9, e100409 (2014).
Zarotsky, V. et al. Systematic literature review of the risk factors, comorbidities, and consequences of hypogonadism in men. Andrology 2, 819–834 (2014).
Yialamas, M. A. et al. Acute sex steroid withdrawal reduces insulin sensitivity in healthy men with idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 92, 4254–4259 (2007).
Pugeat, M. & Nicolino, M. From paediatric to adult endocrinology care: the challenge of the transition period. Pediatr. Endocrinol. Rev. 6 (Suppl. 4), 519–522 (2009).
Godbout, A., Tejedor, I., Malivoir, S., Polak, M. & Touraine, P. Transition from pediatric to adult healthcare: assessment of specific needs of patients with chronic endocrine conditions. Horm. Res. Paediatr. 78, 247–255 (2012).
Crouch, N. S. & Creighton, S. M. Transition of care for adolescents with disorders of sex development. Nat. Rev. Endocrinol. 10, 436–442 (2014).
Acknowledgements
The authors acknowledge support of COST Action BM1105.
Author information
Authors and Affiliations
Contributions
M.T.D., L.D., A.A.D., A.J., N.P., T.R., R.Q. and J.Y. researched data for the article. U.B., M.T.D., L.D., A.A.D., P.G., J.-P.H., A.J., N.P., V.P., T.R., R.Q. and J.Y. wrote the article. All authors provided substantial contributions to discussions of the content and reviewed and/or edited the manuscript before submission. The final Consensus statement was approved by the Clinical Working Group (Corin Badiu, Hermann Behre, Marco Bonomi, Claire Bouvattier, Jean-Claude Carel, Sophie Christin-Maitre, Martine Cools, Waljit Dhillo, Jyothis George, Neoklis Georgopoulos, Christina Ghervan, Jörg Gromoll, Michael Hauschild, Sabine Kliesch, Csilla Krausz, Philip Kumanov, Mariarosaria Lang-Muritano, Beatriz L. Santamaria, Juliane Leger, Madalina Musat, Vassos Neocleous, Irene Netchine, Marek Niedziela, Franziska Phan-Hug, Duarte Pignatelli, Michel Polak, Vera Popovic, Nicos Skordis, Vallo Tillmann, Jorma Toppari, Philippe Touraine and Josanne Vasallo) and the Genetic & Bioinformatics Working Group (James Acierno Jr, Magdalena Avbelj-Stefanija, Jérôme Bouligand, Anne Guichon-Mantel, Sabine Heger, Soo-Hyun Kim, Zoltán Kutalik, Manuel Lemos, Ken Ong, Luca Persani, Serban Radian, Tuula Rinne, Manuela Simoni, Brian Stevenson, Gerasimos Sykiotis, Johanna Tommiska and Ionannis Xenarios).
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Boehm, U., Bouloux, PM., Dattani, M. et al. European Consensus Statement on congenital hypogonadotropic hypogonadism—pathogenesis, diagnosis and treatment. Nat Rev Endocrinol 11, 547–564 (2015). https://doi.org/10.1038/nrendo.2015.112
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrendo.2015.112
This article is cited by
-
Whole-Exome Sequencing Analysis of Idiopathic Hypogonadotropic Hypogonadism: Comparison of Varicocele and Nonobstructive Azoospermia
Reproductive Sciences (2024)
-
The Tuscany Regional Network for rare diseases: from European Reference Networks’ experience to registry based organisation and management model for rare diseases
Orphanet Journal of Rare Diseases (2023)
-
Mutation spectrum of Kallmann syndrome: identification of five novel mutations across ANOS1 and FGFR1
Reproductive Biology and Endocrinology (2023)
-
Multi-omics study identifies that PICK1 deficiency causes male infertility by inhibiting vesicle trafficking in Sertoli cells
Reproductive Biology and Endocrinology (2023)
-
Successful pregnancy and delivery after ovulation induction therapy in a woman with congenital hypogonadotropic hypogonadism: a case report
BMC Pregnancy and Childbirth (2023)
