
Key Points
-
Academic drug discovery offers an opportunity to effectively harness curiosity-driven research to improve human and animal health, but it is not without risk.
-
We believe that the associated risks can be managed by considering at least five factors that affect the success or failure of projects: organization, target selection, assay design, medicinal chemistry and preclinical pharmacology.
-
This manuscript presents guidelines for reducing the risk that can be caused by poor planning in any of these areas.
Abstract
The number of academic drug discovery centres has grown considerably in recent years, providing new opportunities to couple the curiosity-driven research culture in academia with rigorous preclinical drug discovery practices used in industry. To fully realize the potential of these opportunities, it is important that academic researchers understand the risks inherent in preclinical drug discovery, and that translational research programmes are effectively organized and supported at an institutional level. In this article, we discuss strategies to mitigate risks in several key aspects of preclinical drug discovery at academic drug discovery centres, including organization, target selection, assay design, medicinal chemistry and preclinical pharmacology.
Similar content being viewed by others
Main
Over the past decade, the number of academic drug discovery centres that have been established has increased substantially1,2,3. We believe this trend holds great potential for the development of innovative approaches in both the technological and biological aspects of drug discovery and the concomitant benefits to human and animal health, given the rich history of successful contributions to new drugs from academic institutions.
These centres have arisen because of at least four interrelated developments in translational research. First, the contraction in early-stage discovery (preclinical) activities by the pharmaceutical industry has been partly offset by increased academic collaborations3. Second, funding agencies such as the US National Institutes of Health (NIH) appear to have expanded their expectations for evidence of translational potential in many of their funding mechanisms4. Third, many partnerships and consortia involving government, academic institutions, disease foundations and private sector organizations have been created to promote translational and drug discovery research (see Supplementary information S1 (table) for examples), which has increased the level of such research and enhanced the training opportunities for students within academic-based units5. Fourth, declining levels of government funding for basic research are increasing the need for the exploration of alternative sources such as non-governmental organizations and/or product licensing.
The time is therefore right to develop organized support of academic translational research in a manner that does not interfere or compete with either the funding of, or the continued emphasis on, what we term curiosity-driven research. Rather, this support can offer academic researchers the option of broadening the potential applied impact of their research without making translation their overarching goal. Establishment of university-associated centres to foster drug discovery and development is one way to offer this support, and this Review is written with this concept in mind. Most importantly, these units can provide the scientific expertise required in domains such as assay development and compound discovery and optimization — expertise that most biologically oriented principal investigators do not have. They can also lighten the associated organizational burden and provide a venue to conduct translational research outside the constraints of pharmaceutical shareholder demands and the reward systems of academia6. Finally, they can contribute to the innovation of drug discovery methods, which has primarily been undertaken historically in industry.
A challenge facing academic drug discovery is the environment in which it exists. One of the hallmark attributes of academia is principal investigators who are engaged in curiosity-driven research. Although these investigators may collaborate with other laboratories, they are often accustomed to leading projects in a different manner to that of 'product-oriented' organizations, and may be unaware of available translational resources7. By contrast, the core mission of pharmaceutical companies is the translation of basic research into novel and useful therapeutics, resulting in a strong emphasis on multidisciplinary collaboration and a product-oriented approach to project management. Such infrastructural organization and focus is sometimes missing from academic institutions. However, academic research and drug discovery can productively coexist, but it may be most effective if the unique goals of each practice are nurtured in an environment that fosters curiosity-driven research in both arenas while lowering the operational risks of pursuing translational projects8,9.
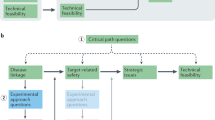
Translational project management is a relatively new concept in academic institutions, and when adopted it is sometimes in the hands of researchers who are unaware of the complexities involved in the effective transitioning of a project through its earliest phases up to a point at which a viable lead compound or series is identified. This inexperience can be an impediment to preclinical drug discovery when combined with the academic mindset or when directed by 'expert' drug discovery consultants who do not appreciate the tension that can exist between academic research and product development. Without the experience of someone who understands and values the need to work at the interface of curiosity-driven research and milestone-driven projects, many of these projects will — using a railway analogy — end up at a dead end or fall off the rails completely (Fig. 1). Such outcomes can be avoided, and there are now examples where an organized, collaborative and strategic approach to academic drug discovery has been successful9.

The figure is based on a railway analogy of translational research as a track, with milestones shown as blue ovals. Although there are several favourable end points in academic drug discovery (for example, early licensing, Phase I studies and rapid no-go decisions; shown as green circles), there are just as many — if not more — opportunities for projects to become derailed or reach a dead end (shown in pink). Practicing sound strategies related to project organization, targets, assays, medicinal chemistry and preclinical pharmacology can help mitigate the risks of unfavourable outcomes. ADMET, absorption, distribution, metabolism, excretion and toxicology; HTS, high-throughput screening; SAR, structure–activity relationship.
But what does success look like? On the purely translational side, success consists of the effective use of limited resources to support projects that culminate in a licensing opportunity, an entry into collaboration with a translational consortium or (most likely) a rapid no-go decision. On the academic side, success can also include better understanding of the disease process, the discovery of a high-quality chemical probe that is made broadly available to the community, the development of new enabling discovery methodologies, increased awareness of underserved diseases, opportunities for enhanced federal funding, and the training of students who can operate in either or both environments. Thus defined, the likelihood of success could be increased by careful mitigation of the operational risks associated with preclinical drug discovery and by academic leaders ensuring that their translation groups incorporate the knowledge of industrial advisors who are experienced in drug discovery prior to its 'commoditization'10.
With this in mind, in this article we propose a set of strategies to mitigate the risk of failure in academic preclinical small-molecule drug discovery (although some of the strategies are also applicable for biologic discovery). Although it is impossible to control for all risk, we discuss risks in five broad categories — organization, target selection, assay design, medicinal chemistry and preclinical pharmacology — and provide strategies that we believe will help academic translational scientists avoid some common pitfalls of, and enhance the impact of, higher-risk, cutting-edge projects. All parties in translational collaborations should accept that the success of a project is not likely to be gauged by the number of first-in-human trials or drugs it produces, given the high rate of project attrition11. However, we envision that many more projects will effectively reach successful end points as highlighted above (such as a better understanding of a disease process) if projects on the drug discovery track are entered into with a strategic plan that clearly outlines milestones and go/no-go decision points aimed at reducing future risk.
Risk mitigation at the organizational level
Once an institution decides that translational research is a priority, risks inherent in the organization must be reduced. The risk that translational research will not be effectively supported by existing bureaucratic, administrative and organizational structures is perhaps the greatest challenge that faces this area. As Mackay et al.12 state in their seminal paper on risk reduction in the pharmaceutical industry: “We must always find better ways to harness the intellectual power of a large company.” The same is true of an academic institution. Large research institutions have enormous intellectual power. For example, the scientific community of the Academic Health Center at the University of Minnesota (UMN), USA, is composed of more than 1,600 research scientists, generates US$380 million of federal funding per year and is housed in 2.4 million square feet of space in 59 buildings on 3 campuses. However, it must be noted that most academic organizations are not set up to perform translational research effectively. The silos in academia are typically the size of individual laboratories — much smaller than those in the pharmaceutical industry. Moreover, universities and colleges are often not set up to effectively handle confidentiality or material-transfer agreements, although the situation has generally improved over the past few years. Finally, when compared to most pharmaceutical companies, academic institutions lack the financial resources to adequately fund all but the most basic discovery operations. Nevertheless, there are several ways to optimize the organization to maximize the benefit to the three key stakeholders in academic translational research — the institution, the researchers and the public — which are summarized in Box 1 and discussed in more depth below.
Establish a culture of collaboration. Translational projects require the integration of many scientific disciplines and breaking down of the 'cultural' barriers that sometimes exist between the disciplines (Fig. 2). Academic drug discovery centres can help bridge the gap between academic researchers and collaborative networks and funding agencies, and medical schools can be an excellent source of translational projects. However, for such interactions to yield results there should be incentives and support available to assemble and fund the critical mass of scientists that a bona fide translational project requires.

An organized drug discovery institute and other specialty centres, together with the academic institute, can help enable a project to meet translational end points. Curiosity-driven researchers collaborating within the academic institute may have translatable projects. Centres within the university can help with the specialized research required for translation and act as intermediaries with contract research organizations for specialized assays and studies, collaborative groups, and funding agencies outside the institution. Academic researchers can also establish collaborative partnerships directly. ADMET, absorption, distribution, metabolism, excretion and toxicology; API, active pharmaceutical ingredient; HTS, high-throughput screening.
Interdisciplinary seminars and meetings can help break down cultural barriers. We believe, however, that one effective way of establishing healthy collaborations is by providing funding opportunities that specifically target lowering of the barriers between different laboratories and also between different institutions. One inter-institutional example of this type of funding mechanism is the Minnesota Partnership for Biotechnology and Medical Genomics, which brings together biomedical researchers at the UMN with scientists at the Mayo Clinic, Rochester, Minnesota, and typically provides 2 years of funding at up to $500,000 per year. On the intra-institutional level, the recent MnDrive grant competition awarded approximately 10 2-year grants of $500,000 for projects, most of which are translational in nature. Funding programmes such as these both encourage interdisciplinary and translational research and help researchers obtain preliminary results on potentially risky projects. This is an important consideration for programmes that will later seek federal or foundation funding.
Play to academic strengths. Attempting to compete with industry is probably counter-productive. For example, it is unlikely that academic research and development can be competitive in areas that pharmaceutical companies are actively investigating (although there are always exceptions) unless the biological targets are extraordinarily novel. However, even though academic institutions often lack the overarching organization and level of funding that industry has to pursue translational research, a major relative strength of academic institutions is the breadth and depth of their basic researchers13. Harnessing such strength in projects to investigate new areas of disease biology, less traditionally druggable targets, rare and neglected diseases, prodrugs, and repurposing of old drugs are probably far more realistic and valuable targets for academic translational efforts.
How a new compound will be different to existing medicines is an important, although often overlooked, first question to ask for academic drug discovery projects. For example, a new anxiolytic will need to be highly differentiated and efficacious to compete with existing drugs or to even gain the attention of biotechnology firms. We believe academia should be a place where high-risk and potentially low-financial-reward projects are accepted, and where one of the goals should be to provide benefit to society overall. However, given the limited availability of translational funding, institutions need to have mechanisms in place to recognize and redirect non-translatable programmes towards an equally valued basic science track. With qualified students, postdoctoral researchers and technical staff, an academic environment surrounding a translational initiative provides an ideal setting to acquire the skills necessary for future interdisciplinary research and communication. For example, the NIH offers joint postdoctoral training between biology and disease-focused NIH laboratories and the translational research environment of the National Center for Advancing Translational Sciences (NCATS).
Mine academic institutions for projects. Departments involved in the support of translational research may wish to consider proactively identifying projects within their institution rather than waiting until the investigator approaches the office of technology transfer or academic drug discovery institute within the organization. Not all basic researchers are able to recognize that their ideas might be amenable to translation and may never think to seek support along these lines.
Active mining for intellectual property and translational opportunities by 'innovation foragers' (designated translational specialists) is an effective first step in addressing this issue14. These foragers can attend and sponsor departmental seminars, meet informally with the faculty and attend scientific meetings where researchers seeking expertise in drug discovery will be present. Text-mining of proposals, literature databases (for example, NIH RePORTER or PubMed) and intra-university websites are an informatics-based means to assess potential opportunities. For promising projects for which the principal investigator is seeking funding, internal proof-of-concept studies or industrial partnerships can be recommended.
Fearlessly evaluate the science. Before embarking on a translational project, a comprehensive examination of its scientific foundations is needed. Just because a researcher has worked on a target for several years does not make it the best possible translational target to enter the academic pipeline15. This comprehensive inventory should include, but not be limited to, rigorous reviews of the following factors: the biological rationale and link to human disease; the approach to identify ligands to modulate the target or pathway; the availability of multiple starting points for chemical matter; the scientific goal or goals of the project; the plan for the confirmation and correlation of in vitro and in vivo activity and potency of compounds; and the potential for licensing, partnering with industry or federal translational programmes, or marketing. Other questions that must be considered include: what are the potential end points? What support is needed to reach these end points? How can these end points be guided by clear go/no-go decisions? For drug discovery projects, it is important to consult experienced medicinal chemists and pharmacologists as early as possible.
Define success broadly. The measure of success in translational research is best not defined in terms of drugs produced or first-in-human trials because the associated timescales and success rates are prohibitive. Although these may be the ultimate goals, other successful outcomes of translational research can be equally compelling. The development of probes to understand biological mechanisms, the enhancement of research funding, the formation of collaborations with an industry partner or the licensing of early-stage projects to industry can all be viewed as successful outcomes. The clear definition and articulation of a 'target product profile' (a detailed description of the candidate drug being aimed for) can also help focus the work of the project team16.
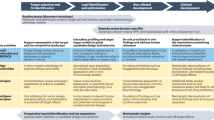
Identify domain expertise and support project teams. The multidisciplinary nature of drug discovery and development requires a broad range of expertise (Fig. 3). Typical project teams in industry are composed of biologists, medicinal chemists, computational experts, pharmacologists, pharmaceutical scientists, patent attorneys and marketing specialists, among others. Many of these skills will also be needed in academic institutions, or they will need to be outsourced. In either case, a central group is required to help support the true collaborative efforts of all these functions. This might take the form of a Director of Discovery or Translational Research and associated project managers, or could be promoted by organizations such as the Clinical and Translational Science Institutes, which have been established at several universities in the United States. It could also take the form of a drug discovery institute with a mandate from the university to organize these activities in a similar manner to existing programmes outside the pharmaceutical industry including, for example, the US National Cancer Institute's Experimental Therapeutics (NExT) programme, the NCATS' Therapeutics for Rare and Neglected Disease programme, the Structural Genomics Consortium's open-access public–private partnerships, and Sanford–Burnham's Florida Translational Research Program.

A large university can have much of the expertise necessary to assist in the translation of basic research into Phase I studies. Shown above is an outline of how this expertise is brought to bear on translational projects at the University of Minnesota, USA. ADME, absorption, distribution, metabolism, excretion; API, active pharmaceutical ingredient; GMP, good manufacturing practice; HTS, high-throughput screening; PD, pharmacodynamic; PK, pharmacokinetic. Adapted with permission from the University of Minnesota Therapeutics Development, Preclinical Testing and Clinical Trial Services brochure, University of Minnesota.
In any of these models, the support of the portfolio of all ongoing drug discovery projects needs to be considered in effective resourcing and funding. Importantly, academic drug discovery can typically afford to have a longer view of translational challenges and may choose to develop a research focus to investigate a difficult technological bottleneck. Therefore, this leadership might also monitor the translational landscape at the institution, helping to remove bottlenecks that might not be under the investigative jurisdiction of a single principal investigator and maintaining the institutional memory of projects that might be worth revisiting as the landscape evolves. Whatever form this overseeing body assumes, it can help ease the tension between individual expertise and overall management in a largely autonomous academic environment, where most principal investigators rightfully view themselves as experts in their niche fields and sometimes have difficulty not being in control. However, these organizations should always consider themselves stewards of translation rather than managers, lest they fall into the trap of treating drug discovery as a process that can be managed like a factory production line, which is considered to have stifled innovation in the pharmaceutical industry17. That is, administrators should focus on promoting the best science rather than simply counting the number of projects that 'leave the station' (using the railway analogy) or reach particular milestones (Fig. 1).
Consult with external experts. When institutional, consortium or philanthropic funds are to be applied to translational research, it is best practice to have projects reviewed by both internal and external experts in drug discovery and development. It is essential to have confidentiality agreements in place prior to seeking the advice of external reviewers and advisors. Early patent filing can help protect intellectual property in these cases, but it must be balanced against the costs and patent-life issues. These project reviews ideally take place in the project selection stage, but could also be useful as projects enter other critical stages, such as lead selection or in vivo studies. Internal experts can serve to recognize the resources available at the institution that would help support the proposed translational research. Some internal experts may also have sufficiently broad experience with drug discovery to point out the scientific, logistical and development issues that might need to be overcome to best prepare the project for success. Expert opinions from outside the institution should typically also be part of the decision-making process.
Maintain scholastic excellence in translational research. There needs to be a path for researchers to undertake longer-term translational projects that might not yield immediate publications or grant applications. Publication requirements for graduate students and postdoctoral collaborators must also be considered and their work not exclusively tied to aspects of the project less likely to be aligned with peer-reviewed publications. This may require some non-numerical evaluation of productivity (for example, evaluating the impact of research rather than counting the number of publications) and evaluation of the value of translational projects by researchers who are both experts in the science and in the particular demands of translational research.
We recommend that a forward-thinking publication and evaluation strategy be put into place for every project, and the pre-competitive nature of work be considered broadly to eliminate unnecessary publication delays. This plan should address the publication requirements of the faculty, students and technical staff involved. Frequently, biological or chemical results that disclose findings related to intellectual property can be published, and they should contain enough details not to compromise the reproducibility of the findings nor limit the complete understanding of the methods and materials published.
Reduce hurdles to industrial collaborations. The institution should ensure that technology transfer and legal offices understand the balance between protecting and offering simplified access to intellectual property. This balancing act can be involved in licensing and commercialization, confidential disclosure agreements, material transfer agreements and sponsored research agreements (SRAs). The SRA probably represents the biggest challenge in the development of intellectual property. Common hurdles in SRAs include balancing confidentiality and the freedom to publish, the difficulty of evaluating projects at an early stage, and developing effective project teams with industry18. Therefore, it should be a strategic imperative to develop a dialogue between stakeholders in the generation and protection of intellectual property to allow these processes to proceed as seamlessly and effectively as possible. Technology transfer offices should serve the academic community by making it as easy as possible to navigate the intellectual property protection and SRA landscape. Additionally, technology transfer offices that are staffed with professionals who understand chemical (and biological) matter protection can both ask the correct questions of the research scientists and evaluate the answers offered by outside experts.
Develop a long-term, stable funding strategy. The pressures associated with securing funding1 do not always ensure the best science and can limit novel approaches to drug discovery. Funding for translational work at a university can come from many sources, including — but not limited to — university initiatives, federal grants, industrial partnerships, foundations and philanthropy. The challenge facing many academic drug discovery centres is how to maintain experienced technical staff and pay for the maintenance and upgrading of extremely costly equipment given the vagaries of the aforementioned funding sources. In 2007, the UMN's Institute for Therapeutics Discovery and Development (ITDD) initiated internal and external fee-for-service activities7; however, this model has not been as good a source of funding as expected. Another method anticipated to support the UMN's ITDD is a percentage of royalty streams (a cost-recovery model) for projects and base support in the future (7–10 years), but this cannot be guaranteed. A longer-term solution may be to provide support via an endowment mechanism, but such funding is often difficult to establish. At NCATS, disease foundation-supported postdoctoral fellows provide a mechanism by which the foundations leverage the Center's resources and expertise to complement foundation-sponsored disease aetiology research. These postdoctoral fellows working at NCATS focus on developing assays and strategies for lead discovery in a strongly translational environment with regular input from the disease foundation's medical advisors and academic grantees19.
Effective institutional organization will facilitate the initiation and progress of translational projects. Although addressing institutional organization is broadly applicable to all types of translational work, the next four sections specifically discuss risk mitigation strategies for academic preclinical drug discovery projects. The key factors that need to be considered are to choose the correct targets, design informative assays to interrogate these targets, involve medicinal chemistry early in the process, and de-risk some of the preclinical pharmacology of lead compounds and series discovered in the processes described above.
Mitigation of risk in target selection
The first risk-mitigating steps on any proposal should be to address whether a target is precedented or unprecedented. For unprecedented targets (which might represent the best opportunities for academic researchers), thorough assessment of target druggability (that is, pharmacological tractability) and the evidence linking the target to the therapeutic hypothesis should be required20,21. Although the number of discrete molecular targets is relatively large22,23,24,25, not all are directly amenable to therapeutic intervention (that is, druggable), by which we mean a protein, peptide or nucleic acid with activity that can be modulated by a drug (typically a small-molecule or a peptide- or nucleic acid-based biologic)26. Targeting a phenotype — for example, one induced by a genetic lesion or infectious agent — should also take into consideration the principles of target-based risk mitigation. Here, target identification would constitute a project goal because although lack of target identification may not preclude advancement, it might severely limit scientific impact and clinical tractability.
Several important benchmarks should be addressed early in an academic drug discovery proposal (Box 2). One of the most important of these is to establish the reproducibility of the key in vitro and in vivo studies27,28,29.
Understand the attributes of an appropriate target. Despite the diversity in targets, human disease and patient populations, there is some consensus on the properties that are common for drug targets. For instance, these targets should have a validated role in disease and, ideally, there should be few consequences from their modulation in the absence of pathophysiology. Druggable targets should be assayable, preferably in both an in vitro and in vivo format to aid in subsequent probe and lead optimization and mechanistic studies. Knowledge of the basic science surrounding the target and its related pathways in both the disease and healthy state is preferred.
Thinking ahead, drug targets should have reliable biomarkers and animal models to assess whether the target is being modulated ('engaged') and to achieve a clear therapeutic effect in a relevant in vivo setting. Druggable also implies the target is amenable to ligand binding from a structural (thermodynamic) perspective. Therefore, having reliable structural models for the target and closely homologous targets is advantageous, as it can facilitate later structure-based design methods and target engagement studies30,31,32. However, judging target druggability is not a purely scientific endeavour. Excessive competition and the potential for overlapping intellectual property claims can diminish target druggability from a financial perspective — a reality that can be unpalatable for those accustomed to curiosity-driven research33.
Establish a tolerable target validation threshold. The strength and nature of the evidence for the importance of a particular target will vary with respect to the proposed therapeutic hypothesis. Strong validation of a target incorporates the existence of evidence tying pharmacological modulation of the target in cellular and animal models to a disease through genetic or epidemiological studies20. Cellular and animal models alone may support chemical probe development; however, lack of human relevance, for example, would sharply increase the risk that a useful therapeutic application would not be discovered. This initial validation process is most frequently accomplished by chemical and/or genetic manipulations of the target (or pathway) in a cellular system to demonstrate that its modulation alters the proposed disease phenotype34,35,36. However, certain genetic modification experiments should be interpreted as correlative rather than causative and should be viewed in the context of other biological data37. A parallel approach could include assessing the role of human genetic variations in the target — so-called 'experiments of nature'20.
The substantial resources involved in a long-term drug discovery project, coupled with recent concerns about scientific reproducibility, suggest that any landmark experiment that forms the foundation of a therapeutic hypothesis should be independently replicated, if feasible29. One criticism of strict target-based approaches is that they may be too reductionist and may neglect other crucial components of the relevant biology38,39. Therefore, taking a more global biological systems-based approach and carefully defining the target pathway or pathways may also be important at this juncture. Finally, the status of the target in humans should be verified in the context of healthy and pathological states, because this can further link the target to the disease state and may also aid in patient stratification and preclinical experimental design40.
Assess and establish assays for discovery and beyond. The available methods for assaying the proposed target should be surveyed and, in the case of novel targets, assay technologies may need to be created and validated. Projects will eventually have a need for appropriate in vivo models and equally valid biomarkers41. As an example, one of the joint projects between the UMN and the Mayo Clinic involves developing antifungal agents against pathogenic moulds such as Aspergillus fumigatus. In addition to assessing mouse survival in an invasive aspergillosis model, we are also measuring biomarkers such as fungal burdens (via PCR) and certain fungal antigens to monitor for fungicidal effects.
Designing assays, animal models and discovering biomarkers — and the equally important validation of these components — are not trivial undertakings and may consume considerable time and resources. Although some of these studies may be distant 'stations' on the translational track (Fig. 1), in terms of risk mitigation strategy, it is worth investing resources early in determining how compounds will be tested, lest a project reaches a critical bottleneck because no suitable test is in place. For discrete molecular targets, a plan to generate biomolecular structural information should also be an early priority to help jumpstart the development of any downstream structure-based experiments. Additionally, structural information can provide important insight about potential molecular target–ligand interactions and binding hypotheses and can inform rational functional mutagenesis strategies42,43,44.
Understand the risks and rewards of phenotype-based approaches. Target-based drug discovery seeks to directly modulate a specific molecular target (or targets), whereas phenotypic approaches more broadly attempt to reveal a desirable pharmacological response in cell-based or whole-organism settings45,46. Despite a past focus on target-based (rational drug design) approaches, therapeutics have been developed without knowledge of the discrete molecular target or targets47. The risks of such mechanistic uncertainty can confound lead selection and development, for example, by optimizing a lead compound on the basis of an assay readout that is not related to the anticipated therapeutic effect.
Such a situation was studied in depth for PTC124 (atalauren), a clinical candidate developed to promote read-through of disease-causing nonsense codon mutations. PTC124 was discovered and probably optimized using a cell-based reporter gene assay in which the luciferase reporter contained a nonsense mutation48. The readout, however, is prone to compound-mediated post-translational stabilization of the firefly luciferase reporter, thus complicating the identification and optimization of nonsense codon-suppressing compounds49,50.
To mitigate risk, it is strongly advisable to have a plan in place for target identification rather than relying on a 'black box' understanding of the phenotype. Identification of a target from such a forward chemical genomics approach is a de-risking strategy worth serious consideration. Otherwise, a series of cross-validating phenotypic assays is needed to assure pathway- or target-directed activity of follow-up compounds.
Pursue novel or higher-risk targets. As highlighted above, academic institutions can play to their strengths by choosing targets that would not normally be pursued by biotechnology and pharmaceutical companies13, in part because of the familiarity of academic researchers with the basic science of novel and high-risk targets and their ability to sustain long-term research projects. Examples of targets many feel are amenable to academic drug discovery include protein–protein and protein–polynucleotide interactions, targets often deemed undruggable, as well as tropical, rare and neglected diseases (for example, the NCATS Therapeutics for Rare and Neglected Diseases programme)51.
Certain epigenetic targets may also be well suited for academics (for example, challenging targets such as histone acetyltransferases). There is considerable interest in epigenetics-based therapeutics, and many seminal basic epigenetic discoveries are being made in academic institutions52. Additionally, with strong industry interest, academic–industry partnerships as exemplified by the Structural Genomics Consortium are becoming a viable model to support academic basic biology research with pharmaceutical medicinal chemistry resources. Nevertheless, academics can still target well-characterized systems provided their approaches are innovative. An example may be drug repurposing or prodrug design, a strategy that has had early success at the UMN53. This natural push to pursue riskier targets and to utilize novel techniques therefore necessitates operational 'risk assessment and mitigation' strategies.
Mitigation of risk in assays
Drug discovery assays combine current knowledge of specific proteins and biological pathways with sensitive measurement techniques to determine whether a chemical or biological entity will be effective in modulating the assay response in a concentration-dependent manner54 (Box 2). As all too many examples continue to show, haphazardly implementing the assay phase of a discovery campaign can lead to the misidentification of irrelevant compounds as hits, failure to identify the true or best hits (if any), and the unnecessary consumption of valuable resources49,55,56.
Guarantee the assay readout is on target. Assays designed for high-throughput screening (HTS) aim to integrate biological fidelity in an efficiently designed experiment measuring the effect of large numbers of substances on the biological readout of interest57. Because these technologies are often automated and conducted in low-volume, high-well-density microtitre plates (for example, 384 or 1,536 wells per plate), the protocols are considerably streamlined compared with standard laboratory practices, often comprising 'mix and read' schemes. The combined effect of testing many thousands of structurally heterogeneous chemical substances in HTS formats based on various fluorescence modalities (for example, fluorescence intensity, fluorescence polarization and fluorescence resonance energy transfer), reporter genes, cell viability measures, morphological and ion current sensors, among others, is to reduce substantially complex response outputs to relatively simple ranking outcomes. Although any given HTS assay may have been fully validated with appropriate controls, assay hits will often not only include compounds (and series) that are modulators of target biology, but also other compounds or their impurities that affect the assay components or signal output intensity (see Supplementary information S2 (table) for common sources of assay artefacts). These apparent compound activities can be notable and often display striking structure–activity relationships (SAR)49.
Consequently, the use of so-called 'orthogonal' assay formats is strongly encouraged, despite the additional effort required at the onset of the project to develop independent systems (see Supplementary information S3 (table) for a summary). The idea of using orthogonal assays is to be as sure as possible that the modulation of the measured activity is due to engagement of the target, pathway or phenotype of interest and not an assay component-dependent readout modulation (assay artefacts). Given that compound activities due to assay artefacts can be reproducible and concentration-dependent from the perspective of the HTS assay — characteristics shared by compounds with genuine activity — they can be difficult to easily triage. There are several well-documented studies and reviews that have explored in detail the underlying causes of some of the most common pitfalls with assay artefacts (Supplementary information S2 (table)). In some cases, solutions have been developed to help reduce the occurrence of these unwanted activities — for example, detergent suppression of aggregating compound effects or reporter configurations addressing compound-induced reporter stabilization58,59. Because chemical libraries are, by their very nature, highly heterogeneous with respect to chemotypes and never devoid of trace impurities, it is currently not possible to completely eliminate assay-dependent artefacts, whether their basis arises from confounding sample pharmacology, fluorescence or other biophysical properties (Supplementary information S2 (table)).
Target-based, phenotypic or both? Two broad categories define common assay approaches for general drug discovery screening: those measuring the activity of compounds on a specific target protein and those measuring a phenotypic response derived from an ensemble of processes (see Supplementary information S4 (table) for examples). A molecular target-based assay can include purified recombinant proteins or expressed cell-surface receptors, whereas phenotypic assays (also known as pathway assays) often rely on the measurement of an activity circumscribed by a cellular sentinel or defined response profile (for example, reporter gene, cellular signature or expressed transcripts). Cellular extracts, co-cultures and various model organisms have all been adapted as screening assays, allowing considerable complexity and context-specificity to be incorporated into an assay design47.
In some cases, particularly with some inherited genetic disorders, the causative gene mutation is defined but does not constitute a druggable target, meaning discovery strategies default to modifiers of the disease as potential drug targets. Regardless of the case, assays are designed with the current state of knowledge for the disease pathology and biology, and therefore can be subject to improvement or redesign as new information arises. In target-based assays, a mechanistic hypothesis for the target's role in disease underlies the rationale for this approach. However, unlike phenotypic assays, target-based screening primarily proceeds outside the disease pathway; therefore, these assays may not accurately reflect or recapitulate the target's molecular mechanism when examined in a more complex biological context. To help account for this lack of accuracy, such assays are best utilized with guidance from secondary assays of increasing physiological relevance20,40.
Mitigation of risk through medicinal chemistry
At their root, small-molecule drug discovery campaigns are based on the search for and optimization of chemical matter with specific physicochemical and pharmacological properties. The underlying basis for these properties is chemical in nature, and it logically follows that medicinal chemistry principles play a central role in this process (Box 2).
Assemble a quality library. The chemical matter identified by screening is predicated on the chemical library composition60. Screening libraries can be judged by several metrics, including size, chemical diversity, chemotypes, chemical integrity and follow-up accessibility. The optimal balance of these library characteristics may vary depending on the discovery project. For instance, smaller (focused) library subsets may be used for specific target classes. A large but non-chemically diverse library that samples only a small fraction of accessible chemical space is less likely to identify a range of chemotypes for follow-up. For example, it is estimated that a well-designed library of 350,000 compounds can adequately represent commercially available chemical space61. To minimize the odds of impurity artefacts, project teams should look for chemical libraries that are well managed, especially with respect to compound purity and identity62.
Another consideration should be the synthetic and commercial availability of the library. Many academic screening libraries are built from the same core group of commercial vendors, and therefore may have considerable overlap in terms of chemical composition and size. Corporate libraries, which are usually larger than academic libraries, may represent attractive sources of alternative or additional chemical matter. Tapping into libraries that arise from diversity-oriented synthesis and purified natural products, and incorporating more stereochemistry are strategies that academic researchers may wish to integrate into their plans for screening library enhancements63,64. It may be that the highest-value libraries reside in the chemistry and medicinal chemistry departments of the university, particularly with respect to novelty, access and ease of re-synthesis65.
Understand and respect the danger of nuisance compounds. Along with many successes, the cumulative experience of academic and industrial drug discovery has also shed light on unfavourable chemical properties and specific problematic chemotypes. As previously mentioned, HTS (both virtual and experimental) is liable to identify interference compounds, artefacts, false positives, promiscuous bioactive compounds and compounds with undesirable physicochemical properties, all of which can be difficult to remedy. Promiscuous compounds, such as certain p-hydroxyarylsulfonamides, show apparent biological activity across multiple assay formats and biological targets (Fig. 4). Such compounds have been called many names, including pan-assay interference compounds (PAINS) and frequent hitters56,66,67. They may show initially tantalizing results, but they usually fail to meet the requirements for chemical probes or lead compounds, although not before considerable resources have been spent on their follow-up. Tools such as cheminformatics, structural filters and literature searches for the 'natural history' of tentative follow-up compounds can rapidly mitigate the risk of pursuing these dead-end molecules68,69. At the UMN, we have successfully implemented a combination of computational resources, such as the PubChem database and BadAPPLE, and experimental counter-screens, such as ALARM NMR, to gauge bioassay promiscuity70,71,72.

There have been several reports in reputable journals of compounds bearing the p-hydroxyarylsulfonamide chemotype as being active in unrelated bioasssays92,93,94,95, despite either being flagged as pan-assay interference compounds (PAINS) or closely resembling the 'sulfonamide_B' PAINS substructure shown in the figure56. The bioassay promiscuity of p-hydroxyarylsulfonamides probably stems from a combination of compound instability, thiol-reactivity and redox activity96. BRAFV600E, B-rapidly accelerated fibrosarcoma V600E; CT-L, chymotrypsin-like; KEAP1, kelch-like ECH-associated protein; MCL1, myeloid cell leukaemia 1; NRF2, NFE2-related factor 2.
Arrive at lead compounds well informed. The choice of follow-up compounds is one of the most crucial steps in any drug discovery campaign73. A common temptation is to select the most potent compounds from a HTS campaign for follow up. However, more often than not, this method selects a non-chemically diverse set of compounds that can be fraught with artefacts, promiscuous bioactive compounds and compounds with poor lead-like properties.
The post-HTS triage phase has two main objectives: first, to identify the screening hits most likely to progress to later stages in the discovery pipeline and, second, to remove those hits that are probably false positives, artefacts, promiscuous inhibitors, possess poor physicochemical properties or are not amenable to synthesis. The triage process typically combines cheminformatics, confirmatory assays and analytical chemistry to prioritize chemical matter. This often entails a statistical analysis and correction of HTS data, followed by structural filters to identify problematic chemotypes (for example, PAINS and undesirable calculated properties). Depending on the activity criteria and specific screening approach, hits can be analysed by a series of cheminformatics techniques, such as scaffold clustering, and desirable calculated properties, such as ligand efficiency and lead-like descriptors. Hits can then be prioritized for additional experiments, such as dose–response confirmation and determination of half-maximal inhibitory concentrations (IC50), and appropriate follow-up screens. Additionally, as in any active area of research, advances in HTS are being made (for example, quantitative HTS)74 and should be incorporated when possible and practical to enhance the depth of information from the primary screen for more informed prioritization of active compounds.
Design a closely coupled screening and follow-up strategy. Although many chemical libraries will not contain compounds with the sought-after target activity, they will typically yield at least some hits in HTS assays. A data-driven, iterative prioritization process — ideally based on experimental cross-validation assays as highlighted above — can mitigate the risks of diverting time from the evaluation of promising though perhaps weakly active compounds owing to the presence of potentially more numerous 'noise' compounds (that is, those with apparent activity that are unlikely to be useful as either chemical probes or leads; Fig. 5). Therefore, a key issue that needs to be addressed is limited follow-up assay capacity after the original HTS assay, which could create pressure to select compounds primarily from statistical and computational methods rather than experimental data. A study by Lyssiotis et al.75 illustrated how 80% of the 3,000 active compounds from a 500,000-compound HTS assay to identify chemical reprogramming factors could be experimentally narrowed to 3 chemotype series with carefully designed high-capacity follow-up assays.

Drug discovery projects typically involve a target or phenotype identification, characterization and validation phase. This is usually followed by the design, optimization and validation of high-throughput screening (HTS) or related assays aimed at lead discovery, along with development of orthogonal assays and key counter-screens. Chemotypes for further investigation should be selected to have the desired physicochemical and experimental parameters relevant to project success. The top chemical entities with confirmed bioactivity and other desirable traits relevant for the project (for example, selectivity and lead-like properties) should then be investigated in higher-order experiments (preclinical pharmacology) such as cell-based assays and mechanism-of-action (MOA) explorations. The dotted arrow represents drug repurposing. HCS, high-content screening. Adapted with permission from Ref. 6, Elsevier.
Forming an early partnership with medicinal chemists, informaticians and molecular pharmacologists can facilitate the timely design and balanced incorporation of statistical analysis with appropriate controls, counter-screens and other follow-up assays. The design of counter-screens for assay interference and selectivity can be aided by understanding the chemical basis of the assays — another reason for partnering early with medicinal chemists. Well-managed screening programmes typically: confirm activity with orthogonal and counter-screens or selectivity assays; de-risk compounds of interest for common sources of non-specific interference such as thiol reactivity; demonstrate activity early on in more complex assays with greater physiological relevance, such as cell-based assays; and confirm that the activity of particular compounds is genuinely due to the compound (rather than an impurity or degradation product) through commercial resupply and independent re-synthesis.
Incorporate the advice of those experienced in HTS triage. Post-HTS triage (compound elimination and prioritization) is an evolving aspect of translational research, because new technologies and chemical libraries often introduce new unknowns in addition to advancing our reach into novel biological and chemical space. We highly recommend that this process be performed by (or alongside) those with extensive training and expertise in this specific process73. By constantly analysing the results of various HTS campaigns, such experts are often intimately familiar with problematic chemotypes and their natural histories, as well as other tacit knowledge not always apparent to medicinal chemists with other expertise (Fig. 6). As the number of compounds is culled, post-HTS triage experts can increasingly review past literature of the most promising compounds to mitigate the risk of pursuing frequent hitters, those with non-obvious chemical liabilities and those with competing intellectual property coverage.

Rhodanines97 (such as 1), polyhydroxylated phytochemicals such as as epigallocatechin gallate (2), tetrahydro-3H-cyclopenta[c]quinolines98 (such as 3) and 2-acetamidothiophene- 3-carboxylates99 (such as 4) are identified as hits in many high-throughput screening campaigns but are most likely false positives. Cell membrane perturbations have recently been shown to be a potential source of assay interference by compounds such as epigallocatechin gallate100 (2).
Ensure your compounds are fully characterized. The literature is rife with examples of academic projects that have been stalled by impurities and misidentification of the lead compounds. The potential significance of this type of misidentification was highlighted by recent news surrounding the misassigned chemical structure of TIC10 (ONC201). The future of funding for the clinical development of TIC10 as an anticancer drug is uncertain, pending resolution of the validity of an Oncoceutics patent based on the structure of an inactive isomer76.
The minimal critera to meet for translation are that compounds have confirmed identity by re-synthesis, and that their purity has been determined by stringent analytical methods, including high-performance liquid chromatography, mass spectrometry, evaporative light scattering detection, nuclear magnetic resonance (typically 1H and 13C NMR) and, in some cases, methods for trace metal analysis77. At a minimum, compound stability should be measured in aqueous media (and also assay and cell culture media) at acidic, neutral and basic pH, and in the assay buffers used in the HTS and any orthogonal assays.
Mitigation of risk through pharmacology
Whether drug development begins with a target or a compound, certain fundamental criteria will aid in acquiring the confidence to move forward. The tenets of pharmacology shape this path and have been well-articulated in several commentaries20,36,78. Although extensive studies of ADMET (absorption, distribution, metabolism, excretion and toxicology) are standard in the development phase, risk can be mitigated in the later stages of the discovery phase by performing some key, high-information experiments. The goals of these strategies are to increase confidence in candidate compounds with respect to their mechanism or mechanisms of action, their ability to meaningfully engage the target in higher-order systems (that is, cell-based and in vivo assays) and their adherence to general ADMET principles (Box 2).
Establish redundant lines of evidence. This strategy is important to connecting and amplifying results along a translational pathway. From an assay perspective, one should consider how stable the assay or assays are to factors that can create system-dependent variation (for example, cell passage number and batch preparations), if the assay could readily be conducted by any qualified scientist or technician with sufficient instruction, and how susceptible the assay design is to confounding compound-dependent interferences55,78. Creating a panel of assays that cross-validate the activity of a compound class is analogous to applying orthogonal chemotypes to crosscheck for the potential effect of off-target activities as the drivers of an observed biological effect.
Demand an interpretable SAR. From the perspective of the compound, obtaining a robust SAR not only demonstrates pharmacological tractability — mandatory evidence for lead optimization — but it also provides access to inactive or low potency analogues crucial to the evaluation of activity in complex disease models. For example, for conventional reversible inhibitors, close structural analogues should have similar activity to the parent compound, and this activity should decline with increasing structural divergence. By contrast, compounds showing either a flat SAR or an abrupt drop in activity with only minor modification (so-called 'activity cliffs') should raise suspicion79 (Fig. 7). The use of analogues enables a more compelling analysis of the necessary correlation between, for example, target- or pathway-based activity and the target- or pathway-mediated cell phenotype80. Although later stages of discovery traditionally focus on SAR and medicinal chemistry optimization, preliminary SAR can be assessed almost immediately following HTS by examining the activity of key analogues in the HTS library, combined with commercial resupply ('SAR-by-commerce') and well-planned parallel syntheses.

Structure–activity relationships (SARs; half-maximal inhibitory concentrations) reported for two chemical series developed for two different targets: aldose reductase101 (part a) and CDK2 (Ref. 102) (part b). The compounds in part a show a flat SAR in which both minor (highlighted in pink) and major (highlighted in blue) changes to the molecular structure lead to insignificant changes in activity, whereas the compounds in part b show a profound increase in potency as rational changes (highlighted in pink) are made within the series. A threefold to tenfold difference in the potency of well-designed analogues at <25 μM activity typically bodes well for series optimization.
Check for mechanistic soundness. Candidate compounds should be investigated for key mechanistic properties, including dose–response quality (for example, Hill slope and efficacy), reversibility and time-dependence. This strategy can serve as an important check on the previous experiments. For many target-based approaches, reversibility and target binding can often be assessed using surface plasmon resonance. An elegant example of the use of surface plasmon resonance to convincingly demonstrate reversible target binding and rule out non-specific binding (by pre-saturating the target with a potent natural ligand) is WEHI-539, a recently reported subnanomolar BCL-XL inhibitor81. These mechanistic details should be interpreted in the context of previous experiments, as well as chemical structures of the candidates, to form a coherent mechanistic hypothesis. For example, compounds showing time-dependent inhibition may be an indicator for covalent modifiers or they could be unstable under the assay conditions. This suggested strategy should be emphasized for compounds with potentially reactive structural components, which are traditionally avoided because of their potential for off-target effects.
Assess biological promiscuity and off-target effects early. One of the key mediators of off-target effects is non-specific activity, either by the parent compound or by a metabolite. This should be assessed early on (certainly by the time a lead series is chosen) by panel screening versus closely related targets. For example, the kinome selectivity of a new kinase inhibitor should be thoroughly understood prior to extensive series optimization. Assays such as this are commercially available. With respect to understanding the risk of toxic side effects, pharmaceutical companies typically profile their lead compounds in panel assays that may consist of >200 off-target enzymes, receptors, ion channels and transporters. An early gauge of the potential of adverse drug reactions could be to assay the activity of candidate compounds in a mini-panel of some of the more crucial anti-targets to avoid, such as the potassium voltage-gated channel subfamily H member 2 (KCNH2; also known as hERG), 5-HT2B receptors, α1B adrenoceptors, σ receptors, M1 muscarinic acetylcholine receptors, β2 adrenoceptors and μ-type opioid receptors82.
Investigate target engagement. For cases in which a target is known or presumed to be the site of action of a specific compound, engagement in a cellular context should be investigated. At the discovery stage, this could first take the form of establishing cell permeability. For early animal data, a natural extension would be to confirm the presence of the compound in the tissue. A recently described method to measure in vitro binding events involves the stabilization of the target to thermal denaturation in the cell or tissue by the bound chemotype83,84. If the approach proves generally applicable, it will be a powerful tool augmenting traditional affinity labelling and current proteomic approaches given its independence from chemical modifications of the target ligand85. If a project has progressed to the point of early animal models, then biomarker modulation should be confirmed.
Given that the majority of clinical failures result from an apparent lack of effect at a tolerable dose, establishing the magnitude of the pharmacological activity needed to achieve efficacy is a key factor. Understanding the extent to which dose-limiting toxicities could preclude reaching an efficacious dose is also an important factor. For example, the mutant BRAFV600E kinase inhibitor PLX4032 (also known as vemurafenib) initially did not result in a clinical response, even though the compound showed significant inhibition of ERK (extracellular signal-regulated kinase) phosphorylation in patient biopsies (IC50 of 31 nM). Dose escalation with a modified formulation, however, achieved >80% inhibition of biopsy-determined ERK phosphorylation and resulted in significant tumour regression. The low toxicity of this drug allowed administration at an oral dose of 960 mg twice a day86. In this striking example, recognizing the large therapeutic window afforded by this compound was crucial to its success in the clinic.
Investigate and understand ADMET issues. ADMET profiles of compounds will need to be understood in increasingly finer detail as they progress towards candidate status. In the early stages of discovery, these qualities can be estimated computationally, but in vitro and in vivo studies will be necessary to de-risk preclinical candidates87,88.
Build a coherent, evidence-based argument. An additional consideration surrounds how contextually independent the assay is (for example, species, tissue type, subunits, co-regulatory proteins and the endogenous substrate) from the definitive in vivo therapeutic environment. Here, establishing a correlation between the target or pathway assay and a measurable phenotype is advised. For example, Frearson et al.89 demonstrated the essential function of Trypanosoma brucei N-myristoyltransferase (TbNMT) by illustrating a direct correspondence between inhibition of the recombinant enzyme and bloodstream T. brucei proliferation for a series of pyrazole sulfonamides. Before proceeding to the costly development stage, the body of evidence should be scientifically and technically compelling. A minimal set of guidelines are that the cell-free, cell-based and in vivo data show correlation, and that the chemical structure fits the proposed mechanism of action79.
Summary and future perspectives
Many academic institutions have invested heavily in drug discovery and development efforts. For example, the Academic Drug Discovery Consortium now lists a membership of more than 100 academic drug discovery centres. If trends continue, more institutions and investigators will enter this arena. The single most important question that needs to be addressed with respect to both existing and emerging translation efforts is: how are these efforts most effectively supported in the local academic environment? We believe that academic drug discovery and development efforts should be built on the foundation of basic, curiosity-driven academic research, and should offer access to the capabilities and expertise required to de-risk potential translational projects.
Here, we have outlined some risk mitigation strategies that can help set the stage for effective engagement at the academic–applied science interface. However, even with the best risk management, >95% of these projects will fail90,91. We have already discussed an alternative goal mindset that redefines success more reasonably and more broadly. The goal cannot only be advancing drugs into the clinic, because in some cases this could take a decade or more. Strategic planning will help project teams or individual researchers imagine many possible successful end points. Quarterly reviews will help projects stay on the translational track. Minimally, these reviews should consider scientific progress, the competitive landscape, potential funding, partnership opportunities and exit strategies, as appropriate.
Working together, the stakeholders in the quest for new therapeutics in academic institutions can help establish an effective environment for drug discovery. The environment that will yield the best results is one that vigorously funds a mainline track of basic science while establishing a secondary, translational track that offers robust opportunities for innovative therapeutic discoveries.
References
Frye, S., Crosby, M., Edwards, T. & Juliano, R. US academic drug discovery. Nature Rev. Drug Discov. 10, 409–410 (2011). This is a comprehensive overview of the state and challenges of US academic drug discovery pre-2011.
Abou-Gharbia, M. & Childers, W. E. Discovery of innovative therapeutics: today's realities and tomorrow's vision. 2. Pharma's challenges and their commitment to innovation. J. Med. Chem. 57, 5525–5553 (2014).
Khanna, I. Drug discovery in pharmaceutical industry: productivity challenges and trends. Drug Discov. Today 17, 1088–1102 (2012).
Zoghbi, H. Y. The basics of translation. Science 339, 250 (2013).
Gurvich, V. J. & Byrn, S. R. NIPTE: a multi-university partnership supporting academic drug development. Drug Discov. Today 18, 916–921 (2013).
Hasson, S. & Inglese, J. Innovation in academic chemical screening: filling the gaps in chemical biology. Curr. Opin. Chem. Biol. 17, 329–338 (2013).
Dosa, P. I. et al. From HTS to Phase I: the Institute for Therapeutics Discovery and Development at the University of Minnesota. Comb. Chem. High Throughput Screen. 17, 231–240 (2014).
Alberts, B., Kirschner, M., Tilghman, S. & Varmus, H. Rescuing US biomedical research from its systemic flaws. Proc. Natl Acad. Sci. USA 111, 5773–5777 (2014).
Macdonald, G. J. & Lindsley, C. W. A unique industrial–academic collaboration towards the next generation of schizophrenia therapeutics. Curr. Top. Med. Chem. 14, 304–312 (2014).
Munos, B. H. & Chin, W. W. How to revive breakthrough innovation in the pharmaceutical industry. Sci. Transl. Med. 3, 1–3 (2011). This paper argues for high-risk research as an avenue towards breakthrough drugs, and examines the conflict between the rewards of pioneering work and the failure of profit-motivated research and development.
Paul, S. M. et al. How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nature Rev. Drug Discov. 9, 203–214 (2010).
Mackay, M., Street, S. D. A. & McCall, J. M. Risk reduction in drug discovery and development. Curr. Top. Med. Chem. 5, 1087–1090 (2005).
Huryn, D. M. Drug discovery in an academic setting: playing to the strengths. ACS Med. Chem. Lett. 4, 313–315 (2013). This paper details the unique strengths of academic drug discovery: the ability to pursue high-risk projects and the depth of expertise across a broad range of scientific fields.
Ungar, T. & Marcus, M. The innovation forager: stimulating academic innovation. Acad. Med. 89, 194 (2014).
Jorgensen, W. L. Challenges for academic drug discovery. Angew. Chem. Int. Ed. Engl. 51, 11680–11684 (2012).
Ashlock, M. A. & Olson, E. R. Therapeutics development for cystic fibrosis: a successful model for a multisystem genetic disease. Annu. Rev. Med. 62, 107–125 (2011).
Cuatrecasas, P. Drug discovery in jeopardy. J. Clin. Invest. 116, 2837–2842 (2006).
Bruneel, J., D'Este, P. & Salter, A. Investigating the factors that diminish the barriers to university–industry collaboration. Res. Policy 39, 858–868 (2010).
Inglese, J. et al. Genome editing-enabled HTS assays expand drug target pathways for Charcot–Marie-tooth disease. ACS Chem. Biol. 9, 2594–2602 (2014).
Plenge, R. M., Scolnick, E. M. & Altshuler, D. Validating therapeutic targets through human genetics. Nature Rev. Drug Discov. 12, 581–594 (2013). This review evaluates the methods used for drug target validation.
Robertson, G. Towards a more robust approach to selecting and prosecuting promising targets and compounds. Future Med. Chem. 2, 25–34 (2010).
Yan, C. & Higgins, P. J. Drugging the undruggable: transcription therapy for cancer. Biochim. Biophys. Acta 1835, 76–85 (2013).
Rual, J.-F. et al. Towards a proteome-scale map of the human protein–protein interaction network. Nature 437, 1173–1178 (2005).
Rask-Andersen, M., Almén, M. S. & Schiöth, H. B. Trends in the exploitation of novel drug targets. Nature Rev. Drug Discov. 10, 579–590 (2011).
Strachan, R. T., Ferrara, G. & Roth, B. L. Screening the receptorome: an efficient approach for drug discovery and target validation. Drug Discov. Today 11, 706–716 (2006).
Gashaw, I., Ellinghaus, P., Sommer, A. & Asadullah, K. What makes a good drug target? Drug Discov. Today 17, S24–S30 (2012). This article provides a pharmaceutical industry description of the evaluation of drug targets.
Peers, I. S., South, M. C., Ceuppens, P. R., Bright, J. D. & Pilling, E. Can you trust your animal study data? Nature Rev. Drug Discov. 13, 560 (2014).
Smith, M. A. & Houghton, P. A. Proposal regarding reporting of in vitro testing results. Clin. Cancer Res. 19, 2828–2833 (2013).
Prinz, F., Schlange, T. & Asadullah, K. Believe it or not: how much can we rely on published data on potential drug targets? Nature Rev. Drug Discov. 10, 712–713 (2011).
Surade, S. & Blundell, T. L. Structural biology and drug discovery of difficult targets: the limits of ligandability. Chem. Biol. 19, 42–50 (2012).
Egner, U. & Hillig, R. C. A structural biology view of target drugability. Exp. Opin. Drug Discov. 3, 391–401 (2008).
Perola, E., Herman, L. & Weiss, J. Development of a rule-based method for the assessment of protein druggability. J. Chem. Inf. Model. 52, 1027–1038 (2012).
Herschel, M. Portfolio decisions in early development: don't throw out the baby with the bathwater. Pharm. Med. 26, 77–84 (2012).
Vidalin, O., Muslmani, M., Estienne, C., Echchakir, H. & Abina, A. M. In vivo target validation using gene invalidation, RNA interference and protein functional knockout models: it is the time to combine. Curr. Opin. Pharmacol. 9, 669–676 (2009).
Wyatt, P. G., Gilbert, I. H., Read, K. D. & Fairlamb, A. H. Target validation: linking target and chemical properties to desired product profile. Curr. Topics Med. Chem. 11, 1275–1283 (2011).
Bunnage, M. E., Chekler, E. L. P. & Jones, L. H. Target validation using chemical probes. Nature Chem. Biol. 9, 195–199 (2013).
Decher, N., Netter, M. F. & Streit, A. K. Putative impact of RNA editing on drug discovery. Chem. Biol. Drug Des. 81, 13–21 (2013).
Vandamme, D., Minke, B. A., Fitzmaurice, W., Kholodenko, B. N. & Kolch, W. Systems biology-embedded target validation: improving efficacy in drug discovery. Wiley Interdiscip. Rev. Syst. Biol. Med. 6, 1–11 (2014).
Cucurull-Sanchez, L., Spink, K. G. & Moschos, S. A. Relevance of systems pharmacology in drug discovery. Drug Discov. Today 17, 665–670 (2012).
Fishman, M. C. Power of rare diseases: found in translation. Sci. Transl. Med. 201, 201ps11 (2013). This article describes examples from small cohort, first-in-human clinical studies, involving mechanistically homogeneous patient groups that have a genetic basis for a disease, as proof-of-concept trials.
Wendler, A. & Wehling, M. The translatability of animal models for clinical development: biomarkers and disease models. Curr. Opin. Pharmacol. 10, 601–606 (2010).
Wanner, J., Fry, D. C., Peng, Z. & Roberts, J. Druggability assessment of protein–protein interfaces. Future Med. Chem. 3, 2021–2038 (2011).
Kozakov, D. et al. Structural conservation of druggable hot spots in protein–protein interfaces. Proc. Natl Acad. Sci. USA 108, 13528–13533 (2011).
Fuller, J. C., Burgoyne, N. J. & Jackson, R. M. Predicting druggable binding sites at the protein–protein interface. Drug Discov. Today 14, 155–161 (2009).
Lee, J. A. & Berg, E. L. Neoclassic drug discovery: the case for lead generation using phenotypic and functional approaches. J. Biomol. Screen. 18, 1143–1155 (2013).
Sams-Dodd, F. Target-based drug discovery: is something wrong? Drug Discov. Today 10, 139–147 (2005).
Swinney, D. Phenotypic versus target-based drug discovery for first-in-class medicines. Clin. Pharm. Ther. 93, 299–301 (2013). This article promotes phenotypic assays for the identification of first-in-class drugs from which the mechanism of action can be determined.
Welch, E. et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 447, 87–91 (2007).
Auld, D. S. et al. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc. Natl Acad. Sci. USA 107, 4878–4883 (2010).
McElroy, S. P. et al. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays PLOS Biol. 11, e1001593 (2013).
Loregian, A. & Palù, G. How academic labs can approach the drug discovery process as a way to synergize with big pharma. Trends Microbiol. 21, 261–264 (2013).
DeWoskin, V. A. & Million, R. P. The epigenetics pipeline. Nature Rev. Drug Discov. 12, 661–662 (2013).
Chugh, R. et al. A preclinical evaluation of Minnelide as a therapeutic agent against pancreatic cancer. Sci. Transl. Med. 4, 156ra139 (2012).
Sittampalam, G. S. et al. Assay Guidance Manual http://www.ncbi.nlm.nih.gov/books/NBK53196/ (National Institutes of Health, 2004).
Thorne, N., Auld, D. S. & Inglese, J. Apparent activity in high-throughput screening: origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 14, 315–324 (2010). This is a comprehensive evaluation of potential interference mechanisms that are commonly encountered in high-throughput screening.
Baell, J. B. & Holloway, G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53, 2719–2740 (2010). This is the seminal manuscript on pan-assay interference compounds.
Inglese, J. et al. High-throughput screening assays for the identification of chemical probes. Nature Chem. Biol. 3, 466–479 (2007).
Hasson, S. A. et al. Chemogenomic profiling of endogenous PARK2 expression using a genome-edited coincidence reporter. ACS Chem. Biol. http://doi:10.1021/cb5010417 (2015).
Feng, B. Y. & Shoichet, B. K. A detergent-based assay for the detection of promiscuous inhibitors. Nature Protoc. 1, 550–553 (2006). This paper describes an important diagnostic assay from the laboratory that first defined a major source of HTS artifacts associated with the colloidal aggregates many library compounds form in assay buffers.
Lushington, G. & Chaguturu, R. To screen or not to screen: an impassioned plea for smarter chemical libraries to improve drug lead finding. Future Med. Chem. 6, 497–502 (2014).
Baell, J. B. Broad coverage of commercially available lead-like screening space with fewer than 350,000 compounds. J. Chem. Inf. Model. 53, 39–55 (2013).
Matson, S. L. et al. Best practices in compound management for preserving compound integrity and accurately providing samples for assays. J. Biomol. Screen. 14, 476–484 (2009).
Lovering, F., Bikker, J. & Humblet, C. Escape from Flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756 (2009).
Schreiber, S. L. Organic synthesis toward small-molecule probes and drugs. Proc. Natl Acad. Sci. USA 108, 6699–6702 (2011).
Hergenrother, P. J. Obtaining and screening compound collections: a user's guide and a call to chemists. Curr. Opin. Cell Biol. 10, 213–218 (2006).
Bruns, R. F. & Watson, I. A. Rules for identifying potentially reactive or promiscuous compounds. J. Med. Chem. 55, 9763–9772 (2012).
Walters, W. P. & Namchuk, M. A guide to drug discovery: designing screens: how to make your hits a hit. Nature Rev. Drug Discov. 2, 259–266 (2003).
Baell, J. & Walters, M. A. Chemistry: chemical con artists foil drug discovery. Nature 513, 481–483 (2014).
Saubern, S., Guha, R. & Baell, J. B. KNIME workflow to assess PAINS filters in SMARTS Format. Comparison of RDKit and Indigo cheminformatics libraries. Mol. Inform. 30, 847–850 (2011).
Han, L., Wang, Y. & Bryant, S. H. A survey of across-target bioactivity results of small molecules in PubChem. Bioinformatics 25, 2251–2255 (2009).
Huth, J. R. et al. ALARM NMR: a rapid and robust experimental method to detect reactive false positives in biochemical screens. J. Am. Chem. Soc. 127, 217–224 (2005).
Yang, J. et al. BioActivity Data Associative Promiscuity Pattern Learning Engine http://pasilla.health.unm.edu/tomcat/badapple/badapple (2014).
Dahlin, J. L. & Walters, M. A. The essential roles of chemistry in high-throughput screening triage. Future. Med. Chem. 6, 1265–1290 (2014).
Inglese, J. et al. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl Acad. Sci. USA 103, 11473–11478 (2006).
Lyssiotis, C. et al. Reprogramming of murine fibroblasts to induced pluripotent stem cells with chemical complementation of Klf4. Proc. Natl Acad. Sci. USA 106, 8912–8917 (2009).
Jacob, N. T., Lockner, J. W., Kravchenko, V. V. & Janda, K. D. Pharmacophore reassignment for induction of the immunosurveillance cytokine TRAIL. Angew. Chem. Int. Ed. 53, 6628–6631 (2014).
Hermann, J. C. et al. Metal impurities cause false positives in high-throughput screening campaigns. ACS Med. Chem. Lett. 4, 197–200 (2013).
Kenakin, T. Quantifying biological activity in chemical terms: a pharmacology primer to describe drug effect. ACS Chem. Biol. 4, 249–260 (2009). This article reviews the fundamental principles of pharmacology that are essential to those involved in any aspect of drug discovery and development.
Baell, J. B. Observations on screening-based research and some concerning trends in the literature. Future Med. Chem. 2, 1529–1546 (2010).
Chung, C. et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J. Med. Chem. 54, 3827–3838 (2011).
Lessene, G. et al. Structure-guided design of a selective BCL-XL inhibitor. Nature Chem. Biol. 9, 390–397 (2013).
Peters, J.-U. et al. Can we discover pharmacological promiscuity early in the drug discovery process? Drug Discov. Today 17, 325–335 (2012).
Molina, D. M. et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87 (2013). This paper describes a highly enabling method to determine the extent to which a compound binds to a target protein by virtue of its capacity to stabilize the target protein from thermal denaturation.
Jafari, R. et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nature Protoc. 9, 2100–2122 (2014).
Lee, J. C. et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372, 739–746 (1994). This is a harbinger of the power of phenotypic assays in what was then the new era of molecular biology.
Bollag, G. et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599 (2010).
Workman, P. & Collins, I. Probing the probes: fitness factors for small molecule tools. Chem. Biol. 17, 561–577 (2010). This is a comprehensive description of the quality attributes that define good probes, leads and drugs.
Kerns, E. H. & Di, L. Drug-like Properties: Concepts, Structure Design and Methods: from ADME to Toxicity Optimization (Academic Press, 2008).
Frearson, J. A. et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 464, 728–732 (2010). This paper contains an excellent example of a SAR correlation between a recombinant molecular target and parasite proliferation.
Nwaka, S. & Ridley, R. G. Science and society: virtual drug discovery and development for neglected diseases through public–private partnerships. Nature Rev. Drug Discov. 2, 919–928 (2003).
Silber, B. M. Driving drug discovery: the fundamental role of academic labs. Sci. Transl. Med. 2, 30cm16 (2010).
Abulwerdi, F. et al. A novel small-molecule inhibitor of Mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther. 13, 565–575 (2014).
Ge, Y. et al. Discovery and synthesis of hydronaphthoquinones as novel proteasome inhibitors. J. Med. Chem. 55, 1978–1998 (2012).
Qin, J. et al. Identification of a novel family of BRAFV600E inhibitors. J. Med. Chem. 55, 5220–5230 (2012).
Zhuang, C., Narayanapillai, S., Zhang, W., Sham, Y. Y. & Xing, C. Rapid identification of Keap1-Nrf2 small-molecule inhibitors through structure-based virtual screening and hit-based substructure search. J. Med. Chem. 57, 1121–1126 (2014).
Dahlin, J. et al. PAINS in the assay: chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J. Med. Chem. http://dx.doi.org/10.1021/jm5019093 (2015).
Sinko, W. et al. Undecaprenyl diphosphate synthase inhibitors: antibacterial drug leads. J. Med. Chem. 57, 5693–5701 (2014).
Johnson, S. et al. A biochemical screen for GroEL/GroES inhibitors. Bioorg. Med. Chem. Lett. 24, 786–789 (2014).
Yan, D. et al. Dual myxovirus screen identifies a small-molecule agonist of the host antiviral response. J. Virol. 87, 11076–11087 (2013).
Ingólfsson, H. I. et al. Phytochemicals perturb membranes and promiscuously alter protein function. ACS Chem. Biol. 9, 1788–1798 (2014).
Kadam, A. et al. Development of novel pyrazolone derivatives as inhibitors of aldose reductase: an eco-friendly one-pot synthesis, experimental screening and in silico analysis. Bioorg. Chem. 53, 67–74 (2014).
Schonbrunn, E. et al. Development of highly potent and selective diaminothiazole inhibitors of cyclin-dependent kinases. J. Med. Chem. 56, 3768–3782 (2013).
Acknowledgements
J.L.D. was supported by the following funding bodies: an NIH predoctoral fellowship (F30 DK092026-01); a Pharmaceutical Research and Manufacturers of America Foundation predoctoral pharmacology/toxicology fellowship; and the Mayo Foundation. J.I. is supported by the intramural programme in the National Center for Advancing Translational Sciences (NCATS), at the NIH. M.A.W. acknowledges research funding from the NIH and the Minnesota Partnership for Biotechnology and Medical Genomics. The authors specifically acknowledge the helpful comments and corrections suggested by the referees. The authors also acknowledge K. Nelson for critical reading of the manuscript and the many other important contributions in the drug discovery community that, regrettably, could not be cited owing to space constraints.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary information S1 (table)
Selected examples of entities that promote translational research (PDF 113 kb)
Supplementary information S2 (table)
Common artefactual compound-mediated biological activities (PDF 118 kb)
Supplementary information S3 (table)
Assay types employed in drug discovery (PDF 81 kb)
Supplementary information S4 (table)
Examples of high-throughput screening assays (PDF 106 kb)
Glossary
- Chemical probe
-
A well-characterized compound showing context-related selectivity that potently modulates a biochemical target or pathway in cells or in vivo.
- Prodrugs
-
Compounds that are the metabolic precursors of drugs. Prodrugs can aid in drug delivery and can be new chemical entities.
- Biomarkers
-
Factors that are objectively measured and evaluated as indicators of biological or pathological processes, or of pharmacological responses to therapeutic intervention.
- Reporter gene assay
-
An assay in which an easily detectable reporter gene such as luciferase is fused to the promoter sequence of downstream target genes of the signalling pathway under investigation. Modulation of the pathway, such as activation or inhibition, will lead to changes in reporter gene expression (in the case of luciferase, these changes will be measured as luminescence).
- Orthogonal assays
-
Corresponding assays used following, or in parallel to, the primary high-throughput screening assay to confirm compound activity that is independent of the primary assay technique.
- Chemotypes
-
Chemical structure motifs or primary substructures that are common to a group of compounds.
- Diversity-oriented synthesis
-
Efficient synthesis of a collection (library) of structurally diverse and complex small molecules that differ in stereochemistry, functional groups and molecular framework.
- Pan-assay interference compounds
-
(PAINS). Compounds containing substructures that give rise to activity in assays irrespective of the biological target.
- Off-target activities
-
Activities of a compound that differ from its designed or annotated mechanism of action. Off-target activities are often undefined.
Rights and permissions
About this article

Cite this article
Dahlin, J., Inglese, J. & Walters, M. Mitigating risk in academic preclinical drug discovery. Nat Rev Drug Discov 14, 279–294 (2015). https://doi.org/10.1038/nrd4578
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd4578
This article is cited by
-
Theoretical insight on antioxidant potency of kanzakiflavone-2 and its derivatives
Structural Chemistry (2021)
-
Early Detection of Toxic Cyanobacteria in Bulgarian Dam Water and In Vitro Evaluation of the Effect of Saponins From Astragalus glycyphyllos and A. glycyphylloides, in Cyanotoxin (Anatoxin-α)-Induced Neurotoxicity
Revista Brasileira de Farmacognosia (2020)
-
Computational advances in combating colloidal aggregation in drug discovery
Nature Chemistry (2019)
-
‘Stealth’ corporate innovation: an emerging threat for therapeutic drug development
Nature Immunology (2019)
-
Arylmethylamino steroids as antiparasitic agents
Nature Communications (2017)
