
Key Points
-
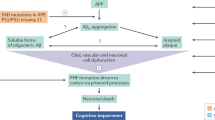
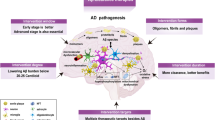
The amyloid cascade hypothesis posits that the deposition of the amyloid-β peptide in the brain parenchyma is a crucial step in Alzheimer's disease (AD). This concept has influenced and guided much of the academic and pharmaceutical research carried out during the past twenty years.
-
However, several therapeutic agents that purport to reduce the production or aggregation of the amyloid-β peptide have failed in Phase III clinical trials and this has brought an increased focus to this area of research.
-
Two crucial questions must therefore be considered: by how much should a therapeutic agent inhibit amyloid-β production, or facilitate amyloid-β clearance; and at what stage in the disease process should such a therapeutic agent be administered, to produce a disease-modifying effect in AD?
-
This article re-evaluates the amyloid cascade hypothesis and reviews relevant preclinical, clinical and genetic data. In particular, autosomal dominant familial AD is used to distinguish between the effects of amyloid-β deposition on the age of disease onset and the duration of the disease.
-
A strong case can be made that the deposition of amyloid-β in the brain parenchyma is crucial for initiating the disease process, but there are no compelling data to support the view that, once initiated, the disease process is continuously driven by or requires amyloid-β deposition.
-
Four scenarios that describe the potential role of amyloid-β in AD are described: amyloid-β trigger; amyloid-β threshold; amyloid-β driver; and amyloid-β irrelevant. Whether current and future amyloid-β-centric therapeutics will show clinical efficacy will crucially depend on which of these scenarios most accurately reflects the AD process
Abstract
The amyloid cascade hypothesis, which posits that the deposition of the amyloid-β peptide in the brain is a central event in Alzheimer's disease pathology, has dominated research for the past twenty years. Several therapeutics that were purported to reduce amyloid-β production or aggregation have failed in Phase III clinical testing, and many others are in various stages of development. Therefore, it is timely to review the science underpinning the amyloid cascade hypothesis, consider what type of clinical trials will constitute a valid test of this hypothesis and explore whether amyloid-β-directed therapeutics will provide the medicines that are urgently needed by society for treating this devastating disease.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
References
Golde, T. E., Petrucelli, L. & Lewis, J. Targeting Aβ and tau in Alzheimer's disease, an early interim report. Exp. Neurol. 223, 252–266 (2010).
Ness, D. K. et al. Reduced β-amyloid burden, increased C-99 concentrations and evaluation of neuropathology in the brains of PDAPP mice given LY450139 dihydrate daily by gavage for 5 months. Neurobiol. Aging 25, S238–S239 (2004).
Bateman, R. J. et al. A γ-secretase inhibitor decreases amyloid-β production in the central nervous system. Ann. Neurol. 66, 48–54 (2009).
Lanz, T. A. et al. Concentration-dependent modulation of amyloid-β in vivo and in vitro using the γ-secretase inhibitor, LY-450139. J. Pharmacol. Exp. Ther. 319, 924–933 (2006).
Wakabayashi, T. & De Strooper, B. Presenilins: members of the γ-secretase quartets, but part-time soloists too. Physiology (Bethesda) 23, 194–204 (2008).
Holtzman, D. M., Morris, J. C. & Goate, A. M. Alzheimer's disease: the challenge of the second century. Sci. Transl. Med. 3, 77sr1 (2011).
Golde, T. E., Schneider, L. S. & Koo, E. H. Anti-Aβ therapeutics in Alzheimer's disease: the need for a paradigm shift. Neuron 69, 203–213 (2011).
Burns, A. & Ilife, S. Dementia. BMJ 338, 405–409 (2009).
Wimo, A. & Prince, M. World Alzheimer Report 2010: The Global Economic Impact of Dementia. Alzheimer's Disease International [online], (2011).
Alzheimer, A. About a peculiar disease of the cerebral cortex. [in German] Centralblatt für Nervenheilkunde Psychiatrie 30, 177–179 (1907).
Goate, A. et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706 (1991). This seminal paper identified that a mutation in the APP gene causes autosomal dominant AD, thereby providing important support for the amyloid cascade hypothesis.
Hussain, I. et al. Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol. Cell Neurosci. 14, 419–427 (1999).
Lin, X. et al. Human aspartic protease memapsin 2 cleaves the β-secretase site of β-amyloid precursor protein. Proc. Natl Acad. Sci. USA 97, 1456–1460 (2000).
Sinha, S. et al. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 402, 537–540 (1999).
Vassar, R. et al. β-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 (1999).
Yan, R. et al. Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature 402, 533–537 (1999). References 12, 13, 14 and 16 identified the BACE enzyme, which is the enzyme responsible for initiating amyloid-β generation.
De Strooper, B. et al. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390 (1998). This work demonstrated that PSEN1 is an essential component of the γ-secretase complex, and required for γ-secretase activity.
Wolfe, M. S. et al. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 398, 513–517 (1999). This was the first paper to suggest that the aspartate residues in transmembrane domains 6 and 7 of PSEN participate in the catalytic function of γ-secretase.
Hardy, J. A. & Higgins, G. A. Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185 (1992). This paper is acknowledged as being the first, complete articulation of the amyloid cascade hypothesis.
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002).
Selkoe, D. J. The molecular pathology of Alzheimer's disease. Neuron 6, 487–498 (1991).
Hardy, J. & Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol. Sci. 12, 383–388 (1991).
Sherrington, R. et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 375, 754–760 (1995). This work identified the PSEN1 gene by linkage analysis.
Levy-Lahad, E. et al. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269, 973–977 (1995).
Rogaev, E. I. et al. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature 376, 775–778 (1995).
Corder, E. H. et al. Gene dosage of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923 (1993). This paper identified APOE4 as a major risk gene for AD.
Strittmatter, W. J. et al. Apolipoprotein E: high avidity binding to β-amyloid and increased frequency of type 4 allele in late onset familial Alzheimer disease. Proc. Natl Acad. Sci. USA 90, 1977–1981 (1993).
Nickerson, D. A. et al. Sequence diversity and large-scale typing of SNPs in the human apolipoprotein E gene. Genome Res. 10, 1532–1545 (2000).
Farrer, L. A. et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. JAMA 278, 1349–1356 (1997).
Holtzman, D. M. et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc. Natl Acad. Sci. USA 97, 2892–2897 (2000). This paper demonstrates the crucial role of APOE in amyloid-β deposition, providing a link between the major genetic risk factor for AD and the amyloid cascade hypothesis.
Kim, J., Basak, J. M. & Holtzman, D. M. The role of apolipoprotein E in Alzheimer's disease. Neuron 63, 287–303 (2009).
Harold, D. et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nature Genet. 41, 1088–1093 (2009).
Lambert, J. C. et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nature Genet. 41, 1094–1099 (2009).
Hollingworth, P. et al. Common variants at ABCA7, MS4Ai6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nature Genet. 43, 429–435 (2011).
Hutton, M. et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393, 702–705 (1998). This paper identified mutations in the tau gene that cause frontotemporal dementia, showing that tau pathology alone is sufficient to cause progressive neurodegeneration.
Roberson, E. D. et al. Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J. Neurosci. 31, 700–711 (2011).
Roberson, E. D. et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer's disease mouse model. Science 316, 750–754 (2007).
Welander, H. et al. Aβ43 is more frequent than Aβ40 in amyloid plaque cores from Alzheimer disease brains. J. Neurochem. 110, 697–706 (2009).
Arnold, S. E., Hyman, B. T., Flory, J., Damasio, A. R. & Van Hoesen, G. W. The topographical and neuro-anatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer's disease. Cereb. Cortex 1, 103–116 (1991).
Azevedo, F. A. et al. Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J. Comp. Neurol. 513, 532–541 (2009).
Gravina, S. A. et al. Amyloid β protein (Aβ) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at Aβ40 or Aβ42(43). J. Biol. Chem. 270, 7013–7016 (1995).
Naslund, J. et al. Correlation between elevated levels of amyloid β-peptide in the brain and cognitive decline. JAMA 283, 1571–1577 (2000).
Delacourte, A. et al. Nonoverlapping but synergetic tau and APP pathologies in sporadic Alzheimer's disease. Neurology 59, 398–407 (2002).
Bateman, R. J. et al. Human amyloid-β synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nature Med. 12, 856–861 (2006). This paper describes the SILK technology.
Iwata, N. et al. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nature Med. 6, 143–150 (2000). This was a systematic analysis of amyloid-β catabolic pathways in vivo , which revealed the role of neprilysin in amyloid-β clearance.
Vekrellis, K. et al. Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 20, 1657–1665 (2000). This paper identified insulin degrading enzyme in vitro as one of the enzymes involved in amyloid-β clearance.
Wang, D.-S., Dickson, D. W. & Malter, J. S. β-Amyloid degradation and Alzheimer's disease. J. Biomed. Biotechnol. 2006, 1–12 (2006).
Tseng, B. P. et al. Deposition of monomeric, not oligomeric, Aβ mediates growth of Alzheimer's disease amyloid plaques in human brain preparations. Biochemistry 38, 10424–10431 (1999).
Shibata, M. et al. Clearance of Alzheimer's amyloid-β1–40 peptide from brain by LDL receptor-related protein-1 at the blood–brain barrier. J. Clin. Invest. 106, 1489–1499 (2000).
Cirrito, J. R. et al. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in amyloid-β metabolism and half-life. J. Neurosci. 23, 8844–8853 (2003).
Redzic, Z. B., Preston, J. E., Duncan, J. A., Chodobski, A. & Szmydynger-Chodobska, J. The choroid plexus-cerebrospinal fluid system: from development to aging. Curr. Top. Dev. Biol. 71, 1–52 (2005).
Esler, W. P. et al. Alzheimer's disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry 39, 6288–6295 (2000).
Maggio, J. E. et al. Reversible in vitro growth of Alzheimer disease β-amyloid plaques by deposition of labeled amyloid peptide. Proc. Natl Acad. Sci. USA 89, 5462–5466 (1992).
Yan, P. et al. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J. Neurosci. 29, 10706–10714 (2009).
Garcia-Alloza, M. et al. Existing plaques and neuritic abnormalities in APP: PS1 mice are not affected by administration of the γ-secretase inhibitor LY-411575. Mol. Neurodegener. 4, 19 (2009).
Hefendehl, J. K. et al. Long-term in vivo imaging of β-amyloid plaque appearance and growth in a mouse model of cerebral β-amyloidosis. J. Neurosci. 31, 624–629 (2011).
Jack, C. R. Jr et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 9, 119–128 (2010). This is a very influential paper that describes the temporal nature of key AD biomarkers.
Abramowski, D. et al., Dynamics of Aβ turnover and deposition in different β-amyloid precursor protein transgenic mouse models following γ-secretase inhibition. J. Pharmacol. Exp. Ther. 327, 411–424 (2008).
Jankowsky, J. L. et al. Persistent amyloidosis following suppression of Aβ production in a transgenic model of Alzheimer disease. PLoS Med. 2, e355 (2005).
McConlogue, L. et al. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J. Biol. Chem. 282, 26326–26334 (2007).
DeMattos, R. B., Bales, K. R., Cummins, D. J., Paul, S. M. & Holtzman, D. M. Brain to plasma amyloid-β efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science 295, 2264–2267 (2002). This was a key paper describing the peripheral sink hypothesis that underpins the development of solanezumab.
DeMattos, R. B. et al. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc. Natl Acad. Sci. USA 98, 8850–8855 (2001).
Schenk, D. et al. Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177 (1999). A key paper demonstrating that the immunization of PDAPP mice with Aβ42 raises antibodies that facilitate the clearance of parenchymal plaques. This work finally led to the development of bapineuzumab and other amyloid-β-specific antibodies.
Bard, F. et al. Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nature Med. 6, 916–919 (2000).
Fagan, A. M. et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann. Neurol. 59, 512–519 (2006).
Price, J. L. et al. Neuropathology of non-demented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol. Aging 30, 1026–1036 (2009).
Shaw, L. M. et al. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann. Neurol. 65, 403–413 (2009).
De Meyer, G. et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Arch. Neurol. 67, 949–956 (2010).
Fagan, A. M. et al. Cerebrospinal fluid tau and ptau181 increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer's disease. EMBO Mol. Med. 1, 371–380 (2009).
Saido, T. C., Yamao-Harigaya, W., Iwatsubo, T. & Kawashima, S. Amino- and carboxyl-terminal heterogeneity of β-amyloid peptides deposited in human brain. Neurosci. Lett. 215, 173–176 (1996).
Saido, T. C. et al. Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(pE), in senile plaques. Neuron 14, 457–466 (1995).
Nicoll, J. A. et al. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nature Med. 9, 448–452 (2003).
De Strooper, B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol. Rev. 90, 465–494 (2010).
Serneels, L. et al. Differential contribution of the three Aph1 genes to γ-secretase activity in vivo. Proc. Natl Acad. Sci. USA 102, 1719–1724 (2005). This was the first evidence that the different γ-secretase complexes differentially contribute to Notch signalling and amyloid-β generation.
Herreman, A. et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl Acad. Sci. USA 96, 11872–11877 (1999).
Baumeister, R. et al. Human presenilin-1, but not familial Alzheimer's disease (FAD) mutants, facilitate Caenorhabditis elegans Notch signalling independently of proteolytic processing. Genes Funct. 1, 149–159 (1997).
Song, W. et al. Proteolytic release and nuclear translocation of Notch-1 are induced by presenilin-1 and impaired by pathogenic presenilin-1 mutations. Proc. Natl Acad. Sci. USA 96, 6959–6963 (1999).
Bentahir, M. et al. Presenilin clinical mutations can affect γ-secretase activity by different mechanisms. J. Neurochem. 96, 732–742 (2006). This paper shows that amyloid-β peptide ratios are more important than absolute levels of amyloid-β in understanding the pathogenicity of FAD-linked PSEN mutations.
De Strooper, B. Loss-of-function presenilin mutations in Alzheimer disease. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 8, 141–146 (2007).
Wolfe, M. S. When loss is gain: reduced presenilin proteolytic function leads to increased Aβ42/Aβ40. Talking point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 8, 136–140 (2007).
Yagishita, S., Morishima-Kawashima, M., Tanimura, Y., Ishiura, S. & Ihara, Y. DAPT-induced intracellular accumulations of longer amyloid β-proteins: further implications for the mechanism of intramembrane cleavage by γ-secretase. Biochemistry 45, 3952–3960 (2006).
Takami, M. et al. γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 29, 13042–13052 (2009).
Saito, T. et al. Potent amyloidogenicity and pathogenicity of Aβ43. Nature Neurosci. 14, 1023–1032 (2011).
Portelius, E. et al. Distinct cerebrospinal fluid amyloid β peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer's disease. Mol. Neurodegener. 5, 2 (2010).
Snider, B. J. et al. Novel presenilin 1 mutation (S170F) causing Alzheimer disease with Lewy bodies in the third decade of life. Arch. Neurol. 62, 1821–1830 (2005).
Hellstrom-Lindahl, E., Viitanen, M. & Marutle, A. Comparison of Aβ levels in the brain of familial and sporadic Alzheimer's disease. Neurochem. Int. 55, 243–252 (2009).
Kim, J. et al. Aβ40 inhibits amyloid deposition in vivo. J. Neurosci. 27, 627–633 (2007).
Meyer-Luehmann, M. et al. Exogenous induction of cerebral β- amyloidogenesis is governed by agent and host. Science 313, 1781–1784 (2006).
Braak, H. & Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259 (1991). This is a landmark paper that describes the temporal and brain-regional evolution of AD pathology.
Gomez-Isla, T. et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer's disease. Ann. Neurol. 41, 17–24 (1997).
Savva, G. M. et al. Age, neuropathology, and dementia. N. Engl. J. Med. 360, 2302–2309 (2009).
Rowe, C. C. et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol. Aging 31, 1275–1283 (2010).
Clavaguera, F. et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nature Cell Biol. 11, 909–913 (2009).
Olzscha, H. et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144, 67–78 (2011).
Thies, W. & Bleiler, L. 2011 Alzheimer's disease facts and figures. Alzheimers Dement. 7, 208–244 (2011).
Reisberg, B. Dementia: a systematic approach to identifying reversible causes. Geriatrics 41, 30–46 (1986).
Helzner, E. P. et al. Survival in Alzheimer disease: a multiethnic, population-based study of incident cases. Neurology 71, 1489–1495 (2008).
Larson, E. B. et al. Survival after initial diagnosis of Alzheimer disease. Ann. Intern. Med. 140, 501–509 (2004).
Ganguli, M., Dodge, H. H., Shen, C., Pandav, R. S. & DeKosky, S. T. Alzheimer disease and mortality: a 15-year epidemiological study. Arch. Neurol. 62, 779–784 (2005).
Holmes, C. Genotype and phenotype in Alzheimer's disease. Br. J. Psychiatry 180, 131–134 (2002).
Swearer, J. M., O'Donnell, B. F., Ingram, S. M. & Drachman, D. A. Rate of progression in familial Alzheimer's disease. J. Geriatr. Psychiatry Neurol. 9, 22–25 (1996).
Holmes, C. & Lovestone, S. The clinical phenotype of familial and sporadic late onset Alzheimer's disease. Int. J. Geriatr. Psychiatry 17, 146–149 (2002).
Kumar-Singh, S. et al. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Aβ42 and decreased Aβ40. Hum. Mutat. 27, 686–695 (2006).
Acosta-Baena, N. et al. Pre-dementia clinical stages in presenilin 1 E280A familial early-onset Alzheimer's disease: a retrospective cohort study. Lancet Neurol. 10, 213–220 (2011).
Godbolt, A. K. et al. The natural history of Alzheimer disease: a longitudinal presymptomatic and symptomatic study of a familial cohort. Arch. Neurol. 61, 1743–1748 (2004).
Meyer, M. R. et al. APOE genotype predicts when — not whether — one is predisposed to develop Alzheimer disease. Nature Genet. 19, 321–322 (1998).
Craft, S. et al. Accelerated decline in apolipoprotein E-epsilon4 homozygotes with Alzheimer's disease. Neurology 51, 149–153 (1998).
Dal Forno, G. et al. APOE genotype and survival in men and women with Alzheimer's disease. Neurology 58, 1045–1050 (2002).
Allan, C. L. & Ebmeier, K. P. The influence of APOE4 on clinical progression of dementia: a meta-analysis. Int. J. Geriatr. Psychiatry 26, 520–526 (2011).
Pastor, P. et al. Apolipoprotein Eɛ4 modifies Alzheimer's disease onset in an E280A PS1 kindred. Ann. Neurol. 54, 163–169 (2003).
Morris, J. C. et al. APOE predicts amyloid-β but not tau Alzheimer pathology in cognitively normal aging. Ann. Neurol. 67, 122–131 (2010).
Hardy, J. Expression of normal sequence pathogenic proteins for neurodegenerative disease contributes to disease risk: 'permissive templating' as a general mechanism underlying neurodegeneration. Biochem. Soc. Trans. 33, 578–581 (2005).
Lambert, M. P. et al. Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc. Natl Acad. Sci. USA 95, 6448–6453 (1998).
Lesne, S. et al. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 440, 352–357 (2006).
Shankar, G. M. et al. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nature Med. 14, 837–842 (2008).
Hepler, R. W. et al. Solution state characterization of amyloid β-derived diffusible ligands. Biochemistry 45, 15157–15167 (2006). This detailed analysis describes the methodological problems associated with the biochemical characterization of amyloid-β.
Jones, L. et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer's disease. PLoS ONE 5, e13950 (2010).
Shen, J. & Kelleher, R. J. The presenilin hypothesis of Alzheimer's disease: evidence for a loss-of-function pathogenic mechanism. Proc. Natl Acad. Sci. USA 104, 403–409 (2007).
Wang, B. et al. γ-secretase gene mutations in familial acne inversa. Science 330, 1065 (2010).
Pink, A. E. et al. PSENEN and NCSTN mutations in familial hidradenitis suppurativa (acne inversa). J. Invest. Dermatol. 131, 1568–1570 (2011).
Blennow, K. et al. Immunotherapy with bapineuzumab lowers CSF tau protein levels in patients with Alzheimer's disease. Alzheimers Dement. 6, S134–S135 (2010).
Rinne, J. O. et al. 11C-PiB PET assessment of change in fibrillar amyloid-β load in patients with Alzheimer's disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol. 9, 363–372 (2010).
Holmes, C. et al. Long-term effects of Aβ42 immunisation in Alzheimer's disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet 372, 216–223 (2008).
Mangialasche, F., Solomon, A., Winblad, B., Mecocci, P. & Kivipelto, M. Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 9, 702–716 (2010).
Braak, H. & Braak, E. Staging of Alzheimer-related cortical destruction. Int. Psychogeriatr. 9 (Suppl. 1), 257–261 (1997). This paper classifies the development of AD pathology into 'Braak stages'.
Jalbert, J. J., Daiello, L. A. & Lapane, K. L. Dementia of the Alzheimer type. Epidemiol. Rev. 30, 15–34 (2008).
Rosen, W. G., Mohs, R. C. & Davis, K. L. A new rating scale for Alzheimer's disease. Am. J. Psychiatry 141, 1356–1364 (1984). This description of the cognition rating scales is used in clinical trials testing AD therapeutics.
Schneider, L. S. & Sano, M. Current Alzheimer's disease clinical trials: methods and placebo outcomes. Alzheimers Dement. 5, 388–397 (2009).
Mohs, R. C. The clinical syndrome of Alzheimer's disease: aspects particularly relevant to clinical trials. Genes Brain Behav. 4, 129–133 (2005).
Morris, J. C. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–2414 (1993).
Galasko, D. et al. An inventory to assess activities of daily living for clinical trials in Alzheimer's disease. The Alzheimer's Disease Cooperative Study. Alzheimer Dis. Assoc. Disord. 11 (Suppl. 2), 33–39 (1997).
Gelinas, I., Gauthier, L., McIntyre, M. & Gauthier, S. Development of a functional measure for persons with Alzheimer's disease: the disability assessment for dementia. Am. J. Occup. Ther. 53, 471–481 (1999).
Schneider, L. S. et al. Validity and reliability of the Alzheimer's disease cooperative study-clinical global impression of change. The Alzheimer's Disease Cooperative Study. Alzheimer Dis. Assoc. Disord. 11 (Suppl. 2), 22–32 (1997).
Hampel, H. et al. Biomarkers for Alzheimer's disease: academic, industry and regulatory perspectives. Nature Rev. Drug Discov. 9, 560–574 (2010).
Leber, P. Slowing the progression of Alzheimer disease: methodologic issues. Alzheimer Dis. Assoc. Disord. 11 (Suppl. 5), 10–21 (1997).
Mohs, R. C., Kawas, C. & Carrillo, M. C. Optimal design of clinical trials for drugs designed to slow the course of Alzheimer's disease. Alzheimers Dement. 2, 131–139 (2006).
Moghekar, A. et al. Large quantities of Aβ peptide are constitutively released during amyloid precursor protein metabolism in vivo and in vitro. J. Biol. Chem. 286, 15989–15997 (2011).
Silverberg, G. D., Mayo, M., Saul, T., Rubenstein, E. & McGuire, D. Alzheimer's disease, normal-pressure hydrocephalus, and senescent changes in CSF circulatory physiology: a hypothesis. Lancet Neurol. 2, 506–511 (2003).
Jack, C. R. Jr et al. Introduction to the recommendations from the National Institute on Aging and the Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 257–262 (2011).
Sperling, R. A. et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 280–292 (2011).
Khachaturian, Z. S. et al. Developing a global strategy to prevent Alzheimer's disease: Leon Thal Symposium 2010. Alzheimers Dement. 7, 127–132 (2011).
Thorvaldsson, V. et al. Onset and rate of cognitive change before dementia diagnosis: findings from two Swedish population-based longitudinal studies. J. Int. Neuropsychol. Soc. 17, 154–162 (2011).
Games, D. et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373, 523–527 (1995). This description of the first transgenic mouse model reliably demonstrated the amyloid plaque pathology of AD.
Glabe, C. G. Common mechanisms of amyloid oligomer pathogenesis in degenerative disease. Neurobiol. Aging 27, 570–575 (2006).
Walsh, D. M. et al. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539 (2002).
Kuperstein, I. et al. Neurotoxicity of Alzheimer's disease Aβ peptides is induced by small changes in the Aβ42 to Aβ40 ratio. EMBO J. 29, 3408–3420 (2010).
Bernstein, S. L. et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease. Nature Chem. 1, 326–331 (2009).
Scheuner, D. et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nature Med. 2, 864–870 (1996). This was the first study linking PSEN mutations to abnormal APP processing.
van Broeckhoven, C. & Kumar-Singh, S. Genetics and pathology of α-secretase site Aβ PP mutations in the understanding of Alzheimer's disease. J. Alzheimers Dis. 9, 389–398 (2006).
Zhang-Nunes, S. X. et al. The cerebral β- amyloid angiopathies: hereditary and sporadic. Brain Pathol. 16, 30–39 (2006).
Acknowledgements
B.D.S. is the Bax-Vanluffelen Chair for Alzheimer's Disease and is supported by a Methusalem grant (FWO-Flanders). The authors would like to thank S. M. Paul and J. Hardy for their careful reading of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Bart De Strooper is a consultant for Janssen Pharmaceutica in Beerse, Belgium; Envivo Pharmaceuticals in Boston, Massachusetts, USA; and Remynd NV in Leuven, Belgium. He also receives research funding from Janssen Pharmaceutica, Beerse.
Mark Mercken is currently employed by Janssen Pharmaceutica, and holds stock in Johnson and Johnson.
Eric Karran is currently employed by Janssen Pharmaceutica, and holds stock in Johnson and Johnson.
Related links
Glossary
- Amyloid-β
-
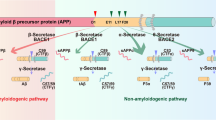
Amyloid-β peptides result from sequential cleavage of the amyloid precursor protein by β-cleaving amyloid precursor protein enzyme (BACE) and γ-secretase. These peptides vary in length, with Aβ40 (the 40-amino acid form of the peptide) being predominant.
- Amyloid plaques
-
Amyloid plaques are deposits of insoluble amyloid-β in the parenchyma of the brain that can be diffuse or compact. If they are associated with dystrophic and degenerating neurons, they are often termed 'neuritic plaques'.
- Neurofibrillary tangles
-
Large deposits of hyperphosphorylated tau (5–9 moles of phosphate per mole of tau) that fill the cell body of the neuron and take its shape. They are composed of both paired helical and straight filaments of hyperphosphorylated tau.
- Tau
-
A protein that binds to and stabilizes microtubules within cells and is abundant in neurons. Humans express six isoforms of tau that result from alternative splicing of exons 2, 3 and 10 of the tau gene. Tau can be multiphosphorylated and this regulates its microtubule-binding properties.
- Apolipoprotein E
-
(APOE). A 34-kDa secreted protein that is synthesized predominantly in the liver but is also produced by glial cells in the brain. It acts as a lipoprotein-binding protein and mediates lipid metabolism by binding to the low-density lipoprotein superfamily of receptors.
- Genome-wide association (GWA) studies
-
This technique enables common, single-nucleotide polymorphic genetic variations to be compared in patients and in controls, to discover whether there is evidence for a genetic predisposition to the disease.
- Familial Alzheimer's disease
-
(FAD). A form of Alzheimer's disease (AD) caused by rare, autosomal dominant mutations that are inherited in a Mendelian fashion within families. Currently identified FAD mutations result in early-onset AD.
- Paired helical filaments
-
Hyperphosphorylated tau filaments that can be visualized in neurons using electron microscopy.
Rights and permissions
About this article
Cite this article
Karran, E., Mercken, M. & Strooper, B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov 10, 698–712 (2011). https://doi.org/10.1038/nrd3505
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd3505
This article is cited by
-
The use of individual-based FDG-PET volume of interest in predicting conversion from mild cognitive impairment to dementia
BMC Medical Imaging (2024)
-
Molecular dynamics simulations to explore the binding mode between the amyloid-β protein precursor (APP) and adaptor protein Mint2
Scientific Reports (2024)
-
A novel mouse model for N-terminal truncated Aβ2-x generation through meprin β overexpression in astrocytes
Cellular and Molecular Life Sciences (2024)
-
Sustained meningeal lymphatic vessel atrophy or expansion does not alter Alzheimer’s disease-related amyloid pathology
Nature Cardiovascular Research (2024)
-
The FDA-approved anti-amyloid-β monoclonal antibodies for the treatment of Alzheimer’s disease: a systematic review and meta-analysis of randomized controlled trials
European Journal of Medical Research (2023)
