
Key Points
-
Flaviviruses are a genus within the Flaviviridae family with a single-stranded, positive-sense RNA genome. Many representatives of the genus, such as dengue, West Nile and Zika viruses, are associated with pathogenic effects in humans.
-
Infections with flaviviruses cause symptoms of varying severity, ranging from flu-like illnesses and fevers to potentially lethal haemorrhagic fevers. Neurological complications include encephalitis (for West Nile virus) or microcephaly and Guillain–Barré syndrome (for Zika virus).
-
Vaccine development for humans was successful for some members of the genus, but no specific, pharmacological antiviral agent has reached the market. Drugs with broad-spectrum antiviral activity would be particularly desirable to tackle outbreaks of emerging or neglected flaviviruses.
-
Molecular targets with promising prospects for broad-spectrum relevance include viral proteins, such as the viral protease or polymerase, and host targets exploited by these viruses, including α-glucosidase and mechanisms involved in nucleoside biosynthesis.
Abstract
Infections with flaviviruses, such as dengue, West Nile virus and the recently re-emerging Zika virus, are an increasing and probably lasting global risk. This Review summarizes and comments on the opportunities for broad-spectrum agents that are active against multiple flaviviruses. Broad-spectrum activity is particularly desirable to prepare for the next flaviviral epidemic, which could emerge from as-yet unknown or neglected viruses. Potential molecular targets for broad-spectrum antiflaviviral compounds include viral proteins, such as the viral protease or polymerase, and host targets that are exploited by these viruses during entry and replication, including α-glucosidase and proteins involved in nucleoside biosynthesis. Numerous compounds with broad-spectrum antiviral activity have already been identified by target-specific or phenotypic assays. For other compounds, broad-spectrum activity can be anticipated because of their mode of action and molecular targets.
Similar content being viewed by others


Main
Three global megatrends — urbanization, climate change and increased intercontinental travel — are facilitating the spread of flaviviruses beyond their habitats in tropical forests. A reversal of these megatrends is highly improbable; thus, it is worthwhile to evaluate the potential of antiviral treatments against known (and unknown) flaviviral pathogens.
Within the Flaviviridae family, the genera Flavivirus and Hepacivirus encompass single-stranded, positive- sense RNA viruses that are pathogenic to humans. Medicinal chemistry has successfully addressed infections with hepatitis C virus (HCV)1, the only Hepacivirus currently known to infect humans. By contrast, drug discovery against members of the genus Flavivirus (here denoted as flaviviruses) has thus far received less attention. This discrepancy can be attributed to the following factors: the availability of effective vaccines against some flaviviruses, such as yellow fever virus (YFV)2 and Japanese encephalitis virus (JEV); the prevalent spread of most flaviviruses in tropical and less affluent regions, which limits the commercial potential of antiviral agents; and the variability of the pathological effects of flaviviral infections, which range from asymptomatic infection to severe and fatal disease3,4. Flaviviruses are predominantly transmitted by insect (arthropod) vectors, in particular mosquitos (of the genera Aedes and Culex) and ticks, and are therefore also classified as arboviruses (arthropod-borne).
Many diseases caused by flaviviral infections, especially dengue fever, tend to be epidemics, causing millions of cases per year5. The recent Zika virus (ZIKV) pandemic6 is therefore not an exception, and highlights the risk potential of flaviviruses as a group, even though the mortality risk of many flaviviral infections is relatively low. The Zika epidemic highlighted the following risk factors. First, globalization and travel can distribute previously obscure viruses into populations with no previous exposure or immunity, which enables the virus to infect a high proportion of the population7. Second, although transmission occurs mainly via arthropod vectors, flaviviruses can also use routes that were previously not considered relevant and these viruses can persist in some tissues for several months after the viraemic period8. Third, infections with flaviviruses can lead to unexpected pathologies, such as microcephaly and Guillain–Barré syndrome associated with ZIKV9. Finally, virus-naive populations have often previously been exposed to other, closely related viruses and may have developed immunity against these. Evidence in vitro suggests that antibodies generated during previous infections (or possibly vaccination) can exacerbate the course of the disease10. However, the clinical relevance of these in vitro observations is uncertain.
Fortunately, much has been learned recently about flavivirus biology, and numerous methods have been devised to characterize potential drug candidates in assay systems that range from isolated molecular targets to mouse and non-human primate models (Box 1). The successes achieved in the treatment of HCV, which can largely be attributed to the availability of cell-based assay systems for viral replication11, have been particularly inspiring for antiviral drug discovery research. These phenotypic assay systems bridged the critical gap between biochemical target-oriented assays and animal models. For flaviviruses, cell-based systems are established and have recently been used to screen for antiviral agents against ZIKV, with a particular focus on drug repurposing12,13.
This Review covers the compound classes, targets and assay methods that currently hold the most promise to develop drugs with broad-spectrum activity. We exclude compounds that are poor starting points for medicinal chemistry efforts owing to issues such as activities at levels ≥50 μM and blatant deviation from commonly accepted medicinal chemistry criteria (for example polyphenolic compounds with molecular masses in the 1,000 Da range). In addition, compounds with evidence of high cytotoxicity, either for the compounds themselves or for close analogues, are excluded.
The most important criterion for the inclusion of a compound or target is proven activity against more than one flavivirus in a cell-based assay or, ideally, in an animal model. In some instances, we also include compounds and targets for which either this proof is still lacking but broad-spectrum activity appears likely, or a promising activity has been reported against viruses of considerable current interest, such as ZIKV and dengue virus (DENV). Although we focus on pharmacological interventions, alternative strategies such as vaccination are also briefly discussed. Priority is given to targets with at least some initial medicinal chemistry exploration or those for which a drug intervention has a high probability of success. Numerous host targets have been proposed on the basis of RNAi screening results, and we restrict our discussion to those that have been validated by follow-up medicinal chemistry or other methods. Results from phenotypic compound screens are included if they fulfil the criteria outlined above. Potential high-throughput biochemical assay methods for viral targets and cellular and animal models are outlined. Ancillary topics such as history and epidemiology, phylogeny and antigenic relationships, as well as controlling flavivirus vectors are provided in Supplementary information S1, S2, S3, S4, S5 (box, box, box, figure, table).
Biology and replication
The replication cycle of flaviviruses in cells is outlined in Fig. 1 along with the most relevant sites for pharmacological intervention. Numerous receptors have been suggested to mediate the binding of flaviviruses to host cells and the subsequent endocytosis. After release of the flaviviral genome from the endosome, the single- stranded, positive-sense RNA is translated by ribosomes to form the viral polyprotein, which is cleaved by host proteases and the viral protease to form the structural and non-structural proteins of the virus. This process and the replication of the viral genome occur in a multi-molecular complex located at the endoplasmic reticulum (ER), known as the replication complex, that contains membranes, viral RNA, lipid droplets, and viral and host proteins. Most viral and many host targets — such as the viral protease, polymerase, helicase, as well as host kinases and glucosidases — are localized in or near the replication complex. The microenvironment of the replication complex is likely to be a major factor that influences the biophysical properties of the targets and the ligand binding behaviour. An important step in virion maturation is the sequential trimming of glucose residues on the surface of glycoproteins by host glucosidases within the ER. This process is a prerequisite for the proper folding of the glycoproteins by the ER chaperones calnexin and/or calreticulin. For incompletely folded proteins, the reglycosylation process is launched by UDP-glucosyltransferase 1, which acts as a sensor for correct protein folding14. After budding into the ER, the assembled progeny viruses are processed further in the trans-Golgi network. Key factors for this final maturation are a decrease in pH, which induces the conformational reorganization of the viral envelope (E) glycoprotein and the pre-membrane (prM) glycoprotein, as well as the proteolytic cleavage of prM by the host protease furin. Finally, mature virions egress from the infected cell via exocytosis mediated by the exocyst complex15.

Several putative host cell receptors for flaviviruses are indicated at the cellular membrane. The strongest evidence has, thus far, been found for the involvement of DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin). Processes that occur during the viral replication cycle are shown in green boxes, and antiflaviviral compounds being investigated are indicated in orange boxes. The inset in the lower left corner shows the sequential and structural organization of the flaviviral polyprotein at the endoplasmic reticulum membrane, with the cleavage sites of the host and viral proteases indicated. Note the colour coding of the viral proteins, indicated at the bottom. C, capsid protein; E, envelope glycoprotein; ER, endoplasmic reticulum; GRP78, 78 kDa glucose-regulated protein; HSP, heat shock protein; NS, non-structural protein; prM, pre-membrane glycoprotein; SAH, S-adenosylhomocysteine; ss, single-stranded; TAM, TYRO3, AXL (also known as UFO) and MERTK; TIM, T cell immunoglobulin and mucin.
Similar to other RNA viruses, flaviviruses rapidly accumulate mutations because of the low fidelity of the viral RNA polymerase16. This high mutation capacity leads to the formation of intragenic variation or quasispecies in the infected host; these quasispecies are the primary cause of drug resistance in RNA viruses. To prevent the development of this relatively rapid resistance, classes of compounds active against various viral and host targets should be developed and used in a combination antiviral treatment17. Vaccines generated against specific strains might be ineffective against diverse viral populations, and vaccine resistance can also develop (Box 2). The persistent efficiency of the YFV 17D-204 live vaccine strain can be explained by the genetic stability of the YFV wild-strain population18, which might not happen in other flaviviruses. Thus, the formation of flaviviral quasispecies should be taken into consideration in broad-spectrum antiviral drug design and polyvalent vaccine development.
Severe cases of flaviviral infections are frequently detected only after the viraemia has peaked. Therefore, antiviral compounds that target the viral life cycle are most useful for prophylaxis and treating early-stage or persistent subclinical infections. Alternative treatment options, which may be identified by drug repurposing, could be directed against the pathological immune response that frequently has an important role in severe cases of acute disease.
Pathology and tissue tropism
The incubation period of flaviviral infections usually ranges from 3 days to 2 weeks, with incubation periods of up to 4 weeks reported for Murray Valley encephalitis virus (MVEV). Flaviviral infections often remain asymptomatic, and for some viruses only a very small percentage of infected persons develop symptoms. Initial symptoms such as fever, rash, headache, nausea and fatigue are often nonspecific. Symptomatic cases are often self-limiting and resolve within approximately 1 week. Potentially lethal or permanently damaging pathologies develop in up to 25–30% of the symptomatic cases depending on the virus type and on the age, immune status, co-morbidity and previous heterologous infections of patients.
Flaviviruses preferentially infect particular cells and tissues. The tissue tropism of flaviviruses determines the pathology of severe cases and human-to-human viral transmission patterns. Neurotropic flaviviruses (including West Nile virus (WNV), tick-borne encephalitis virus (TBEV), JEV, MVEV and ZIKV) cause numerous neurological pathologies, from myelitis and encephalitis to seizures, permanent brain damage and paralysis, as occurs in MVEV infection19. Some neurological pathologies, such as Guillain–Barré syndrome, are caused by the host immune response to the viral pervasion, and may occur in infections by neurotropic viruses, such as ZIKV20,21, or in severe cases of non-neurotropic viral infections, such as DENV and YFV22,23. The mechanisms by which viruses enter the CNS vary and are not well studied for all flaviviruses, but two main routes are suggested: haematogenous transport and axonal transport4. The viral E glycoprotein is the main neurovirulence determinant, and mutation of a single amino acid in its structure can lead to loss of neuroinvasiveness24. The viral loads of some neurotropic viruses, such as JEV and WNV, can persist in the CNS, especially in immunocompromised patients25,26. Flaviviral neurotropism directly influences the design of antiflaviviral compounds, as penetration of the blood–brain barrier becomes a crucial pharmacokinetic property, particularly in cases of persistent neuroinfection.
The haemorrhagic fever observed in other flaviviral infections, such as DENV, YFV, Omsk haemorrhagic fever virus, Kyasanur forest disease virus or Alkhurma haemorrhagic fever virus, is linked to the host immune response27. Consequently, immunomodulatory and antihaemorrhagic properties are important for the development of symptomatic agents; however, antiviral agents would be effective only if designed to prevent the onset of fever. Specific viral determinants for the development of haemorrhagic fever have not yet been determined.
For most flaviviruses, there are contradictory or no data regarding their ability to cross the placental barrier. ZIKV, however, causes teratogenic effects in developing fetuses, and is associated with microcephaly, CNS lesions and fetal death. The first reports appeared during the recent outbreaks in Brazil28,29 and, retrospectively, for an outbreak in French Polynesia30. Teratogenic effects of ZIKV have also been observed in animal models31,32. Thus, compounds that prevent the penetration of ZIKV through the placental barrier could be explored as emergency measures, whereas the development of an effective vaccine that prevents maternal infections seems to be the best option from a medical perspective.
Animals infected with JEV33 and individuals or animals infected with ZIKV34,35,36,37 can exhibit high viral load in semen and testes. ZIKV reaches high loads of infectious viral particles (1 × 104–1 × 105 times the blood or urine viral loads) both in human semen34,35 and in animal testes36,37. The presence and persistence of ZIKV RNA in cervical mucus has also been reported, increasing the probability of sexual and vertical transmission of the virus38. Multiple cases of sexual transmission of ZIKV have been reported recently39,40,41, and prolonged persistence of ZIKV in semen and cervical mucus could make sexual transmission the main ZIKV distribution route in vector-free regions. High loads of viral RNA have also been detected in breast milk (DENV, WNV, YFV and ZIKV)42,43,44,45 and saliva (ZIKV)46, which suggest that these routes can also contribute to vertical and sexual transmission. These factors make it crucial for newly developed antivirals to penetrate and accumulate in the respective tissues and organs, especially in persistent infections (such as the testes in ZIKV infection), while not affecting their physiological functions.
Viral targets
A viral target protein should ideally combine two attributes: it should be essential for the viral cell cycle and should have a low rate of 'allowed' (non-lethal, but resistance-conferring) mutations. The latter attribute is particularly important for RNA viruses such as HCV and flaviviruses, the RNA polymerases of which do not have a proofreading function. The high mutation rate leads to a relatively large proportion of non-functional progeny virions, but this issue is balanced by other advantages, such as efficient immune evasion and the development of drug resistance. With respect to essentiality, the genome of flaviviruses is minimal and does not contain non- essential proteins or duplicate functions; therefore, this issue does not need to be considered here.
In practical terms, the target protein should have a straightforward biochemical assay that correlates well with a phenotypic effect. Enzymatic targets, such as the flaviviral protease, are therefore highly attractive and have been extensively pursued in high-throughput screening campaigns; however, success has been variable. The clinical success of inhibitors of other enzymatic targets, notably the proteases and RNA polymerases from HIV and HCV47, has probably spurred the interest in flaviviral enzymes. By contrast, few antiviral agents target non-enzymatic viral proteins (for example, M2 and gp41), many of which are associated with declining efficacy or other severe limitations. One caveat of high-throughput screening for enzymatic inhibitors is that the microenvironment of the replication complex, in which all enzymatic viral targets are localized48, probably differs substantially from the in vitro conditions of biochemical assays of isolated targets. This caveat may, in addition to other confounding factors such as pharmacokinetics, lead to discrepancies between biochemical, cellular and whole-organism assays.
Structural or functional features that have a high genetic barrier to mutations show a high degree of evolutionary conservation. This conservation enables us to assess the likelihood of resistance-inducing mutations arising by comparing viral genomes. A high degree of conservation also indicates target structures that have the largest potential for broad-spectrum relevance. A multiple sequence alignment of 50 flaviviral polyproteins demonstrates a high degree of conservation for amino acid residues with structural functions, such as Gly, Pro and Cys. For example, Cys residues are required for many of the conserved disulfide bridges in the E protein (Supplementary information S6 (figure)). Several of the enzymatic motifs involved in protein and RNA processing — such as the catalytic motifs of the NS3 protease, the ATP- and RNA-binding regions of NS3 helicase, and the substrate or metal recognition motifs of NS5 — are highly conserved. These conserved regions have biochemical functions that may be modified by drug-like, small-molecule inhibitors. The interspecies variability between some non-structural proteins is remarkably high, particularly for NS2 and portions of NS4, rendering these proteins less promising as targets for antiviral drugs with broad activity or that are resistant to the development of resistance.
Allosteric ligands and inhibitors can be identified by high-throughput screening campaigns directed at enzyme targets, and these ligands probably constitute a considerable fraction of initial screening hits. However, hits obtained from these campaigns are frequently difficult to optimize and may therefore not be useful for further development. Exploiting allosteric sites has some advantages, but is also associated with several inherent limitations and risks. Most importantly, the mutational barrier is low in most allosteric regions that are not related to substrate binding, so allosteric ligands are unlikely to be 'resistance-robust', broad-spectrum antiflaviviral agents. By contrast, the orthosteric binding sites for natural substrates such as RNA or the polyprotein cleavage sites are highly conserved, both during evolution of a single viral species and across flaviviral species. Chemical functionalities and their geometric distribution in the orthosteric substrate-recognition and catalytic regions have a high barrier against mutations because their substrates remain unchanged.
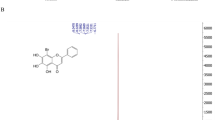
In the following sections, we focus on the targets with the greatest potential for broad-spectrum activity, considering their genetic variability and experiences from other viruses. The most promising viral targets are the NS3 protease and the NS5 polymerase, and, to a lesser degree, the E glycoprotein, the capsid protein, NS4B, NS3 helicase and NS5 methyltransferase. Other targets, such as NS5 guanylyltransferase and the NS3–NS5 interaction, currently seem to have limited potential and are not discussed further in the text, but are included in Supplementary information S7 (table). The structures of selected compounds that target viral proteins are presented in Fig. 2.

Chemical structures of selected potential broad-spectrum antiflaviviral drugs that target viral proteins.
NS3 protease inhibitors. Targeting the protease has been successful for the treatment of HCV and HIV, for which numerous peptidic and pseudopeptidic inhibitors are currently in clinical use47. The substrate-binding site in HCV and flaviviral proteases is relatively shallow and therefore not easily amenable to inhibition by small-molecule compounds. The flaviviral proteases have a strong preference for substrates with dibasic or polybasic recognition sequences49,50,51. Consequently, the recognition motifs in inhibitors also tend to incorporate basic or polar functionalities, which may partially explain the lower efficacy often observed in cell-based assays relative to biochemical assays, as the compounds have low passive membrane permeability52,53,54,55,56. The substrate-binding residues and the substrate recognition patterns of the protease are well conserved across the flaviviruses and therefore hold promise for the development of inhibitors with broad activity. The extensive experience with inhibitor development for other serine proteases with basic recognition preferences (for example, thrombin and factor Xa), for which arginine mimetics57 or prodrug strategies58 were devised, may provide valuable inspiration for the development of clinically effective inhibitors of flaviviral proteases.
The amino-terminal domain of NS3 is a trypsin-like serine protease59 that interacts with the core hydrophilic region of NS2B and processes the viral polyprotein60,61. In the catalytically active 'closed' form, NS2B contributes to the S2 and S3 sub-pockets of the binding site62,63,64,65,66. The protease was the first ZIKV target whose structure has been solved in complex with a high-affinity inhibitor67. Given the importance of protease inhibitors for the treatment of other viral infections, the straightforward and robust enzymatic assay, and the availability of structural data62,64,68,69,70,71,72, it is not surprising that numerous studies report high-throughput and virtual screening results for this target70. Reported activities, however, are often low, and follow-up hit-to-lead development is frequently missing for these hits. These weak reported activities indicate that the flaviviral protease is, similar to the related HCV protease, not an easy target. This circumstance can be explained by the factors discussed above; that is, the molecular recognition properties of the protease and its cellular microenvironment (reviewed in Ref. 70).
Numerous studies have focused on the development of protease inhibitors derived from a substrate-mimicking peptide. Strategies included incorporation of a carboxy- terminal electrophile52,54,73,74,75, optimization of the N-terminal capping moiety53,56,76,77, and modulation of the P1 and P2 basic residues through non-natural building blocks53,78. Aldehydic inhibitors displayed low micromolar to nanomolar affinity for DENV serotype 2 (DENV-2)74 and WNV52,75 proteases. An analogue, compound 3a, was reported to be stable in serum and cell permeant, and suppressed WNV replication without detected cytotoxicity; however, its antiviral activity against other flaviviruses was not assessed52. Another peptide aldehyde, compound 3b, inhibits DENV-2 (Ref. 73) and WNV proteases62,75 in biochemical assays and co-crystallized with DENV-3 (Ref. 64) and WNV proteases62. A recent study of this derivative, however, reported no passive permeability in a parallel artificial membrane permeability assay (PAMPA) and no reduction of DENV-2 titres in cellular assays55. The in vitro target affinities of the two peptide aldehydes differ by three orders of magnitude, which could explain their divergent activities in cell culture52,55.
An evaluation of tetrapeptides with different C-terminal electrophiles identified a boronic acid analogue with low nanomolar affinity towards the DENV-2 protease73. Replacement of the arginine in the P2 position by non-natural arginine mimetics78 generated benzoyl- capped dipeptides with activity against DENV-2, WNV and ZIKV proteases in biochemical assays54. The two analogues with the highest affinity, compound 4 and compound 7, reduced DENV-2 and WNV titres in plaque assays54. Notably, compound 4 has high affinity for the ZIKV protease, and the crystal structure of the complex was recently published67. The main limitation of the boronic acid inhibitors appears to be their low selectivity against off-targets such as thrombin and trypsin54. This problem may be addressed by extension towards the prime site or optimization of the N-terminal cap, as shown for other peptide-based compounds53,56,76,77 and for the aldehyde compound 3a52. Although the clinical potential of peptide aldehydes and peptide boronic acids remains uncertain, they are crucial for understanding the molecular recognition and structural transitions of the flaviviral protease54,62,64,67.
Fortunately, high affinity is not restricted to protease inhibitors that incorporate electrophilic groups. Using a fragment-merging strategy, a class of N-capped tripeptides was developed that incorporates 4-hydroxyphenylglycine benzyl ethers as non-natural C-terminal residues53. By varying the N-terminal moiety and the benzyl ether substituent, these competitive inhibitors achieved in vitro affinities in the low nanomolar range for DENV-2 and WNV proteases53. These compounds display remarkable selectivity against thrombin and trypsin, and representative analogues inhibit DENV-2 and WNV replication in plaque assays at low micromolar concentrations without detected cytotoxicity53. A discrepancy between the most active congener in enzymatic assays (compound 83) and in cellular assays (compound 104) may be due to the higher metabolic stability and passive permeability of compound 104 (Ref. 53).
In addition to peptidic inhibitors, palmatine, an isoquinoline alkaloid from Coptis chinensis, a medicinal plant, was investigated for its antiviral effects against flaviviruses79. Palmatine can reduce the viral titres of WNV, YFV and DENV-2 (but not the Rhabdoviridae vesicular stomatitis Indiana virus) without cytotoxicity79. Enzymatic assays of the WNV protease showed a non-competitive mechanism of inhibition with relatively low potency compared with the antiviral effects in cells79. The same phenomenon was observed previously for two inhibitors of DENV protease identified from phenotypic assays, BP2109 (Ref. 80) and BP13944 (Ref. 81), in which the discrepancy between cell- and target-based results was explained by the artificiality of the protease construct and the assay conditions80,81. BP2109 and BP13944 were active against DENV serotypes 1–4 (DENV-1–4) but not JEV in viral yield reduction assays80,81.
Meanwhile, the inhibitory potential of HIV and HCV protease inhibitors against DENV-2 and chikungunya virus (CHIKV) was explored82. Because of the weak antiviral activity against DENV-2 and CHIKV, all drugs had a much lower selectivity index for these viruses than for HIV or HCV. Against DENV-2, nelfinavir showed similar antiviral activity to compound 104 in cellular assays, but displayed much higher cytotoxicity53,82. Identifying broad-spectrum protease inhibitors with sufficient activity via drug repurposing appears challenging.
NS5 polymerase inhibitors. The flaviviral RNA-dependent RNA polymerase (RdRp) is located at the C-terminal portion of the NS5 protein83,84,85. The structure of WNV and DENV-3 RdRp shows a typical right-hand orientation with three subdomains: fingers, palm and thumb84,85. The catalytic site is positioned at the intersection of two tunnels; one provides access to the active site for the single-stranded RNA template and the second tunnel allows entry of NTPs at one end and exit of the nascent double-stranded RNA at the other end84,85. Recently, the crystal structures of the ZIKV full-length NS5 protein86,87 and RdRp88 were reported.
The flaviviral polymerase RdRp is essential for the viral replication cycle, is highly conserved and lacks a eukaryotic homologue, so is therefore an attractive target for drug development. This requirement is further underlined by the clinical success of polymerase inhibitors for HCV and HIV47.
Nucleoside inhibitors are substrate analogues. Upon phosphorylation, these agents inhibit RdRp activity by competing with endogenous NTPs; their incorporation into the nascent RNA leads to either chain termination or a lethal accumulation of mutations in a process denoted as 'error catastrophe' (Ref. 89).
Nucleoside inhibitors for flaviviruses based on the structure of the four natural nucleotides have been reported90,91,92,93,94,95. 7-Deaza-2′-C-methyl adenosine (7DMA), a potent HCV inhibitor, exerted broad-spectrum antiviral effects against WNV, YFV, DENV-2 (Ref. 90), TBEV96 and ZIKV in cell-based assays97,98. 7DMA reduced viraemia in AG129 mice infected with DENV or ZIKV97,99. Another nucleoside inhibitor, 2′-C-methyladenosine (2′CMA), was also reported to inhibit ZIKV98 and TBEV96 in titre reduction assays, indicating the potential of 2′-C-methylated nucleosides96,98.
Replacing the 2′-C-methyl by an ethynyl group provided another potent nucleoside inhibitor, NITD008 (Refs 91,100,101), which inhibited DENV-1–4 at submicromolar to micromolar concentrations in numerous assays and cell lines91. NITD008 also inhibited HCV and other flaviviruses such as WNV, YFV, Powassan virus, TBEV, Kyasanur forest disease virus, Alkhurma haemorrhagic fever virus, Omsk haemorrhagic fever virus and ZIKV12,91,102. Despite efficacy in a DENV mouse model and favourable pharmacokinetic properties, the compound was not pursued further owing to failure at the preclinical stage during in vivo toxicity studies91,101,103.
A C-nucleoside analogue of adenosine, BCX4430, originally developed against filoviruses, exerted broad-spectrum activity against numerous viruses, including YFV, DENV-2 and JEV92. BCX4430 had a favourable pharmacokinetic profile and efficacy against Ebola virus (EBOV) and YFV in animal models92,104, and a phase I clinical trial (ClinicalTrials.gov identifier: NCT02319772) for the compound has been completed105.
Efforts to develop nucleoside inhibitors against the flaviviral RdRp are complicated by several challenges. Unfortunately, none of the reported examples could be further developed as a drug owing to low efficacy, toxicity or — for nucleoside inhibitors repurposed from HCV or HIV — differences in the cellular tropism. The first crucial issue is the conversion of the nucleoside inhibitors into the biologically active triphosphate by host kinases, which is not assessed in the initial biochemical assays. The capacity to optimize the nucleoside scaffold is often limited by the restricted substrate specificity of cellular kinases106. Furthermore, the phosphorylation efficiency of kinases is influenced by several factors, resulting in variations of the EC50 (effector concentration for half-maximum response) value of a nucleoside inhibitor in different cell types used for the assay. For example, EC50 values were reportedly higher in immortalized cell lines, such as the frequently used Huh-7 cells, compared with primary hepatic cells107. Based on these considerations, the tropism exerted by different flaviviruses may also have an important role in the potency of nucleoside inhibitors.
In most cases, the rate-limiting step for the intracellular formation of NTPs is the first phosphorylation. It is therefore tempting to use partially activated nucleoside monophosphates as drugs. These compounds, however, are poorly permeable and prone to degradation by phosphatases103,108. Prodrug strategies were developed to overcome these limitations108, and the phosphoramide prodrug approach was successful for the HCV nucleoside inhibitor sofosbuvir109.
The activity of host kinases is influenced by viral infection. HIV-1 and DENV increase cytokine levels, causing activation of peripheral blood mononuclear cells, which results in lower phosphorylation efficiency of some nucleoside inhibitors110,111. This effect, in addition to lower potency in DENV-infected hepatocytes, explains the efficacy failure of balapiravir against DENV in clinical trials, despite its established activity in vitro94,110. Notably, the influence of peripheral blood mononuclear cell activation on phosphorylation appears to be scaffold specific, because the cytidine-based balapiravir was more affected than the adenosine-based nucleoside inhibitor NITD008 (Ref. 110). Both peripheral blood mononuclear cells and hepatocytes are key host cell types targeted by DENV, which highlights the role of flaviviral tropism in the efficacy of the tested nucleoside inhibitors.
The second important issue related to nucleoside inhibitors is the toxicity resulting from insufficient selectivity against off-target polymerases, which is partly caused by the inhibition of the mitochondrial DNA polymerase-γ112. Indeed, the primary reason for failure of nucleoside inhibitors at the clinical stage is the inability to predict the side effects of nucleoside inhibitors during in vitro testing113.
To minimize toxicity, a combination strategy may prove useful. Antiviral synergy was observed for a combination of INX-08189 with ribavirin in cell-based assays of DENV114, and for NITD008 with vorinostat (a histone deacetylase inhibitor also known as suberanilohydroxamic acid (SAHA)), in WNV-infected C57BL/6 mice115.
Recently, allosteric inhibitors of the DENV NS5 polymerase were reported to be active against DENV-1–4 in biochemical and cell-based assays116. The inhibitors target the 'N pocket' near the active site, thus interfering with the conformational changes required for RdRp to transition from initiation to elongation116. Residues lining the N pocket are conserved across other flaviviruses116; however, the activity of these residues has only been assessed in DENV so far. One potential problem with this allosteric strategy is the expected low genetic barrier to resistance, as observed for allosteric inhibitors of HCV polymerase117.
Entry or fusion inhibitors. Studies aiming to inhibit flaviviral entry by interfering with host receptors did not yield notable results, with the exception of heat shock protein 70 (HSP70) ligands118, which are discussed in the section on host targets. Heparan sulfate proteoglycans have been considered as host cell receptors for viral attachment and potential drug discovery targets, but the validity of this approach is disputed (Box 3).
The flaviviral E glycoprotein mediates the first steps of viral infection by attaching to the host cell and mediating entry and membrane fusion119,120. The E glycoprotein is composed of three ectodomains and the stem anchor that provides a link to the viral membrane, as first elucidated for TBEV120,121,122.
A hydrophobic site between domains I and II of E glycoprotein binds to n-octyl-β-d-glucoside (β-OG) — which was present in very high concentrations in the crystallization buffer — in one of the crystal structures of DENV-2 E protein122, and is therefore referred to as the β-OG pocket. Compounds binding this pocket are suggested to interfere with conformational changes of the E protein that are required for fusion122. However, the validity of the β-OG pocket as target for antiviral drug discovery is doubtful. Several other crystallization experiments with flaviviral E proteins also included β-OG and similar detergents in high concentrations, but found no occupation of the hydrophobic pocket by these compounds120,121,123,124,125. The conservation of the residues lining this pocket is limited, so the pocket does not appear to be a viable target for broad-spectrum antiviral agents. Nevertheless, virtual screening of the β-OG pocket identified two drug-like compounds with nanomolar to low micromolar antiviral potency in cell-based assays126,127. The supposed interaction with the β-OG pocket could not be confirmed by structural, biochemical or resistance selection studies. Phenotypic assays for viral entry or fusion and time-of-addition studies are therefore the only confirmation for the assumed mechanism of action of these compounds, and they probably act through another mechanism. The first compound, a thiophene-quinazoline derivative, compound 6, exhibited broad-spectrum activity against DENV-1–4, YFV, WNV and JEV126. Time-of-addition studies of DENV verified an effect of compound 6 at an early stage of the viral life cycle126, but the compound was not assessed in animal models. The second compound, compound A5, a phenyl hydrazone derivative, was active in plaque assays against DENV-2, WNV and YFV127, but the anticipated toxic liabilities associated with the phenyl hydrazone moiety could hinder its further development.
Natural products such as griffithsin and squalamine were reported as entry and/or fusion inhibitors for several viruses, including flaviviruses, with efficacy in mouse models128. Griffithsin, a 13 kDa lectin isolated from algae128, mediates its effects by binding to oligosaccharides at the surface of enveloped viruses128,129,130,131 and was tolerated as a systemic antiviral with minimal toxicity following subcutaneous administration in mice132. However, griffithsin is a xenogeneic protein, and therefore may trigger an immune-mediated response of variable severity. Therefore, immunogenic regions must be modified before long-term treatment would be possible132. Squalamine, a cationic aminosterol, is proposed to disturb the electrostatic interaction between virus and host membranes during the early steps (entry and/or fusion) in the viral life cycle and also the late stages of virion assembly and budding133. The proposed mechanism has not been confirmed by time-of-addition studies.
T cell immunoglobulin and mucin (TIM) proteins are receptors for the apoptotic markers phosphatidylserine and phosphatidylethanolamine134. TIM receptors can promote viral entry through an 'apoptotic mimicry' mechanism by binding to virion-associated phosphatidylserine and phosphatidylethanolamine135,136. Duramycin-biotin inhibits TIM1-mediated entry of DENV-2, WNV and EBOV at submicromolar concentrations without detectable cytotoxicity134. Although duramycin-biotin has less profound haemolytic effects than duramycin itself, the suitability of the biotin derivative for clinical use in the case of haemorrhagic viral infections requires assessment. Furthermore, the strategy of interfering with phosphatidylserine or phosphatidylethanolamine to inhibit viral entry should be tested in animal models to evaluate the safety profile, as other cellular processes that depend on TIM-mediated binding to phosphatidylserine or phosphatidylethanolamine in host cells could also be affected.
Capsid inhibitors. Compared with E protein, the capsid protein has received minimal attention to date. The flaviviral capsid is a dimeric protein with a high density of positively charged residues at the surface and a hydrophobic core pocket. The monomer contains four α-helices and an N-terminal disordered region as elucidated for the WNV137 and DENV138 capsid structures. The N-terminal region has been implicated in interactions with lipid droplets139 and very-low-density lipoprotein140. A single small-molecule inhibitor, ST-148, has been identified by phenotypic high-throughput screening followed by resistance selection141. This compound has antiviral effects in cell-based assays against DENV-1–4, Modoc virus, YFV and HCV (but not JEV) with favourable CC50 (50% cytotoxic concentration) values141. Despite its poor oral bioavailability, ST-148 displayed efficacy in the AG129 mouse model for DENV infection. ST-148 interfered with both assembly and/or release and entry of DENV infectious particles, probably by stabilizing the capsid protein structure and enhancing capsid self-interaction142.
NS4B inhibitors. Numerous studies have examined inhibitors of the flaviviral NS4B protein143,144, a highly hydrophobic integral membrane protein145. NS4B mediates several interactions with other non-structural viral proteins and host proteins to modulate viral replication146,147. Compounds targeting NS4B lacked broad-spectrum antiviral activity, and inhibitory effects were limited to a single virus or even specific serotypes143,144,148,149. The only exception is lycorine, which reduced viral titres for WNV, DENV-2 and YFV150. WNV resistance to lycorine was conferred by V9M mutations in the 2K peptide located between NS4A and NS4B150. A modification of the structure to 1-acetyl-2-oxo-lycorine provided a slightly enhanced potency at WNV, with remarkable improvement in cytotoxicity150,151.
NS3 helicase inhibitors. In contrast to targeting the flaviviral capsid protein or NS4B, targeting NS3, which functions as an RNA helicase and ATPase, or NS5, which is a methyltransferase and guanylyltransferase (discussed below), is often complicated by the need to achieve selectivity against host enzymes with similar functions.
The flaviviral helicase belongs to the helicase superfamily 2 (SF2) and is located at the C-terminal domain of NS3 (Refs 152,153). NS3 is responsible for unwinding viral RNA during replication, and its activity is driven by an intrinsic NTPase activity152,153. The structure of NS3 helicase has been elucidated for many flaviviruses154, including ZIKV155. NS3 helicase comprises three subdomains, with the well-conserved ATP-binding pocket located between subdomains 1 and 2. A long tunnel runs across the protein and is expected to accommodate the viral RNA154,155.
A benzoxazole analogue, ST-610, was reported as an inhibitor of helicase activity156. The compound had low cytotoxicity, inhibited viral replication of DENV-1–4 and YFV (but not WNV or JEV) in various cell types, and reduced viral load in a mouse model of DENV infection156. A pyrrolone derivative (compound 25) inhibited WNV and DENV replication in cell culture, albeit with low selectivity index values, by targeting helicase- catalysed ATP hydrolysis, but had no effect on HCV helicase157. Ivermectin has also demonstrated inhibitory activity against YFV, DENV-2 and WNV helicases in the upper nanomolar range158, and is a weak inhibitor of DENV protease159, as discussed in detail below.
NS5 methyltransferase inhibitors. The N-terminal domain of the flaviviral NS5 protein functions as a guanylyltransferase160,161 and a methyltransferase162,163. NS5 catalyses N7 and 2′-O methylation reactions using S-adenosylmethionine (SAM) as a methyl donor162,163, and is inhibited by the non-selective competitive inhibitors S-adenosylhomocysteine and sinefungin164. In addition to the SAM-binding pocket, the crystal structure of the DENV-3 and WNV methyltransferases revealed a conserved hydrophobic cavity next to the SAM-binding site, which could be used to design specific inhibitors against flaviviruses165,166. Structural evidence for the binding of compounds to this hydrophobic pocket is currently missing. Introduction of a silyl group at the 5′-position of azidothymidine-based triazoles, a class of compounds with potent antiviral activity against HIV-1, resulted in inhibitors of DENV and WNV methyltransferases167. These compounds showed antiviral effects in DENV and WNV replicon assays, and in a DENV plaque assay, but had relatively high cytotoxicity. Docking studies suggested that the bulky 5′-silyl group is positioned in the hydrophobic cavity167. Using virtual screening of compounds binding to the WNV methyltransferase, NSC 12155 was identified, which inhibits the methyltransferase activity of NS5 from WNV, DENV-2 and DENV-3, and YFV in enzymatic assays. NSC 12155 reduced viral titres for WNV, DENV-2, JEV and Saint Louis encephalitis virus168.
Host targets
Flaviviruses interfere with the host cells in numerous ways. Some cellular pathways may be upregulated to promote replication, whereas other functions, particularly those related to the cellular immune response, are suppressed by the virus. Interfering with processes exploited or controlled by the virus has therefore long been considered a conceptually promising route towards antiviral treatment, albeit with limited success so far.
Because certain host factors are usurped by a large number of viruses, these targets should confer broad-spectrum antiviral activity. In addition, compounds targeting host factors are anticipated to be less prone to development of resistance, although some cases of resistance have already been reported (see below). Broad-spectrum activity must be carefully confirmed on a case-by-case basis for each antiviral agent to avoid activation of other viral co-infections and to cover all co-circulating flaviviruses. A further obvious complication is that affecting host factors involved in normal physiological function has a higher potential for side effects, and striking a balance between antiviral activity and toxicity is not easy.
A general, cautionary remark must be made with respect to the repurposing of host targets from one virus to another. For example, antagonists of C-C chemokine receptor type 5 (CCR5) were developed for anti-HIV-1 therapy and suggested for treatment of DENV infection169. However, there is conflicting evidence for the role of CCR5 in JEV170 and WNV171 infections, and anti-HIV CCR5 ligands could be inefficient or aggravate these infections. Even approved therapies targeting host factors should be cautiously evaluated for each group of viruses, including those that may be present as unapparent co-infections in the patient.
Some of the most promising or interesting classes of compounds that act against host factors will be discussed here based on their mechanism of action. The structures of selected compounds are presented in Fig. 3. Other classes, such as S-adenosylhomocysteine hydrolase and autophagy inhibitors (Supplementary information S7 (table)), have limited broad-spectrum activity or their high toxicity precludes in vivo testing, and will not be discussed further. Inhibition of nuclear transport by fenretinide (N-(4-hydroxyphenyl)retinamide; 4-HPR) and ivermectin is discussed in later sections.

Chemical structures of selected potential broad-spectrum antiflaviviral drugs that target host proteins.
α-Glucosidase inhibitors. α-Glucosidase removes glucose units from N-linked glycans and thereby participates in the maturation and folding of flaviviral glycoproteins172. Glucosidase inhibitors have broad-spectrum antiviral activity in vitro and in vivo against a multitude of enveloped viruses173,174, including flaviviruses, and have a confirmed high genetic barrier against escape mutations in vivo175. The most promising glucosidase inhibitors are the iminosugars, such as castanospermine and 1-deoxynojirimycin. Their main disadvantages are the high dosages required, relative toxicity and weak activity during the post-infection period. Previous studies of iminosugars in vivo and in the clinical treatment of infections caused by other enveloped viruses (for example, HIV, HCV and influenza)174,176,177 have demonstrated that most of their disadvantages can be overcome by derivatization into prodrugs, association with other antivirals and/or by early (possibly prophylactic) treatment.
Castanospermine is a potent antiviral compound in vitro and in vivo against all DENV serotypes, but it has much lower activity against YFV and no effect on WNV172,178. Its 6-O-butanoyl derivative, celgosivir, is an oral prodrug that is 100-fold more active in vitro and 2 times more active in vivo179,180. Celgosivir failed in a proof-of-concept clinical trial in patients with dengue fever181, and is probably more effective if treatment starts on the day of infection180. Increased doses of celgosivir initiated on the second or third day post infection significantly reduce viraemia, and a phase II clinical trial (NCT02569827) with an optimized celgosivir regimen was recently approved in Singapore180.
A N-nonyl-derivative of 1-deoxynojirimycin inhibits in vitro replication of DENV-2 and JEV182, and further structural modifications afforded derivatives with lower toxicity and higher activity. The introduction of a cyclohexyl group in the N-alkyl chain resulted in enhanced potency against DENV, WNV and bovine viral diarrhoea virus (BVDV), as well as an improved safety profile183. Modification by oxygen-containing functionalities in the N-alkyl side chain increased the activity against DENV-2 and, to a lesser extent, against WNV and BVDV184. Increased cellular uptake probably accounts for the improved activity in this compound class. Further optimization of the pharmacokinetic profile led to derivatives with low toxicity and good oral bioavailability, but a narrow therapeutic window in AG129 mice (limited to the first 48 hours post infection)185,186,187.
α-Glucosidase inhibitors can therefore be considered as broad-spectrum, drug-like antivirals. Their main limitation — the necessity of initiating treatment very soon after infection — will probably also apply to other treatments that interfere with flaviviral replication, such as helicase and protease inhibitors. In this respect, the exploration of iminosugar antivirals may have yielded a generally applicable conclusion: emergency prophylaxis under epidemic conditions may be more promising than post-infection treatment. If the treatment is started after symptomatic diagnosis of the infection, a more aggressive dosing and treatment regimen, as for celgosivir, appears to be necessary to reduce viraemia. As α-glucosidase is a host factor, and high doses of iminosugars are not well tolerated in humans, drugs targeting this protein could be more useful as a low-dose emergency prophylaxis regimen than as a treatment for verified infections with respect to both efficacy and safety.
Nucleoside biosynthesis inhibitors. Despite the potential of nucleoside biosynthesis inhibitors to have broad- spectrum antiviral activity and the considerable knowledge on this class of compounds, the results obtained so far are modest and largely restricted to one compound in clinical use: ribavirin. Ribavirin was one of the first broad-spectrum antivirals on the market and is commonly used to treat HCV infections188,189. The antiviral activity of ribavirin has been attributed to inhibition of the host inosine monophosphate dehydrogenase (IMPDH), inhibition of viral polymerase and RNA capping, a mutagenic effect on viral RNA and/or immunomodulation190. Ribavirin is active against flaviviruses in vitro only in high concentrations191, whereas in vivo or clinical studies often yielded negative results192,193 or showed activity only in early phases of the infection194,195.
Development of more active IMPDH inhibitors (to enhance antiviral potency) resulted in compounds with higher cytotoxic or immunosuppressive activity. Example compounds include 5-ethynyl-1-β-d-ribofuranosylimidazole-4-carboxamide (EICAR; 5-ethynylribavirin)196, which had substantially higher activity against Modoc virus, YFV and DENV than did ribavirin. Cytostatic effects comparable to those of 5-fluoruracil197 and a narrow therapeutic window prohibited the clinical use of IMPDH inhibitors as antivirals. A non-nucleoside mycophenolic acid196 has reportedly high activity against DENV196, YFV196, JEV198 and ZIKV12, but its immunosuppressive activity limits its potential as an antiviral compound199.
Brequinar is an inhibitor of dihydroorotate dehydrogenase (DHODH), a host enzyme responsible for pyrimidine nucleoside biosynthesis. Brequinar has potent antiviral activity in vitro against DENV, WNV, YFV and Powassan virus200, but was not approved for clinical use owing to a low therapeutic index201. Other promising non-nucleoside DHODH inhibitors are the indole derivative compound A3 (Ref. 202) and 2-(4-benzyl- 3-ethoxy-5-methyl-1H-pyrazol-1-yl)pyrimidine203. The latter compound has been described as a low-nanomolar DHODH inhibitor203; however, its activity against flaviviruses is currently unknown. Compound A3 demonstrated broad-spectrum antiviral activity against multiple viruses in vitro in the submicromolar range and no resistance development in influenza virus202,204, but its in vivo activity or toxicity data are not available200.
The main obstacles to the development of nucleoside biosynthesis inhibitors as antiviral agents are a narrow therapeutic window and the potential for immunosuppressive effects, properties that are incompatible with expected co-infections, pregnancy and extended (prophylactic) dosage regimens. Another problem is that resistance can develop, and could occur via various mechanisms depending on the target and virus. Resistance could be addressed by using compounds that inhibit multiple biosynthesis steps or by combination therapy with antivirals that act via other mechanisms.
Cyclophilin inhibitors. Cyclophilins are peptidyl-prolyl isomerases that facilitate protein folding and have an important role in viral replication. Inhibition of cyclophilin A by cyclosporine reduces its interaction with flaviviral NS5 and has an antiviral effect against DENV-2, WNV and YFV in cells205. Cyclosporine was more effective against DENV-2 and YFV than against WNV205. Cyclosporine demonstrated effectiveness against ZIKV in vitro12. Cyclosporine is also immunosuppressive owing to inhibition of the protein phosphatase calcineurin206. As the binding domains for cyclophilin A and calcineurin are located at different sites of the cyclosporine molecule207, non-immunosuppressive cyclophilin A inhibitors can be designed. The non-immunosuppressive (non-calcineurin inhibiting) cyclophilin A inhibitor alisporivir (also known as Debio 025)208 was developed as an anti-HCV agent, but its efficacy against flaviviruses is currently unknown.
Lipid-related processes. Lipid biosynthesis, signalling and metabolism209 have long been investigated in other diseases, such as atherosclerosis, resulting in numerous well-characterized drugs and drug candidates that can potentially be repurposed as broad-spectrum antiviral drugs. For example, inhibition of acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN)210,211 can have antiviral effects. The FASN inhibitor 4-methylene-2- octyl-5-oxotetrahydrofuran-3-carboxylic acid (C75)210 exhibited dose-dependent inhibition of DENV-2, YFV and WNV replication, and reportedly suppressed the formation of intracellular lipid droplets that occurs in cell infected with DENV139,210. However, FASN inhibitors can cause severe anorexia and weight loss212, and inhibition of ACC appears to have a higher clinical potential for the treatment of viral infections212. The ACC inhibitors TOFA (5-(tetradecyloxy)-2-furoic acid) and MEDICA 16 (3,3,14,14-tetramethylhexadecanedioic acid) induced a dose-dependent reduction of WNV and Usutu virus replication213.
Pharmacological interference with the biosynthesis of host sphingomyelin has also been studied, with varying results214,215. The tricyclic antidepressants amitriptyline and imipramine — inhibitors of acid sphingomyelinase, which hydrolyses sphingomyelin to ceramide — reportedly decrease the infectivity of pseudotype JEV in pretreated Huh-7 cells216. However, inhibition of neutral sphingomyelinase by GW4869 suppressed the release of WNV particles from HeLa, Vero and C6/36 cells, as well as of Usutu virus from HeLa cells, but had the opposite effect for the alphavirus Sindbis virus217. The sphingomyelin synthase inhibitors SPK-601 and MS-209 reduced the production of infectious viral particles in WNV-infected Vero cells217. Fenretinide (4-HPR), an inhibitor of ceramide synthase and dihydroceramide desaturase, may have other functions and is discussed below.
Conflicting results have also been obtained for inhibitors of cholesterol intracellular transport and biosynthesis, but these results could be explained by differences in experimental procedures218,219. Similar to inhibitors against other targets, lipid-altering therapies often have reduced efficiency in vivo in post-infection treatment. The HMG-CoA reductase inhibitor lovastatin increased survival rates for all treatment regimens in vivo against DENV-2 infection220, but reduced viraemia could only be observed in pretreated animals. Considering the broad usage and good tolerability of statins, they could be candidates for an emergency prophylactic antiviral regimen.
Kinase inhibitors. Flaviviruses, similar to most other viruses, usurp a large number of host kinases at various steps in their cellular life cycle. Flaviviral proteins from the RNA replication complex, such as JEV NS3 (Ref. 221) and DENV NS5 (Ref. 222), are phosphorylated by host kinases. Kinase inhibitors, extensively explored for applications in oncology, therefore offer opportunities for antiviral repurposing. However, several challenges and caveats must be considered.
First, drug resistance — which is usually considered less probable for antivirals acting at host targets — can occur via mutations in the viral proteins that act as kinase substrates. For example, the DENV-2 NS4B-T108I mutation confers resistance against RNAi-mediated depletion of the FYN kinase or its inhibition by AZD0530 or dasatinib223. Although this type of resistance is unlikely to develop during normal transmission of DENV between human and mosquito hosts, it nevertheless demonstrates the potential for resistance to kinase inhibitors.
Second, substantial adverse effects of dasatinib and its analogues can also arise from the reactivation of latent, silent viral infections, such as hepatitis B virus (HBV)224,225. Therefore, these compounds should not be the first-line treatment option for individuals with chronic and latent viral infections, for long-term prophylaxis or during pregnancy. Moreover, inhibition of lymphocyte-specific protein tyrosine kinase by dasatinib and AZD0530 can have a detrimental effect on the cellular immune response226.
Third, kinases have varying expression levels and functions, and some of them (such as those in the Janus kinase (JAK)–signal transducer and activator of transcription (STAT) pathway) participate in the cellular immune response. For example, interferon-induced inhibitor of κB kinase ɛ signalling, via STAT1 phosphorylation and induction of interferon-induced protein with tetratricopeptide repeats 2 (IFIT2) expression, restricts WNV infection and pathogenesis227. Therefore, each potential antiviral kinase inhibitor should be evaluated for inhibition of immunologically important kinases.
The outlook for kinase inhibitors as broad-spectrum antiflavivirals is mixed: resistance could develop; repurposing anticancer kinase inhibitors seems risky; and multi-target kinase inhibitors may activate dormant, unrelated viral infections. Alternatively, inhibitors with increased selectivity or activity against both viral and host targets (possibly at the expense of broad-spectrum activity) or kinase inhibitors that modulate the host immune response and therefore the severity of symptoms (but without intrinsic antiviral activity) could be developed. Activators of pro-immunogenic kinases, which would counteract the virus-mediated suppression of the cellular immune response, might also be useful to treat viral infections.
Polyamine synthesis inhibitors. Inhibiting the synthesis of polyamines, which have an important role in the transcription and translation of ZIKV and CHIKV, could have broad-spectrum antiviral potential228,229. Numerous viruses, including flaviviruses, are sensitive to compounds that alter polyamine levels. Examples of polyamine inhibitors with therapeutic potential include eflornithine, an ornithine decarboxylase inhibitor, and diethylnorspermine, an activator of the spermidine/spermine N1-acetyltransferase228. Post-infection administration of eflornithine showed efficacy against coxsackievirus B3 and CHIKV in animal models228. Eflornithine has low toxicity and good stability, is approved for the treatment of African trypanosomiasis, and was proposed for cancer chemoprevention230. Its main disadvantage is the requirement for high doses. More potent derivatives or a combination with other therapeutics may enhance its antiviral activity228.
Other host targets. Several groups of compounds reported to target pleiotropic host targets, including ribosomes (lactimidomycin and its derivatives), the proteasome (bortezomib and its derivatives), DDX3 or HSP70, have demonstrated broad-spectrum antiflaviviral activity in vitro12,15,118,231,232. However, most of these compounds have side effects that severely limit their potential use as (prophylactic) antiviral agents. The 60S ribosome inhibitor lactimidomycin is highly cytotoxic233,234, and bortezomib, a covalent reversible inhibitor of the 26S proteasome, has been labelled as class D for potential teratogenic effects235. Inhibitors of targets that are contested between viruses and the immune system (for example, DDX3, the proteasome and HSP70), can reactivate chronic co-infections such as HCV (as observed for DDX3 inhibition)236 and varicella zoster virus and HBV (proteasome inhibition by bortezomib)147,237. Moreover, inhibition of HSP70 could reduce protection against other infections238,239,240 and tumours241, and inhibition of the proteasome could even enhance some infections242,243,244. Although these difficulties prohibit the use of these compounds in long-term treatment of persistent infections, prophylaxis or pregnancy, they may be tolerable for the short-term treatment of flaviviral infections.
Other and unknown mechanisms of action
Numerous compounds with antiviral activity against flaviviruses have been identified from phenotypic assays. Although the molecular target (or targets) of these compounds has not been identified in many cases, the spectrum of activity of these examples could be a promising starting point for further investigations and drug development. Of particular interest are repurposed drugs, as they have established safety and pharmacokinetic profiles. The structures of selected compounds are provided in Fig. 4.

Chemical structures of selected potential broad-spectrum antiflaviviral drugs with other mechanisms. Many of these compounds were discovered through phenotypic screens.
Nitazoxanide, an antiparasitic ester prodrug used for the treatment of diarrhoea caused by Cryptosporidium parvum and Giardia intestinalis infections245, exerted antiviral effects against a broad range of RNA and DNA viruses246,247,248,249. Nitazoxanide (or its active metabolite tizoxanide) inhibits replication of JEV248, DENV-2 and YFV249 in cell culture, and provides protection against JEV in a mouse model248. The antiviral efficacy and lack of adverse effects of nitazoxanide treatment, either alone or in combination with other antiviral agents, was demonstrated in patients infected with HCV250, rotavirus251, norovirus251 or influenza252. The drug is currently under investigation in a phase III clinical trial (NCT02612922) for the treatment of influenza. Nitazoxanide interferes with the glycosylation of viral proteins and the production of mature viral particles253,254, and induces antiviral innate immunity255,256. However, these effects were observed for other viruses, and investigations with flaviviruses were not reported. Analogues without the nitro group, such as RM-5038 (or its active metabolite RM-4848), were synthesized to increase drug-like features245. Another antiparasitic drug, niclosamide, was identified from a phenotypic screen257 as a potent inhibitor of ZIKV in numerous cell types, although it displayed some degree of cytotoxicity257.
Bromocriptine, an agonist of dopamine receptors D2 and D3, was identified from a screen of pharmacologically active compounds against DENV in focus reduction assays258. This compound inhibited DENV-1–4, and, to a lesser extent, TBEV258. Antiviral effects were not observed with other dopamine agonists, namely quinpirole and rotigotine. A mutation in the NS3 helicase domain was identified in escape mutants, but could only confer minimal drug resistance, suggesting the involvement of other viral or host proteins. Unfortunately, bromocriptine lacked efficacy in the AG129 mouse model258.
The antiviral activity of the dihydrodibenzothiepines can probably not be explained by a single mechanism of action. A representative of this group, SKI-417616, inhibits DENV-1–4, WNV and Sindbis virus by antagonizing the dopamine receptor D4, which inhibits the downstream phosphorylation of epidermal growth factor receptor-related kinase (ERK)259. However, cellular signalling pathways other than ERK also seem to be involved, and further investigations are necessary.
Several antimalarial drugs that contain a quinoline scaffold were evaluated for their antiviral activity against DENV260,261,262 or WNV260. Amodiaquine inhibited DENV-2, DENV-4 and WNV replication but with relatively poor selectivity260. Interestingly, different quinoline-based antimalarial drugs reportedly interfere with different steps of the DENV life cycle: amodiaquine is proposed to affect the initial steps of RNA replication and (to a lesser extent) entry260; hydroxychloroquine activates the host immune system262; and chloroquine is suggested to act during entry or assembly, as it lacked activity in the replicon assay260. Despite the efficacy of chloroquine against DENV-2 in monkeys261, the compound failed to reduce viraemia in patients with dengue fever263,264. A positive effect on acute dengue symptoms was observed, which could be related to the anti-inflammatory effect of chloroquine, a pharmacological property of the drug that forms the basis for its medical use in rheumatic diseases265. Chloroquine may be a candidate for prophylactic use, considering the previous, extensive clinical experience with this drug in the context of malaria.
A cardiac glycoside, lanatoside C, was reported to exert potent antiviral effects against DENV-1–4, Kunjin virus and other RNA viruses266. Analysis of the mechanism of action of lanatoside C suggests that this compound possibly targets viral RNA synthesis266. Digoxin, another cardiac glycoside, was recently evaluated against ZIKV and displayed antiviral effects12. However, considering the narrow therapeutic index of digoxin, the authors suggested that the concentrations needed for anti-ZIKV effects may be toxic12.
Screening of a compound library in a DENV-2 replicon assay identified a lead compound (compound 15a) with antiviral activity against DENV-2 and YFV in the low micromolar range267. Systematic optimization of the aromatic rings in the original imidazole 4,5-dicarboxamide scaffold of compound 15a facilitated modulation of the inhibitory potency and cytotoxicity of the obtained analogues267,268. The most promising antiviral profile against DENV-2 and YFV was observed for compound 7 g268.
A group of hydroxyquinoline derivatives can activate interferon regulatory factor 3 (IRF3) through mitochondrial antiviral signalling and drive antiviral gene expression in cells. This upregulation of the innate immune response leads to an antiviral effect against multiple viruses, including DENV-2, WNV, HCV, EBOV and Lassa virus269,270. However, the molecular target of the compounds has not been identified. Moreover, in vivo studies are necessary to exclude that activation of the immune response produces undesirable side effects.
Multiple mechanisms of antiviral activity have been suggested for ivermectin, which is active in vitro against multiple flaviviruses, including DENV-1–4 (Ref. 271) and ZIKV12. The compound blocks the interaction of DENV-1–4 NS5 with importin α/β1 (IMPα/β1), a nuclear protein import receptor271, which was proposed to be the main mechanism responsible for its antiviral activity. Ivermectin was active in a cell-based flavivirus immunodetection assay for DENV-1–4 in the low-micromolar range271. As demonstrated previously, DENV NS5 contains a nuclear localization sequence that confers interaction with the IMPα/β1 dimer and the exportin receptor CRM1 (Refs 272,273). The nuclear localization sequence is highly conserved in the flavivirus genus, and is consequently a highly attractive target for broad-spectrum antiviral development273. Furthermore, inhibition of CRM1 by leptomycin B caused an increase in the nuclear accumulation of NS5, suppression of interleukin-8 induction, and augmentation of DENV-2 production in cells273. Ivermectin also inhibits helicase unwinding activity for YFV, DENV-2 and WNV in the upper nanomolar range158, and shows weak inhibition of the DENV protease159. Although these effects are probably not the main mechanisms of antiviral activity of ivermectin, their contribution should be considered.
Fenretinide, a retinoic acid derivative, is another compound with multiple mechanisms of antiviral activity. Fenretinide inhibits replication of DENV-1–4 (Ref. 274) and demonstrated anti-WNV274,275 and anti-HCV275 activity in cells and anti-DENV-2 activity in AG129 mice274,275. The main mechanism of fenretinide activity appears to be inhibition of the interaction of viral proteins with the IMPα/β1 receptor. The compound also induces phosphorylation of eukaryotic translation initiation factor 2α, thereby controlling translation attenuation and promoting an antiviral state276. Moreover, fenretinide alters ceramide homeostasis by inhibiting ceramide synthase and dihydroceramide desaturase. However, these latter two activities do not appear to contribute to the antiviral effect of the compound275.
Phosphorodiamidate morpholino oligomers (PMOs) are uncharged, water-soluble compounds that contain nucleobases attached to a backbone of morpholine rings that are connected to each other via phosphorodiamidate linkages. PMOs sterically block the interaction of complementary viral RNA with ribosomes, suppressing the formation of the 43S preinitiation complex and ribosome scanning that occurs during viral translation, thereby blocking RNA replication277,278. Arg-rich peptidic conjugates of PMOs (PPMOs) were reported to have a higher permeability across cell membranes279, and positively charged PMOs (PMO+) were more efficient owing to improved binding kinetics280. PMOs, PPMOs and PMO+ have several important advantages over small interfering RNAs, which are similar in structure and mechanism of action: PMOs have improved resistance to enzymatic degradation and demonstrated safety in clinical trials, as shown for eteplirsen (also known as AVI-4658) and AVI-7288 (Refs 281,282,283). PMOs or PPMOs complementary to the viral untranslated region (UTR) have relatively specific antiviral activity and, with respect to the antiviral spectrum, best cover closely related viruses277,284. Although structural features of the viral UTR are relatively well-conserved, the sequences are divergent, which limits the broad-spectrum potential of these compounds. The therapeutic window in mice is very small for some compounds; however, the toxicity and activity of PPMOs are generally higher than for PMOs owing to better pharmacokinetics278. Treatment with PMOs and PPMOs is effective if started as early as possible, might be totally ineffective at later stages, and requires parenteral administration277,278,284. Nonetheless, experiments in non-human primates and phase I clinical trials in humans of a PMO+, AVI-7288, specifically targeting the mRNA sequence of the nucleoprotein of a filovirus, Marburg virus, achieved survival rates of 83–100% when the drug was administered up to 4 days after animals were infected with this virus; the drug had good pharmacokinetic and toxicity profiles282,283. This observation demonstrates a good level of activity and low toxicity for this group of compounds.
Conclusion and outlook
The current upsurge of interest in antiflaviviral drug discovery and flavivirus biology, triggered largely by the ZIKV epidemic, will certainly lead to an increased understanding of these important pathogens. As in other topical areas of drug discovery, some of the currently proposed targets, pathways and compounds may lack sufficient maturity for further development. This Review attempts to provide some indication as to which approaches currently appear, or have already been shown, to be most promising.
In addition to the flaviviral protease and polymerase, other viral targets have shown varying potential for broad-spectrum antiviral effects. In these cases, the promising activity profile covering more than one flavivirus was generally observed for few examples within a particular class. Most host targets have a high potential for broad-spectrum antiviral activity, and several benefit from advanced progress at the clinical level; for example, α-glucosidase inhibitors. However, some of the host targets are differently used by closely related viruses or have relevance for both the viral replication and the immune system and, therefore, require a careful case-by-case assessment.
The design and development of new antiflaviviral compounds must take their activity spectrum into consideration, and a strong preference must be given to drug candidates that are active against the largest number of co-circulating viruses. This consideration is particularly important for compounds that target host factors, as even closely related flaviviruses interact differently with cellular components. Somewhat unexpectedly, drugs that target host factors are not exempt from the development of resistance. Furthermore, targeting host factors with multiple functions — in addition to their involvement in viral replication — may be associated with severe side effects.
Antiviral treatments should generally be initiated as soon as possible after infection or as prophylactic measures. This scenario is particularly true for acute flaviviral infections, for which the most severe sequelae occur after the peak viral load. Antiviral agents that target the viral non-structural proteins or other replication-relevant factors will probably be most efficient in prophylaxis and treatment of early-stage or persistent subclinical infections, and are less promising to treat the advanced stages of acute disease. Compounds that target the entry of the virus into the cell could be the most effective prophylactics, but investigations in this direction have not yet produced any tangible results.
For persisting flaviviral infections, it is important to adjust the pharmacokinetic parameters of the antiviral compound to ensure its penetration, or even accumulation, in the most affected organs or tissues. In cases of severe disease, which are frequently caused by a pathological immune response, pharmacological interference with this ill-directed host reaction could be promising. For this particular approach, established immunomodulatory drugs could potentially be repurposed.
We anticipate a two-tiered approach to antiflaviviral drug development. In the near-to-midterm, drug repurposing from phenotypic screens has the potential to yield 'emergency' antivirals, for which a higher incidence of side effects and limited broad-spectrum activity can be tolerated. In the longer term, and considering the recent developments in the treatment of HCV and HIV, novel compounds acting at viral targets, in particular the evolutionarily well-conserved enzymatic functions localized within NS3 and NS5, will probably enable us to counter the persistent public health risk that is posed by the already prevalent, as well as the clandestine, flaviviruses.
References
Meanwell, N. A. 2015 Philip S. Portoghese medicinal chemistry lectureship. Curing hepatitis C virus infection with direct-acting antiviral agents: the arc of a medicinal chemistry triumph. J. Med. Chem. 59, 7311–7351 (2016).
Theiler, M. & Smith, H. H. The use of yellow fever virus modified by in vitro cultivation for human immunization. J. Exp. Med. 65, 787–800 (1937). This classical article discusses the development of the YFV vaccine, which is still being used today and for which Theiler was awarded the Nobel Prize in 1951.
Fernandez-Garcia, M.-D., Mazzon, M., Jacobs, M. & Amara, A. Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe 5, 318–328 (2009).
Sips, G. J., Wilschut, J. & Smit, J. M. Neuroinvasive flavivirus infections. Rev. Med. Virol. 22, 69–87 (2012).
Bhatt, S. et al. The global distribution and burden of dengue. Nature 496, 504–507 (2013).
Zanluca, C. et al. First report of autochthonous transmission of Zika virus in Brazil. Mem. Inst. Oswaldo Cruz 110, 569–572 (2015).
Mackenzie, J. S., Gubler, D. J. & Petersen, L. R. Emerging flaviviruses: the spread and resurgence of Japanese encephalitis, West Nile and dengue viruses. Nat. Med. 10, S98–S109 (2004).
Weaver, S. C. & Barrett, A. D. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat. Rev. Microbiol. 2, 789–801 (2004).
Weaver, S. C. et al. Zika virus: history, emergence, biology, and prospects for control. Antiviral Res. 130, 69–80 (2016).
Hadinegoro, S. R. et al. Efficacy and long-term safety of a dengue vaccine in regions of endemic disease. N. Engl. J. Med. 373, 1195–1206 (2015). This paper reports the efficacy and long-term safety studies of Dengvaxia, the dengue tetravalent vaccine that recently entered clinical practice.
Wakita, T. et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 11, 791–796 (2005).
Barrows, N. J. et al. A screen of FDA-approved drugs for inhibitors of Zika virus infection. Cell Host Microbe 20, 259–270 (2016). This seminal study demonstrates the possible efficacy of repurposed US FDA-approved drugs against ZIKV.
Shum, D. et al. High-content assay to identify inhibitors of dengue virus infection. Assay Drug Dev. Technol. 8, 553–570 (2010).
Idris, F., Muharram, S. H. & Diah, S. Glycosylation of dengue virus glycoproteins and their interactions with carbohydrate receptors: possible targets for antiviral therapy. Arch. Virol. 161, 1751–1760 (2016).
Choy, M. M. et al. Proteasome inhibition suppresses dengue virus egress in antibody dependent infection. PLoS Negl. Trop. Dis. 9, e0004058 (2015).
Holmes, E. C. The Evolution and Emergence of RNA Viruses (Oxford Univ. Press, 2009).
Metzner, K. J. et al. Minority quasispecies of drug-resistant HIV-1 that lead to early therapy failure in treatment-naive and-adherent patients. Clin. Infect. Dis. 48, 239–247 (2009).
Beck, A. et al. Comparison of the live attenuated yellow fever vaccine 17D-204 strain to its virulent parental strain Asibi by deep sequencing. J. Infect. Dis. 209, 334–344 (2014).
Selvey, L. A., Speers, D. J. & Smith, D. W. Long-term outcomes of Murray Valley encephalitis cases in Western Australia: what have we learnt? Intern. Med. J. 46, 193–201 (2016).
Oehler, E. et al. Zika virus infection complicated by Guillain–Barre syndrome — case report, French Polynesia, December 2013. Euro Surveill. 19, 20720 (2014).
Fontes, C. A. P., dos Santos, A. A. S. D. & Marchiori, E. Magnetic resonance imaging findings in Guillain–Barré syndrome caused by Zika virus infection. Neuroradiology 58, 837–838 (2016).
McMahon, A. W. et al. Neurologic disease associated with 17D-204 yellow fever vaccination: a report of 15 cases. Vaccine 25, 1727–1734 (2007).
Puccioni-Sohler, M. et al. Neurologic dengue manifestations associated with intrathecal specific immune response. Neurology 73, 1413–1417 (2009).
McMinn, P. C. The molecular basis of virulence of the encephalitogenic flaviviruses. J. Gen. Virol. 78, 2711–2722 (1997).
Ravi, V. et al. Persistence of Japanese encephalitis virus in the human nervous system. J. Med. Virol. 40, 326–329 (1993).
Penn, R. G. et al. Persistent neuroinvasive West Nile virus infection in an immunocompromised patient. Clin. Infect. Dis. 42, 680–683 (2006).
Paessler, S. & Walker, D. H. Pathogenesis of the viral hemorrhagic fevers. Annu. Rev. Pathol. 8, 411–440 (2013).
de Oliveira, W. K. Increase in reported prevalence of microcephaly in infants born to women living in areas with confirmed Zika virus transmission during the first trimester of pregnancy — Brazil, 2015. MMWR Morb. Mortal. Wkly Rep. 65, 242–247 (2016).
Brasil, P. et al. Zika virus infection in pregnant women in Rio de Janeiro — preliminary report. N. Engl. J. Med. 375, 2321–2334 (2016).
Cauchemez, S. et al. Association between Zika virus and microcephaly in French Polynesia, 2013-15: a retrospective study. Lancet 387, 2125–2132 (2016).
Miner, J. J. et al. Zika virus infection during pregnancy in mice causes placental damage and fetal demise. Cell 165, 1081–1091 (2016).
Cugola, F. R. et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534, 267–271 (2016).
Habu, A., Murakami, Y., Ogasa, A. & Fujisaki, Y. Disorder of spermatogenesis and viral discharge into semen in boars infected with Japanese encephalitis virus (author's transl). Uirusu 27, 21–26 (in Japanese) (1977).
Musso, D. et al. Potential sexual transmission of Zika virus. Emerg. Infect. Dis. 21, 359–361 (2015).
Mansuy, J. M. et al. Zika virus: high infectious viral load in semen, a new sexually transmitted pathogen. Lancet Infect. Dis. 16, 405 (2016).
Rossi, S. L. et al. Characterization of a novel murine model to study Zika virus. Am. J. Trop. Med. Hyg. 94, 1362–1369 (2016). This article provides a description of the first murine model for ZIKV pathogenesis.
Lazear, H. M. et al. A mouse model of Zika virus pathogenesis. Cell Host Microbe 19, 720–730 (2016).
Prisant, N. et al. Zika virus in the female genital tract. Lancet Infect. Dis. 16, 1000–1001 (2016).
Foy, B. D. et al. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg. Infect. Dis. 17, 880–882 (2011).
Frank, C. et al. Sexual transmission of Zika virus in Germany, April 2016. Euro Surveill. 21, 30252 (2016).
Deckard, D. T. Male-to-male sexual transmission of Zika virus — Texas, January 2016. MMWR Morb. Mortal. Wkly Rep. 65, 372–374 (2016).
Barthel, A. et al. Breast milk as a possible route of vertical transmission of dengue virus? Clin. Infect. Dis. 57, 415–417 (2013).
Kuhn, S., Twele-Montecinos, L., MacDonald, J., Webster, P. & Law, B. Case report: probable transmission of vaccine strain of yellow fever virus to an infant via breast milk. CMAJ 183, E243–E245 (2011).
Centers for Disease Control and Prevention. Possible West Nile virus transmission to an infant through breast-feeding — Michigan, 2002. MMWR Morb. Mortal. Wkly Rep. 51, 877–878 (2002).
Dupont-Rouzeyrol, M., Biron, A., O'Connor, O., Huguon, E. & Descloux, E. Infectious Zika viral particles in breastmilk. Lancet 387, 1051 (2016).
Musso, D. et al. Detection of Zika virus in saliva. J. Clin. Virol. 68, 53–55 (2015).
De Clercq, E. & Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 29, 695–747 (2016).
Klema, V. J., Padmanabhan, R. & Choi, K. H. Flaviviral replication complex: coordination between RNA synthesis and 5′-RNA capping. Viruses 7, 4640–4656 (2015).
Chambers, T. J., Nestorowicz, A. & Rice, C. M. Mutagenesis of the yellow fever virus NS2B/3 cleavage site: determinants of cleavage site specificity and effects on polyprotein processing and viral replication. J. Virol. 69, 1600–1605 (1995).
Li, J. et al. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J. Biol. Chem. 280, 28766–28774 (2005).
Chappell, K. J., Stoermer, M. J., Fairlie, D. P. & Young, P. R. Insights to substrate binding and processing by West Nile virus NS3 protease through combined modeling, protease mutagenesis, and kinetic studies. J. Biol. Chem. 281, 38448–38458 (2006).
Stoermer, M. J. et al. Potent cationic inhibitors of West Nile virus NS2B/NS3 protease with serum stability, cell permeability and antiviral activity. J. Med. Chem. 51, 5714–5721 (2008).
Behnam, M. A. M., Graf, D., Bartenschlager, R., Zlotos, D. P. & Klein, C. D. Discovery of nanomolar dengue and West Nile virus protease inhibitors containing a 4-benzyloxyphenylglycine residue. J. Med. Chem. 58, 9354–9370 (2015). This paper describes the discovery of DENV-2 and WNV protease inhibitors with nanomolar activity based on 4-hydroxyphenylglycine ethers.
Nitsche, C. et al. Peptide-boronic acid inhibitors of flaviviral proteases: medicinal chemistry and structural biology. J. Med. Chem. 60, 511–516 (2017).
Chu, J. J. et al. Antiviral activities of 15 dengue NS2B-NS3 protease inhibitors using a human cell-based viral quantification assay. Antiviral Res. 118, 68–74 (2015).
Nitsche, C. et al. Thiazolidinone-peptide hybrids as dengue virus protease inhibitors with antiviral activity in cell culture. J. Med. Chem. 56, 8389–8403 (2013).
Peterlin-Mašic˘, L. & Kikelj, D. Arginine mimetics. Tetrahedron 57, 7073–7105 (2001).
Gustafsson, D. et al. The direct thrombin inhibitor melagatran and its oral prodrug H 376/95: intestinal absorption properties, biochemical and pharmacodynamic effects. Thromb. Res. 101, 171–181 (2001).
Bazan, J. F. & Fletterick, R. J. Detection of a trypsin-like serine protease domain in flaviviruses and pestiviruses. Virology 171, 637–639 (1989).
Falgout, B., Pethel, M., Zhang, Y. M. & Lai, C. J. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol. 65, 2467–2475 (1991). This paper describes the NS2B cofactor dependence of the flaviviral NS3 protease and provides the foundation for development of in vitro protease assays.
Chambers, T. J., Grakoui, A. & Rice, C. M. Processing of the yellow fever virus nonstructural polyprotein: a catalytically active NS3 proteinase domain and NS2B are required for cleavages at dibasic sites. J. Virol. 65, 6042–6050 (1991).
Erbel, P. et al. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 13, 372–373 (2006).
Su, X.-C. et al. NMR analysis of the dynamic exchange of the NS2B cofactor between open and closed conformations of the West Nile virus NS2B-NS3 protease. PLoS Negl. Trop. Dis. 3, e561 (2009).
Noble, C. G., Seh, C. C., Chao, A. T. & Shi, P. Y. Ligand-bound structures of the dengue virus protease reveal the active conformation. J. Virol. 86, 438–446 (2012).
Chen, W.-N., Loscha, K. V., Nitsche, C., Graham, B. & Otting, G. The dengue virus NS2B–NS3 protease retains the closed conformation in the complex with BPTI. FEBS Lett. 588, 2206–2211 (2014).
Gupta, G., Lim, L. & Song, J. NMR and MD studies reveal that the isolated dengue NS3 protease is an intrinsically disordered chymotrypsin fold which absolutely requests NS2B for correct folding and functional dynamics. PLoS ONE 10, e0134823 (2015).
Lei, J. et al. Crystal structure of Zika virus NS2B-NS3 protease in complex with a boronate inhibitor. Science 353, 503–505 (2016). This paper provides the first description of the ZIKV protease crystal structure with an inhibitor.
Steuer, C., Heinonen, K. H., Kattner, L. & Klein, C. D. Optimization of assay conditions for dengue virus protease: effect of various polyols and nonionic detergents. J. Biomol. Screen. 14, 1102–1108 (2009).
Nitsche, C. & Klein, C. D. in Antiviral Methods and Protocols (ed. Gong, E. Y.) 221–236 (Humana Press, 2013).
Nitsche, C., Holloway, S., Schirmeister, T. & Klein, C. D. Biochemistry and medicinal chemistry of the dengue virus protease. Chem. Rev. 114, 11348–11381 (2014). This paper describes the medicinal chemistry of the DENV protease, including assay procedures, structural biology and an overview of existing inhibitors.
Nall, T. A. et al. Enzymatic characterization and homology model of a catalytically active recombinant West Nile virus NS3 protease. J. Biol. Chem. 279, 48535–48542 (2004).
Adamek, R. N., Maniquis, R. V., Khakoo, S., Bridges, M. D. & Salzameda, N. T. A FRET-based assay for the discovery of West Nile virus NS2B-NS3 protease inhibitors. Bioorg. Med. Chem. Lett. 23, 4848–4850 (2013).
Yin, Z. et al. Peptide inhibitors of dengue virus NS3 protease. Part 1: warhead. Bioorg. Med. Chem. Lett. 16, 36–39 (2006).
Yin, Z. et al. Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg. Med. Chem. Lett. 16, 40–43 (2006). References 73 and 74 are fundamental studies of the recognition of inhibitors with covalent binding modes by flaviviral proteases.
Knox, J. E. et al. Peptide inhibitors of West Nile NS3 protease: SAR study of tetrapeptide aldehyde inhibitors. J. Med. Chem. 49, 6585–6590 (2006).
Behnam, M. A. M., Nitsche, C., Vechi, S. M. & Klein, C. D. C-Terminal residue optimization and fragment merging: discovery of a potent peptide-hybrid inhibitor of dengue protease. ACS Med. Chem. Lett. 5, 1037–1042 (2014).
Bastos Lima, A. et al. Dual inhibitors of the dengue and West Nile virus NS2B–NS3 proteases: synthesis, biological evaluation and docking studies of novel peptide-hybrids. Bioorg. Med. Chem. 23, 5748–5755 (2015).
Weigel, L. F., Nitsche, C., Graf, D., Bartenschlager, R. & Klein, C. D. Phenylalanine and phenylglycine analogs as arginine mimetics in dengue protease inhibitors. J. Med. Chem. 58, 7719–7733 (2015).
Jia, F., Zou, G., Fan, J. & Yuan, Z. Identification of palmatine as an inhibitor of West Nile virus. Arch. Virol. 155, 1325–1329 (2010).
Yang, C. C. et al. Novel dengue virus-specific NS2B/NS3 protease inhibitor, BP2109, discovered by a high-throughput screening assay. Antimicrob. Agents Chemother. 55, 229–238 (2011).
Yang, C. C. et al. A novel dengue virus inhibitor, BP13944, discovered by high-throughput screening with dengue virus replicon cells selects for resistance in the viral NS2B/NS3 protease. Antimicrob. Agents Chemother. 58, 110–119 (2014).
Bhakat, S. et al. Reaching beyond HIV/HCV: nelfinavir as a potential starting point for broad-spectrum protease inhibitors against dengue and chikungunya virus. RSC Adv. 5, 85938–85949 (2015).
Rice, C. et al. Nucleotide sequence of yellow fever virus: implications for flavivirus gene expression and evolution. Science 229, 726–733 (1985).
Malet, H. et al. Crystal structure of the RNA polymerase domain of the West Nile virus non-structural protein 5. J. Biol. Chem. 282, 10678–10689 (2007).
Yap, T. L. et al. Crystal structure of the dengue virus RNA-dependent RNA polymerase catalytic domain at 1.85-angstrom resolution. J. Virol. 81, 4753–4765 (2007).
Zhao, B. et al. Structure and function of the Zika virus full-length NS5 protein. Nat. Commun. 8, 14762 (2017).
Wang, B. et al. The structure of Zika virus NS5 reveals a conserved domain conformation. Nat. Commun. 8, 14763 (2017).
Godoy, A. S. et al. Crystal structure of Zika virus NS5 RNA-dependent RNA polymerase. Nat. Commun. 8, 14764 (2017).
Malet, H. et al. The flavivirus polymerase as a target for drug discovery. Antiviral Res. 80, 23–35 (2008).
Olsen, D. B. et al. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob. Agents Chemother. 48, 3944–3953 (2004).
Yin, Z. et al. An adenosine nucleoside inhibitor of dengue virus. Proc. Natl Acad. Sci. USA 106, 20435–20439 (2009).
Warren, T. K. et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508, 402–405 (2014).
Vernachio, J. H. et al. INX-08189, a phosphoramidate prodrug of 6-O-methyl-2′-C-methyl guanosine, is a potent inhibitor of hepatitis C virus replication with excellent pharmacokinetic and pharmacodynamic properties. Antimicrob. Agents Chemother. 55, 1843–1851 (2011).
Nguyen, N. M. et al. A randomized, double-blind placebo controlled trial of balapiravir, a polymerase inhibitor, in adult dengue patients. J. Infect. Dis. 207, 1442–1450 (2013).
De Burghgraeve, T. et al. 3′,5′Di-O-trityluridine inhibits in vitro flavivirus replication. Antiviral Res. 98, 242–247 (2013).
Eyer, L. et al. Structure–activity relationships of nucleoside analogues for inhibition of tick-borne encephalitis virus. Antiviral Res. 133, 119–129 (2016).
Zmurko, J. et al. The viral polymerase inhibitor 7-deaza-2′-C-methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl. Trop. Dis. 10, e0004695 (2016).
Eyer, L. et al. Nucleoside inhibitors of Zika virus. J. Infect. Dis. 214, 707–711 (2016).
Schul, W., Liu, W., Xu, H.-Y., Flamand, M. & Vasudevan, S. G. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J. Infect. Dis. 195, 665–674 (2007).
Latour, D. R. et al. Biochemical characterization of the inhibition of the dengue virus RNA polymerase by beta-D-2′-ethynyl-7-deaza-adenosine triphosphate. Antiviral Res. 87, 213–222 (2010).
Chen, Y.-L. et al. Inhibition of dengue virus RNA synthesis by an adenosine nucleoside. Antimicrob. Agents Chemother. 54, 2932–2939 (2010).
Lo, M. K., Shi, P.-Y., Chen, Y.-L., Flint, M. & Spiropoulou, C. F. In vitro antiviral activity of adenosine analog NITD008 against tick-borne flaviviruses. Antiviral Res. 130, 46–49 (2016).
Chen, Y.-L., Yokokawa, F. & Shi, P.-Y. The search for nucleoside/nucleotide analog inhibitors of dengue virus. Antiviral Res. 122, 12–19 (2015).
Julander, J. G. et al. BCX4430, a novel nucleoside analog, effectively treats yellow fever in a hamster model. Antimicrob. Agents Chemother. 58, 6607–6614 (2014). This paper describes the discovery of BCX4430, a nucleoside analogue with broad-spectrum antiviral activity.
Taylor, R. et al. BCX4430 — a broad-spectrum antiviral adenosine nucleoside analog under development for the treatment of Ebola virus disease. J. Infect. Public Health 9, 220–226 (2016).
Golitsina, N. L., Danehy, F. T. Jr, Fellows, R., Cretton-Scott, E. & Standring, D. N. Evaluation of the role of three candidate human kinases in the conversion of the hepatitis C virus inhibitor 2′-C-methyl-cytidine to its 5′-monophosphate metabolite. Antiviral Res. 85, 470–481 (2010).
Berke, J. M. et al. Antiviral activity and mode of action of TMC647078, a novel nucleoside inhibitor of the hepatitis C virus NS5B polymerase. Antimicrob. Agents Chemother. 55, 3812–3820 (2011).
Sofia, M. J. in Antiviral Agents Vol. 67 (ed. de Clercq, E.) 39–73 (Academic Press, 2013).
Sofia, M. J. et al. Discovery of a β-D-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 53, 7202–7218 (2010).
Chen, Y.-L. et al. Activation of peripheral blood mononuclear cells by dengue virus infection depotentiates balapiravir. J. Virol. 88, 1740–1747 (2014).
Gao, W. Y., Shirasaka, T., Johns, D. G., Broder, S. & Mitsuya, H. Differential phosphorylation of azidothymidine, dideoxycytidine, and dideoxyinosine in resting and activated peripheral blood mononuclear cells. J. Clin. Invest. 91, 2326–2333 (1993).
Kohler, J. J. & Lewis, W. A brief overview of mechanisms of mitochondrial toxicity from NRTIs. Environ. Mol. Mutagen. 48, 166–172 (2007).
Coats, S. J. et al. Chutes and ladders in hepatitis C nucleoside drug development. Antiviral Res. 102, 119–147 (2014).
Yeo, K. L. et al. Synergistic suppression of dengue virus replication using a combination of nucleoside analogs and nucleoside synthesis inhibitors. Antimicrob. Agents Chemother. 59, 2086–2093 (2015).
Nelson, J., Roe, K., Orillo, B., Shi, P.-Y. & Verma, S. Combined treatment of adenosine nucleoside inhibitor NITD008 and histone deacetylase inhibitor vorinostat represents an immunotherapy strategy to ameliorate West Nile virus infection. Antiviral Res. 122, 39–45 (2015).
Lim, S. P. et al. Potent allosteric dengue virus NS5 polymerase inhibitors: mechanism of action and resistance profiling. PLoS Pathog. 12, e1005737 (2016). This paper is the first report on allosteric inhibitors of the DENV NS5 polymerase.
Eltahla, A. A., Luciani, F., White, P. A., Lloyd, A. R. & Bull, R. A. Inhibitors of the hepatitis C virus polymerase; mode of action and resistance. Viruses 7, 5206–5224 (2015).
Taguwa, S. et al. Defining Hsp70 subnetworks in dengue virus replication reveals key vulnerability in Flavivirus infection. Cell 163, 1108–1123 (2015).
Smit, J., Moesker, B., Rodenhuis-Zybert, I. & Wilschut, J. Flavivirus cell entry and membrane fusion. Viruses 3, 160–171 (2011).
Zhang, Y. et al. Conformational changes of the flavivirus E glycoprotein. Structure 12, 1607–1618 (2004).
Rey, F. A., Heinz, F. X., Mandl, C., Kunz, C. & Harrison, S. C. The envelope glycoprotein from tick-borne encephalitis virus at 2 Å resolution. Nature 375, 291–298 (1995).
Modis, Y., Ogata, S., Clements, D. & Harrison, S. C. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl Acad. Sci. USA 100, 6986–6991 (2003).
Modis, Y., Ogata, S., Clements, D. & Harrison, S. C. Structure of the dengue virus envelope protein after membrane fusion. Nature 427, 313–319 (2004).
Kanai, R. et al. Crystal structure of West Nile virus envelope glycoprotein reveals viral surface epitopes. J. Virol. 80, 11000–11008 (2006).
Nybakken, G. E., Nelson, C. A., Chen, B. R., Diamond, M. S. & Fremont, D. H. Crystal structure of the West Nile virus envelope glycoprotein. J. Virol. 80, 11467–11474 (2006).
Wang, Q.-Y. et al. A small-molecule dengue virus entry inhibitor. Antimicrob. Agents Chemother. 53, 1823–1831 (2009).
Kampmann, T. et al. In silico screening of small molecule libraries using the dengue virus envelope E protein has identified compounds with antiviral activity against multiple flaviviruses. Antiviral Res. 84, 234–241 (2009).
Martinez, J. P., Sasse, F., Bronstrup, M., Diez, J. & Meyerhans, A. Antiviral drug discovery: broad-spectrum drugs from nature. Nat. Prod. Rep. 32, 29–48 (2015).
Ishag, H. Z. A. et al. Griffithsin inhibits Japanese encephalitis virus infection in vitro and in vivo. Arch. Virol. 158, 349–358 (2013).
Meuleman, P. et al. Griffithsin has antiviral activity against hepatitis C virus. Antimicrob. Agents Chemother. 55, 5159–5167 (2011).
Mori, T. et al. Isolation and characterization of griffithsin, a novel HIV-inactivating protein, from the red alga Griffithsia sp. J. Biol. Chem. 280, 9345–9353 (2005).
Barton, C. et al. Activity of and effect of subcutaneous treatment with the broad-spectrum antiviral lectin Griffithsin in two laboratory rodent models. Antimicrob. Agents Chemother. 58, 120–127 (2014).
Zasloff, M. et al. Squalamine as a broad-spectrum systemic antiviral agent with therapeutic potential. Proc. Natl Acad. Sci. USA 108, 15978–15983 (2011).
Richard, A. S. et al. Virion-associated phosphatidylethanolamine promotes TIM1-mediated infection by Ebola, dengue, and West Nile viruses. Proc. Natl Acad. Sci. USA 112, 14682–14687 (2015).
Jemielity, S. et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 9, e1003232 (2013).
Meertens, L. et al. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 12, 544–557 (2012).
Dokland, T. et al. West Nile virus core protein; tetramer structure and ribbon formation. Structure 12, 1157–1163 (2004).
Ma, L., Jones, C. T., Groesch, T. D., Kuhn, R. J. & Post, C. B. Solution structure of dengue virus capsid protein reveals another fold. Proc. Natl Acad. Sci. USA 101, 3414–3419 (2004).
Samsa, M. M. et al. Dengue virus capsid protein usurps lipid droplets for viral particle formation. PLoS Pathog. 5, e1000632 (2009).
Faustino, A. F. et al. Dengue virus capsid protein interacts specifically with very low-density lipoproteins. Nanomedicine 10, 247–255 (2012).
Byrd, C. M. et al. A novel inhibitor of dengue virus replication that targets the capsid protein. Antimicrob. Agents Chemother. 57, 15–25 (2013).
Scaturro, P. et al. Characterization of the mode of action of a potent dengue virus capsid inhibitor. J. Virol. 88, 11540–11555 (2014).
Zmurko, J., Neyts, J. & Dallmeier, K. Flaviviral NS4b, chameleon and jack-in-the-box roles in viral replication and pathogenesis, and a molecular target for antiviral intervention. Rev. Med. Virol. 25, 205–223 (2015).
Xie, X., Zou, J., Wang, Q.-Y. & Shi, P.-Y. Targeting dengue virus NS4B protein for drug discovery. Antiviral Res. 118, 39–45 (2015).
Miller, S., Sparacio, S. & Bartenschlager, R. Subcellular localization and membrane topology of the dengue virus type 2 non-structural protein 4B. J. Biol. Chem. 281, 8854–8863 (2006).
Youn, S. et al. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J. Virol. 86, 7360–7371 (2012).
Li, X.-D. et al. Genetic interaction between NS4A and NS4B for replication of Japanese encephalitis virus. J. Gen. Virol. 96, 1264–1275 (2015).
Xie, X. et al. Inhibition of dengue virus by targeting viral NS4B protein. J. Virol. 85, 11183–11195 (2011).
Wang, Q.-Y. et al. Discovery of dengue virus NS4B inhibitors. J. Virol. 89, 8233–8244 (2015).
Zou, G. et al. A single-amino acid substitution in West Nile virus 2K peptide between NS4A and NS4B confers resistance to lycorine, a flavivirus inhibitor. Virology 384, 242–252 (2009).
Wang, P. et al. Anti-dengue-virus activity and structure–activity relationship studies of lycorine derivatives. ChemMedChem 9, 1522–1533 (2014).
Luo, D. et al. Insights into RNA unwinding and ATP hydrolysis by the flavivirus NS3 protein. EMBO J. 27, 3209–3219 (2008).
Mastrangelo, E., Bolognesi, M. & Milani, M. Flaviviral helicase: insights into the mechanism of action of a motor protein. Biochem. Biophys. Res. Commun. 417, 84–87 (2012).
Lescar, J. et al. Towards the design of antiviral inhibitors against flaviviruses: the case for the multifunctional NS3 protein from dengue virus as a target. Antiviral Res. 80, 94–101 (2008).
Tian, H. et al. The crystal structure of Zika virus helicase: basis for antiviral drug design. Protein Cell 7, 450–454 (2016).
Byrd, C. M. et al. Novel benzoxazole inhibitor of dengue virus replication that targets the NS3 helicase. Antimicrob. Agents Chemother. 57, 1902–1912 (2013).
Sweeney, N. L. et al. Benzothiazole and pyrrolone flavivirus inhibitors targeting the viral helicase. ACS Infect. Dis. 1, 140–148 (2015).
Mastrangelo, E. et al. Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: new prospects for an old drug. J. Antimicrob. Chemother. 67, 1884–1894 (2012).
Tomlinson, S. M. & Watowich, S. J. Use of parallel validation high-throughput screens to reduce false positives and identify novel dengue NS2B-NS3 protease inhibitors. Antiviral Res. 93, 245–252 (2012).
Egloff, M.-P. et al. Structural and functional analysis of methylation and 5′-RNA sequence requirements of short capped RNAs by the methyltransferase domain of dengue virus NS5. J. Mol. Biol. 372, 723–736 (2007).
Issur, M. et al. The flavivirus NS5 protein is a true RNA guanylyltransferase that catalyzes a two-step reaction to form the RNA cap structure. RNA 15, 2340–2350 (2009).
Egloff, M. P., Benarroch, D., Selisko, B., Romette, J. L. & Canard, B. An RNA cap (nucleoside-2′-O-)-methyltransferase in the flavivirus RNA polymerase NS5: crystal structure and functional characterization. EMBO J. 21, 2757–2768 (2002).
Dong, H. et al. West Nile virus methyltransferase catalyzes two methylations of the viral RNA cap through a substrate-repositioning mechanism. J. Virol. 82, 4295–4307 (2008).
Chung, K. Y. et al. Higher catalytic efficiency of N-7-methylation is responsible for processive N-7 and 2′-O methyltransferase activity in dengue virus. Virology 402, 52–60 (2010).
Dong, H. et al. Structural and functional analyses of a conserved hydrophobic pocket of flavivirus methyltransferase. J. Biol. Chem. 285, 32586–32595 (2010).
Lim, S. P. et al. Small molecule inhibitors that selectively block dengue virus methyltransferase. J. Biol. Chem. 286, 6233–6240 (2011).
Vernekar, S. K. V. et al. 5′-silylated 3′-1,2,3-triazolyl thymidine analogues as inhibitors of West Nile virus and dengue virus. J. Med. Chem. 58, 4016–4028 (2015).
Brecher, M. et al. Identification and characterization of novel broad-spectrum inhibitors of the flavivirus methyltransferase. ACS Infect. Dis. 1, 340–349 (2015).
Marques, R. E. et al. Dengue virus requires the CC-chemokine receptor CCR5 for replication and infection development. Immunology 145, 583–596 (2015).
Larena, M., Regner, M. & Lobigs, M. The chemokine receptor CCR5, a therapeutic target for HIV/AIDS antagonists, is critical for recovery in a mouse model of Japanese encephalitis. PLoS ONE 7, e44834 (2012).
Glass, W. G. et al. Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J. Exp. Med. 202, 1087–1098 (2005).
Courageot, M.-P., Frenkiel, M.-P., Dos Santos, C. D., Deubel, V. & Desprès, P. α-Glucosidase inhibitors reduce dengue virus production by affecting the initial steps of virion morphogenesis in the endoplasmic reticulum. J. Virol. 74, 564–572 (2000).
Zhao, X. et al. Inhibition of endoplasmic reticulum-resident glucosidases impairs severe acute respiratory syndrome coronavirus and human coronavirus NL63 spike protein-mediated entry by altering the glycan processing of angiotensin I-converting enzyme 2. Antimicrob. Agents Chemother. 59, 206–216 (2015).
Stavale, E. J., Vu, H., Sampath, A., Ramstedt, U. & Warfield, K. L. In vivo therapeutic protection against influenza A (H1N1) oseltamivir-sensitive and resistant viruses by the iminosugar uv-4. PLoS ONE 10, e0121662 (2015).
Plummer, E. et al. Dengue virus evolution under a host-targeted antiviral. J. Virol. 89, 5592–5601 (2015).
Fischl, M. A. et al. The safety and efficacy of combination N-butyl-deoxynojirimycin (SC-48334) and zidovudine in patients with HIV-1 Infection and 200–500 CD4 cells/mm. J. Acquir. Immune Defic. Syndr. 7, 139–147 (1994).
Durantel, D. Celgosivir, an alpha-glucosidase I inhibitor for the potential treatment of HCV infection. Curr. Opin. Investig. Drugs 10, 860–870 (2009).
Whitby, K. et al. Castanospermine, a potent inhibitor of dengue virus infection in vitro and in vivo. J. Virol. 79, 8698–8706 (2005).
Rathore, A. P. et al. Celgosivir treatment misfolds dengue virus NS1 protein, induces cellular pro-survival genes and protects against lethal challenge mouse model. Antiviral Res. 92, 453–460 (2011).
Watanabe, S. et al. Optimizing celgosivir therapy in mouse models of dengue virus infection of serotypes 1 and 2: the search for a window for potential therapeutic efficacy. Antiviral Res. 127, 10–19 (2016). This paper describes the development steps leading to the approval of an α-glucosidase inhibitor (celgosivir) for a phase II trial.
Low, J. G. et al. Efficacy and safety of celgosivir in patients with dengue fever (CELADEN): a phase 1b, randomised, double-blind, placebo-controlled, proof-of-concept trial. Lancet Infect. Dis. 14, 706–715 (2014). This article describes the results of a phase I trial of a α-glucosidase inhibitor (celgosivir) in patients with dengue fever.
Wu, S.-F. et al. Antiviral effects of an iminosugar derivative on flavivirus infections. J. Virol. 76, 3596–3604 (2002).
Gu, B. et al. Antiviral profiles of novel iminocyclitol compounds against bovine viral diarrhea virus, West Nile virus, dengue virus and hepatitis B virus. Antivir. Chem. Chemother. 18, 49–59 (2007).
Chang, J. et al. Novel imino sugar derivatives demonstrate potent antiviral activity against flaviviruses. Antimicrob. Agents Chemother. 53, 1501–1508 (2009).
Yu, W. et al. Design, synthesis, and biological evaluation of N-alkylated deoxynojirimycin (DNJ) derivatives for the treatment of dengue virus infection. J. Med. Chem. 55, 6061–6075 (2012).
Perry, S. T. et al. An iminosugar with potent inhibition of dengue virus infection in vivo. Antiviral Res. 98, 35–43 (2013).
Chang, J. et al. Small molecule inhibitors of ER α-glucosidases are active against multiple hemorrhagic fever viruses. Antiviral Res. 98, 432–440 (2013).
Mondelli, M. U. The multifaceted functions of ribavirin: antiviral, immunomodulator, or both? Hepatology 60, 1126–1129 (2014).
Reichard, O., Yun, Z. B., Sönnerborg, A. & Weiland, O. Hepatitis C viral RNA titers in serum prior to, during, and after oral treatment with ribavirin for chronic hepatitis C. J. Med. Virol. 41, 99–102 (1993).
Graci, J. D. & Cameron, C. E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 16, 37–48 (2006).
Crance, J. M., Scaramozzino, N., Jouan, A. & Garin, D. Interferon, ribavirin, 6-azauridine and glycyrrhizin: antiviral compounds active against pathogenic flaviviruses. Antiviral Res. 58, 73–79 (2003).
Kumar, R. et al. Randomized, controlled trial of oral ribavirin for Japanese encephalitis in children in Uttar Pradesh, India. Clin. Infect. Dis. 48, 400–406 (2009).
Chowers, M. Y. et al. Clinical characteristics of the West Nile fever outbreak, Israel, 2000. Emerg. Infect. Dis. 7, 675–678 (2001).
Sbrana, E. et al. Efficacy of post-exposure treatment of yellow fever with ribavirin in a hamster model of the disease. Am. J. Trop. Med. Hyg. 71, 306–312 (2004).
Colombo, G. et al. Brain distribution of ribavirin after intranasal administration. Antiviral Res. 92, 408–414 (2011).
Leyssen, P. et al. A novel model for the study of the therapy of flavivirus infections using the Modoc virus. Virology 279, 27–37 (2001).
Minakawa, N. & Matsuda, A. Mechanism-based design of inosine 5′-monophosphate dehydrogenase inhibitors: synthesis and biological activities of 5-ethynyi-1-β-D-ribofuranosylimidazole-4-carboxamide (EICAR) and its derivatives. Curr. Med. Chem. 6, 615–628 (1999).
Sebastian, L., Madhusudana, S. N., Ravi, V. & Desai, A. Mycophenolic acid inhibits replication of Japanese encephalitis virus. Chemotherapy 57, 56–61 (2011).
Bentley, R. Mycophenolic acid: a one hundred year odyssey from antibiotic to immunosuppressant. Chem. Rev. 100, 3801–3826 (2000).
Qing, M. et al. Characterization of dengue virus resistance to brequinar in cell culture. Antimicrob. Agents Chemother. 54, 3686–3695 (2010).
First, M. R. An update on new immunosuppressive drugs undergoing preclinical and clinical trials: potential applications in organ transplantation. Am. J. Kidney Dis. 29, 303–317 (1997).
Hoffmann, H.-H., Kunz, A., Simon, V. A., Palese, P. & Shaw, M. L. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl Acad. Sci. USA 108, 5777–5782 (2011).
Munier-Lehmann, H. et al. Original 2-(3-alkoxy-1H-pyrazol-1-yl) pyrimidine derivatives as inhibitors of human dihydroorotate dehydrogenase (DHODH). J. Med. Chem. 58, 860–877 (2015).
Ortiz-Riaño, E. et al. Inhibition of arenavirus by A3, a pyrimidine biosynthesis inhibitor. J. Virol. 88, 878–889 (2014).
Qing, M. et al. Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral NS5 protein. Antimicrob. Agents Chemother. 53, 3226–3235 (2009).
Borel, J. F. et al. In vivo pharmacological effects of ciclosporin and some analogues. Adv. Pharmacol. 35, 115–246 (1996).
Hansson, M. J. et al. The nonimmunosuppressive cyclosporin analogs NIM811 and UNIL025 display nanomolar potencies on permeability transition in brain-derived mitochondria. J. Bioenerg. Biomembr. 36, 407–413 (2004).
Flisiak, R. et al. The cyclophilin inhibitor Debio 025 combined with PEG IFNα2a significantly reduces viral load in treatment-naïve hepatitis C patients. Hepatology 49, 1460–1468 (2009).
Munger, J. et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26, 1179–1186 (2008). This paper suggests fatty acid synthesis as a target for antiviral therapy.
Heaton, N. S. et al. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl Acad. Sci. USA 107, 17345–17350 (2010).
Martín-Acebes, M. A., Blázquez, A.-B., De Oya, N. J., Escribano-Romero, E. & Saiz, J.-C. West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS ONE 6, e24970 (2011).
Loftus, T. M. et al. Reduced food intake and body weight in mice treated with fatty acid synthase inhibitors. Science 288, 2379–2381 (2000).
Merino-Ramos, T. et al. Modification of the host cell lipid metabolism induced by hypolipidemic drugs targeting the acetyl coenzyme a carboxylase impairs West Nile Virus replication. Antimicrob. Agents Chemother. 60, 307–315 (2016).
Perera, R. et al. Dengue virus infection perturbs lipid homeostasis in infected mosquito cells. PLoS Pathog. 8, e1002584 (2012).
Martín-Acebes, M. A. et al. Host sphingomyelin increases West Nile virus infection in vivo. J. Lipid Res. 57, 422–432 (2016).
Tani, H. et al. Involvement of ceramide in the propagation of Japanese encephalitis virus. J. Virol. 84, 2798–2807 (2010).
Martín-Acebes, M. A. et al. The composition of West Nile virus lipid envelope unveils a role of sphingolipid metabolism in flavivirus biogenesis. J. Virol. 88, 12041–12054 (2014).
Poh, M. K. et al. U18666A, an intra-cellular cholesterol transport inhibitor, inhibits dengue virus entry and replication. Antiviral Res. 93, 191–198 (2012).
Aktepe, T. E., Pham, H. & Mackenzie, J. M. Differential utilisation of ceramide during replication of the flaviviruses West Nile and dengue virus. Virology 484, 241–250 (2015).
Martinez-Gutierrez, M., Correa-Londoño, L. A., Castellanos, J. E., Gallego-Gómez, J. C. & Osorio, J. E. Lovastatin delays infection and increases survival rates in AG129 mice infected with dengue virus serotype 2. PLoS ONE 9, e87412 (2014). This article reports on the protective action of lovastatin against DENV-2 infection in an animal model.
Raung, S.-L., Chen, S.-Y., Liao, S.-L., Chen, J.-H. & Chen, C.-J. Japanese encephalitis virus infection stimulates Src tyrosine kinase in neuron/glia. Neurosci. Lett. 419, 263–268 (2007).
Bhattacharya, D., Best, S., Perera, R., Kuhn, R. & Striker, R. Protein kinase G phosphorylates mosquito-borne flavivirus NS5. J. Virol. 83, 9195–9205 (2009).
de Wispelaere, M., LaCroix, A. J. & Yang, P. L. The small molecules AZD0530 and dasatinib inhibit dengue virus RNA replication via Fyn kinase. J. Virol. 87, 7367–7381 (2013). This article reports that the DENV-2 NS4B-T108I mutation confers resistance against the host kinase inhibitors AZD0530 and dasatinib, thus demonstrating that antiviral agents targeting host factors are not exempt from resistance development.
Ando, T. et al. Reactivation of resolved infection with the hepatitis B virus immune escape mutant G145R during dasatinib treatment for chronic myeloid leukemia. Int. J. Hematol. 102, 379–382 (2015).
Haile, W. B. et al. The Janus kinase inhibitor ruxolitinib reduces HIV replication in human macrophages and ameliorates HIV encephalitis in a murine model. Neurobiol. Dis. 92, 137–143 (2016).
Sharma, N., Akhade, A. S. & Qadri, A. Src kinases central to T-cell receptor signaling regulate TLR-activated innate immune responses from human T cells. Innate Immun. 22, 238–244 (2016).
Perwitasari, O., Cho, H., Diamond, M. S. & Gale, M. Inhibitor of κB kinase ɛ (IKKɛ), STAT1, and IFIT2 proteins define novel innate immune effector pathway against West Nile virus infection. J. Biol. Chem. 286, 44412–44423 (2011).
Mounce, B. C. et al. Inhibition of polyamine biosynthesis is a broad-spectrum strategy against RNA viruses. J. Virol. 90, 9683–9692 (2016). This work suggests that polyamine biosynthesis inhibitors, such as eflornithine, are broad-spectrum antivirals against RNA viruses.
Mounce, B. C. et al. Interferon-induced spermidine-spermine acetyltransferase and polyamine depletion restrict Zika and chikungunya viruses. Cell Host Microbe 20, 167–177 (2016).
Meyskens, F. L. & Gerner, E. W. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. 5, 945–951 (1999).
Carocci, M. & Yang, P. L. Lactimidomycin is a broad-spectrum inhibitor of dengue and other RNA viruses. Antiviral Res. 128, 57–62 (2016).
Brai, A. et al. Human DDX3 protein is a valuable target to develop broad spectrum antiviral agents. Proc. Natl Acad. Sci. USA 113, 5388–5393 (2016).
Sugawara, K. et al. Lactimidomycin, a new glutarimide group antibiotic. Production, isolation, structure and biological activity. J. Antibiot. (Tokyo) 45, 1433–1441 (1992).
Larsen, B. J. et al. Synthesis and biological evaluation of lactimidomycin and its analogues. Chemistry 21, 19159–19167 (2015).
Bross, P. F. et al. Approval summary for bortezomib for injection in the treatment of multiple myeloma. Clin. Cancer Res. 10, 3954–3964 (2004).
Lai, M. C., Sun, H. S., Wang, S. W. & Tarn, W. Y. DDX3 functions in antiviral innate immunity through translational control of PACT. FEBS J. 283, 88–101 (2016).
Chanan-Khan, A. et al. Analysis of herpes zoster events among bortezomib-treated patients in the phase III APEX study. J. Clin. Oncol. 26, 4784–4790 (2008).
Sugiyama, R. et al. Induction of heat-shock protein 70 by prostaglandin A 1 inhibits HIV-1 Vif-mediated degradation of APOBEC3G. Antiviral Res. 99, 307–311 (2013).
Kumar, M. et al. Reciprocal regulation of human immunodeficiency virus-1 gene expression and replication by heat shock proteins 40 and 70. J. Mol. Biol. 410, 944–958 (2011).
Kim, M. Y. et al. Hsp70 and a novel axis of type I interferon-dependent antiviral immunity in the measles virus-infected brain. J. Virol. 87, 998–1009 (2013).
Chao, C.-H. et al. DDX3, a DEAD box RNA helicase with tumor growth–suppressive property and transcriptional regulation activity of the p21waf1/cip1 promoter, is a candidate tumor suppressor. Cancer Res. 66, 6579–6588 (2006).
Raaben, M. et al. The ubiquitin–proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 84, 7869–7879 (2010).
Raaben, M., Grinwis, G. C., Rottier, P. J. & de Haan, C. A. The proteasome inhibitor velcade enhances rather than reduces disease in mouse hepatitis coronavirus-infected mice. J. Virol. 84, 7880–7885 (2010).
Basler, M., Lauer, C., Beck, U. & Groettrup, M. The proteasome inhibitor bortezomib enhances the susceptibility to viral infection. J. Immunol. 183, 6145–6150 (2009).
Rossignol, J.-F. Thiazolides: a new class of antiviral drugs. Expert Opin. Drug Metab. Toxicol. 5, 667–674 (2009).
Korba, B. E. et al. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antiviral Res. 77, 56–63 (2008).
Rossignol, J.-F. Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antiviral Res. 110, 94–103 (2014).
Shi, Z. et al. Nitazoxanide inhibits the replication of Japanese encephalitis virus in cultured cells and in a mouse model. Virol. J. 11, 10 (2014).
Meneses, M. D. F., Duarte, R. S., Migowski, E. R. & Ferreira, D. F. In vitro study of the effect of nitazoxanide on the replication of Dengue virus and yellow fever virus in Program and Abstracts of 26th International Conference on Antiviral Research (ICAR) 101 (International Society for Antiviral Research, 2013).
Rossignol, J. F., Elfert, A., El–Gohary, Y. & Keeffe, E. B. Improved virologic response in chronic hepatitis C genotype 4 treated with nitazoxanide, peginterferon, and ribavirin. Gastroenterology 136, 856–862 (2009).
Rossignol, J. F. & El-Gohary, Y. M. Nitazoxanide in the treatment of viral gastroenteritis: a randomized double-blind placebo-controlled clinical trial. Aliment. Pharmacol. Ther. 24, 1423–1430 (2006).
Haffizulla, J. et al. Effect of nitazoxanide in adults and adolescents with acute uncomplicated influenza: a double-blind, randomised, placebo-controlled, phase 2b/3 trial. Lancet Infect. Dis. 14, 609–618 (2014).
Santoro, M. G. et al. Thiazolides: a new class of broad-spectrum antiviral drugs targeting virus maturation. Antiviral Res. 74, A31 (2007).
Rossignol, J. F., La Frazia, S., Chiappa, L., Ciucci, A. & Santoro, M. G. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post-translational level. J. Biol. Chem. 284, 29798–29808 (2009).
Elazar, M. et al. The anti-hepatitis C agent nitazoxanide induces phosphorylation of eukaryotic initiation factor 2a via protein kinase activated by double-stranded RNA activation. Gastroenterology 137, 1827–1835 (2009).
Trabattoni, D. et al. Thiazolides elicit anti-viral innate immunity and reduce HIV replication. Sci. Rep. 6, 27148 (2016).
Xu, M. et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 22, 1101–1107 (2016).
Kato, F. et al. Novel antiviral activity of bromocriptine against dengue virus replication. Antiviral Res. 131, 141–147 (2016).
Smith, J. L. et al. Inhibition of dengue virus replication by a class of small-molecule compounds that antagonize dopamine receptor d4 and downstream mitogen-activated protein kinase signaling. J. Virol. 88, 5533–5542 (2014).
Boonyasuppayakorn, S., Reichert, E. D., Manzano, M., Nagarajan, K. & Padmanabhan, R. Amodiaquine, an antimalarial drug, inhibits dengue virus type 2 replication and infectivity. Antiviral Res. 106, 125–134 (2014).
Farias, K. J. S., Machado, P. R. L., Muniz, J. A. P. C., Imbeloni, A. A. & da Fonseca, B. A. L. Antiviral activity of chloroquine against dengue virus type 2 replication in Aotus monkeys. Viral Immunol. 28, 161–169 (2015).
Wang, L.-F. et al. Hydroxychloroquine-inhibited dengue virus is associated with host defense machinery. J. Interferon Cytokine Res. 35, 143–156 (2015).
Tricou, V. et al. A randomized controlled trial of chloroquine for the treatment of dengue in Vietnamese adults. PLoS Negl. Trop. Dis. 4, e785 (2010).
Borges, M. C., Castro, L. A. & da Fonseca, B. A. L. Chloroquine use improves dengue-related symptoms. Mem. Inst. Oswaldo Cruz 108, 596–599 (2013).
Al-Bari, M. A. A. Chloroquine analogues in drug discovery: new directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 70, 1608–1621 (2015).
Cheung, Y. Y., Chen, K. C., Chen, H., Seng, E. K. & Chu, J. J. H. Antiviral activity of lanatoside C against dengue virus infection. Antiviral Res. 111, 93–99 (2014).
Saudi, M. et al. Synthesis and evaluation of imidazole-4,5 -and pyrazine-2,3-dicarboxamides targeting dengue and yellow fever virus. Eur. J. Med. Chem. 87, 529–539 (2014).
Saudi, M. et al. Synthetic strategy and antiviral evaluation of diamide containing heterocycles targeting dengue and yellow fever virus. Eur. J. Med. Chem. 121, 158–168 (2016).
Pattabhi, S. et al. Targeting innate immunity for antiviral therapy through small molecule agonists of the RLR pathway. J. Virol. 90, 2372–2387 (2016).
Green, R. R. et al. Transcriptional analysis of antiviral small molecule therapeutics as agonists of the RLR pathway. Genom. Data 7, 290–292 (2016).
Tay, M. Y. F. et al. Nuclear localization of dengue virus (DENV) 1–4 non-structural protein 5; protection against all 4 DENV serotypes by the inhibitor Ivermectin. Antiviral Res. 99, 301–306 (2013). This article describes how ivermectin blocks the nuclear transport of the NS5 protein of DENV-1–4.
Forwood, J. K. et al. The 37-amino-acid interdomain of dengue virus NS5 protein contains a functional NLS and inhibitory CK2 site. Biochem. Biophys. Res. Commun. 257, 731–737 (1999).
Rawlinson, S. M., Pryor, M. J., Wright, P. J. & Jans, D. A. CRM1-mediated nuclear export of dengue virus RNA polymerase NS5 modulates interleukin-8 induction and virus production. J. Biol. Chem. 284, 15589–15597 (2009).
Fraser, J. E. et al. A nuclear transport inhibitor that modulates the unfolded protein response and provides in vivo protection against lethal dengue virus infection. J. Infect. Dis. 210, 1780–1791 (2014). This article reports that fenretinide protects AG129 mice against lethal DENV infection.
Carocci, M. et al. The bioactive lipid 4-hydroxyphenyl retinamide inhibits flavivirus replication. Antimicrob. Agents Chemother. 59, 85–95 (2015). This work reports on the in vivo activity of fenretinide against DENV infection.
Fraser, J., Wang, C., Chan, K., Vasudevan, S. & Jans, D. Novel dengue virus inhibitor 4-HPR activates ATF4 independent of protein kinase R–like endoplasmic reticulum kinase and elevates levels of eIF2α phosphorylation in virus infected cells. Antiviral Res. 130, 1–6 (2016).
Deas, T. S. et al. Inhibition of flavivirus infections by antisense oligomers specifically suppressing viral translation and RNA replication. J. Virol. 79, 4599–4609 (2005).
Stein, D. A. et al. Treatment of AG129 mice with antisense morpholino oligomers increases survival time following challenge with dengue 2 virus. J. Antimicrob. Chemother. 62, 555–565 (2008).
Moulton, H. M., Nelson, M. H., Hatlevig, S. A., Reddy, M. T. & Iversen, P. L. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjug. Chem. 15, 290–299 (2004).
Swenson, D. L. et al. Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob. Agents Chemother. 53, 2089–2099 (2009).
Cirak, S. et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 378, 595–605 (2011).
Heald, A. E. et al. AVI-7288 for Marburg virus in nonhuman primates and humans. N. Engl. J. Med. 373, 339–348 (2015).
Warren, T. K. et al. Delayed time-to-treatment of an antisense morpholino oligomer is effective against lethal marburg virus infection in cynomolgus macaques. PLoS Negl. Trop. Dis. 10, e0004456 (2016).
Deas, T. S. et al. In vitro resistance selection and in vivo efficacy of morpholino oligomers against West Nile virus. Antimicrob. Agents Chemother. 51, 2470–2482 (2007).
Dean, C. H. et al. Cutaneous delivery of a live, attenuated chimeric flavivirus vaccines against Japanese encephalitis (ChimeriVaxTM-JE) in non-human primates. Hum. Vaccin. 1, 106–111 (2005).
Widman, D. G. et al. Evaluation of RepliVAX WN, a single-cycle flavivirus vaccine, in a non-human primate model of West Nile virus infection. Am. J. Trop. Med. Hyg. 82, 1160–1167 (2010).
Ito, M., Mukai, R.-Z., Takasaki, T., Kotaki, A. & Kurane, I. Antibody-dependent enhancement of dengue virus infection in vitro by undiluted sera from monkeys infected with heterotypic dengue virus. Arch. Virol. 155, 1617–1624 (2010).
Lee, Y.-R. et al. Suckling mice were used to detect infectious dengue-2 viruses by intracerebral injection of the full-length RNA transcript. Intervirology 48, 161–166 (2005).
Shresta, S., Sharar, K. L., Prigozhin, D. M., Beatty, P. R. & Harris, E. Murine model for dengue virus-induced lethal disease with increased vascular permeability. J. Virol. 80, 10208–10217 (2006). This article describes the first experimental murine model of vascular leakage in DENV infection.
Weiskopf, D. et al. Insights into HLA-restricted T cell responses in a novel mouse model of dengue virus infection point toward new implications for vaccine design. J. Immunol. 187, 4268–4279 (2011).
Thibodeaux, B. A. et al. A small animal peripheral challenge model of yellow fever using interferon-receptor deficient mice and the 17D-204 vaccine strain. Vaccine 30, 3180–3187 (2012).
Thibodeaux, B. A. et al. A humanized IgG but not IgM antibody is effective in prophylaxis and therapy of yellow fever infection in an AG129/17D-204 peripheral challenge mouse model. Antiviral Res. 94, 1–8 (2012).
Calvert, A. E., Dixon, K. L., Delorey, M. J., Blair, C. D. & Roehrig, J. T. Development of a small animal peripheral challenge model of Japanese encephalitis virus using interferon deficient AG129 mice and the SA14-14-2 vaccine virus strain. Vaccine 32, 258–264 (2014).
Mota, J. & Rico-Hesse, R. Humanized mice show clinical signs of dengue fever according to infecting virus genotype. J. Virol. 83, 8638–8645 (2009).
Mota, J. & Rico-Hesse, R. Dengue virus tropism in humanized mice recapitulates human dengue fever. PLoS ONE 6, e20762 (2011).
Frias-Staheli, N. et al. Utility of humanized BLT mice for analysis of dengue virus infection and antiviral drug testing. J. Virol. 88, 2205–2218 (2014).
Holbrook, M. R. et al. An animal model for the tickborne flavivirus — Omsk hemorrhagic fever virus. J. Infect. Dis. 191, 100–108 (2005).
Tigabu, B., Juelich, T., Bertrand, J. & Holbrook, M. R. Clinical evaluation of highly pathogenic tick-borne flavivirus infection in the mouse model. J. Med. Virol. 81, 1261–1269 (2009).
Xiao, S.-Y., Guzman, H., Zhang, H., Da Rosa, A. P. T. & Tesh, R. B. West Nile virus infection in the golden hamster (Mesocricetus auratus): a model for West Nile encephalitis. Emerg. Infect. Dis. 7, 714 (2001).
Tesh, R. B. et al. Experimental yellow fever virus infection in the golden hamster (Mesocricetus auratus). I. Virologic, biochemical, and immunologic studies. J. Infect. Dis. 183, 1431–1436 (2001).
Tesh, R. B. et al. Persistent West Nile virus infection in the golden hamster: studies on its mechanism and possible implications for other flavivirus infections. J. Infect. Dis. 192, 287–295 (2005). This article describes the first small-animal model showing long-term persistence of WNV infection, which is useful for the study of antivirals active against persistent flaviviruses.
Ferguson, N. M. et al. Benefits and risks of the Sanofi–Pasteur dengue vaccine: modeling optimal deployment. Science 353, 1033–1036 (2016).
López-Gatell, H., Alpuche-Aranda, C. M., Santos-Preciadob, J. I. & Hernández-Ávilaa, M. Dengue vaccine: local decisions, global consequences. Bull. World Health Organ. 94, 850 (2016).
Davidson, M. M., Williams, H. & Macleod, J. A. Louping ill in man: a forgotten disease. J. Infect. 23, 241–249 (1991).
Holbrook, M. R. Kyasanur forest disease. Antiviral Res. 96, 353–362 (2012).
Dejnirattisai, W. et al. Cross-reacting antibodies enhance dengue virus infection in humans. Science 328, 745–748 (2010). This seminal study demonstrates the antibody-dependent enhancement of DENV infection in vivo.
Barba-Spaeth, G. et al. Structural basis of potent Zika–dengue virus antibody cross-neutralization. Nature 536, 48–53 (2016).
Dejnirattisai, W. et al. A new class of highly potent, broadly neutralizing antibodies isolated from viremic patients infected with dengue virus. Nat. Immunol. 16, 170–177 (2015).
Rouvinski, A. et al. Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature 520, 109–113 (2015).
Orlinger, K. K. et al. A tick-borne encephalitis virus vaccine based on the European prototype strain induces broadly reactive cross-neutralizing antibodies in humans. J. Infect. Dis. 203, 1556–1564 (2011).
Firbas, C. & Jilma, B. Product review on the JE vaccine IXIARO. Hum. Vaccin. Immunother. 11, 411–420 (2015).
Sabchareon, A. et al. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: a randomised, controlled phase 2b trial. Lancet 380, 1559–1567 (2012).
Ishikawa, T., Yamanaka, A. & Konishi, E. A review of successful flavivirus vaccines and the problems with those flaviviruses for which vaccines are not yet available. Vaccine 32, 1326–1337 (2014).
Dejnirattisai, W. et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with Zika virus. Nat. Immunol. 17, 1102–1108 (2016). This seminal ( in vitro ) study implicates possible enhancement of ZIKV in individuals previously exposed to DENV.
Plentz, A., Jilg, W., Schwarz, T. F., Kuhr, H. B. & Zent, O. Long-term persistence of tick-borne encephalitis antibodies in adults 5 years after booster vaccination with Encepur® Adults. Vaccine 27, 853–856 (2009).
Monath, T. P. 17D yellow fever virus vaccine. Am. J. Trop. Med. Hyg. 89, 1225 (2013).
Green, N., Ott, R. D., Isaacs, R. J. & Fang, H. Cell-based assays to identify inhibitors of viral disease. Expert Opin. Drug Discov. 3, 671–676 (2008).
Payne, A. F., Binduga-Gajewska, I., Kauffman, E. B. & Kramer, L. D. Quantitation of flaviviruses by fluorescent focus assay. J. Virol. Methods 134, 183–189 (2006).
Vasilakis, N. et al. Transfection-independent production of alphavirus replicon particles based on poxvirus expression vectors. Nat. Biotechnol. 21, 932–935 (2003).
Drake, J. W. & Holland, J. J. Mutation rates among RNA viruses. Proc. Natl Acad. Sci. USA 96, 13910–13913 (1999).
Klimstra, W. B., Ryman, K. D. & Johnston, R. E. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 72, 7357–7366 (1998).
Heil, M. L., Albee, A., Strauss, J. H. & Kuhn, R. J. An amino acid substitution in the coding region of the E2 glycoprotein adapts Ross River virus to utilize heparan sulfate as an attachment moiety. J. Virol. 75, 6303–6309 (2001).
Bernard, K. A., Klimstra, W. B. & Johnston, R. E. Mutations in the E2 glycoprotein of Venezuelan equine encephalitis virus confer heparan sulfate interaction, low morbidity, and rapid clearance from blood of mice. Virology 276, 93–103 (2000).
Smit, J. M. et al. Adaptation of alphaviruses to heparan sulfate: interaction of Sindbis and Semliki forest viruses with liposomes containing lipid-conjugated heparin. J. Virol. 76, 10128–10137 (2002).
Lee, E., Pavy, M., Young, N., Freeman, C. & Lobigs, M. Antiviral effect of the heparan sulfate mimetic, PI-88, against dengue and encephalitic flaviviruses. Antiviral Res. 69, 31–38 (2006).
Hidari, K. I. et al. Structure and anti-dengue virus activity of sulfated polysaccharide from a marine alga. Biochem. Biophys. Res. Commun. 376, 91–95 (2008).
Lim, S. P., Noble, C. G. & Shi, P.-Y. The dengue virus NS5 protein as a target for drug discovery. Antiviral Res. 119, 57–67 (2015).
Poh, M. K. et al. A small molecule fusion inhibitor of dengue virus. Antiviral Res. 84, 260–266 (2009).
Acknowledgements
The authors are grateful for the highly valuable comments received from the reviewers of this work, who have chosen to remain anonymous. The authors also thank D. Graf and R. Bartenschlager for useful discussions on flaviviral biology and cell-based assays. V.B. appreciates funding by the Alexander von Humboldt Foundation. M.A.M.B. appreciates financial support from the German Academic Exchange Service (DAAD). N.V. receives funding from the National Institute of Allergy and Infectious Diseases, US National Institutes of Health (grants R24AI120942 and 1U01AI115577-01). C.D.K. acknowledges support by the Deutsche Forschungsgemeinschaft (grant KL 1356/3).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
N.V. is a consultant for BioProtection Systems/NewLink Genetics. The other authors declare no conflicting interests.
Related links
FURTHER INFORMATION
Supplementary information
Supplementary information
Supplementary Figures Tables and Boxes (PDF 18174 kb)
Glossary
- Microcephaly
-
A developmental defect that is characterized by a considerably reduced head circumference and brain volume of the affected person relative to the normal population. Microcephaly is frequently associated with severe mental retardation and can be lethal in extreme cases.
- Guillain–Barré syndrome
-
An autoimmune disorder affecting the peripheral nervous system. Initial symptoms are weakness and numbness in the extremities, which eventually develop into paralysis.
- Viral envelope (E) glycoprotein
-
A structural protein of flaviviruses that is embedded in the host-cell-derived membrane of mature virions. It encompasses the viral capsid and helps viruses escape the host immune system. E glycoprotein also serves to identify and bind to host cell receptors on target cells, which initiates the viral entry process.
- Pre-membrane (prM) glycoprotein
-
A component of the structural proteins of flaviviruses. prM is a scaffold protein that ensures proper folding of the viral envelope protein and prevents virion maturation before release from the cell.
- Haematogenous transport
-
The spread of viruses via circulating blood.
- Axonal transport
-
The spread of viruses via existing neuronal transport pathways.
- Haemorrhagic fever
-
A general term for a severe illness associated with bleeding and caused by infection of four families of viruses (Arenaviridae, Bunyaviridae, Filoviridae and Flaviviridae).
- Allosteric ligands
-
Molecules that bind to a protein at a site other than the orthosteric site, do not compete directly with orthosteric ligands or substrates but influence their binding behaviour by eliciting-conformational changes in the target protein.
- Orthosteric binding sites
-
Binding sites of the natural ligand or substrate.
- Prime site
-
Subsite within the protease-binding cavity located towards the carboxyl terminus of the substrate as seen from the cleavage site.
- Innate immunity
-
Nonspecific immunity consisting of cells and mechanisms that are the initial host defence against infection by other organisms.
Rights and permissions
About this article

Cite this article
Boldescu, V., Behnam, M., Vasilakis, N. et al. Broad-spectrum agents for flaviviral infections: dengue, Zika and beyond. Nat Rev Drug Discov 16, 565–586 (2017). https://doi.org/10.1038/nrd.2017.33
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd.2017.33
This article is cited by
-
Development of flavivirus subviral particles with low cross-reactivity by mutations of a distinct antigenic domain
Applied Microbiology and Biotechnology (2023)
-
Searching for plant-derived antivirals against dengue virus and Zika virus
Virology Journal (2022)
-
Antiviral drug research for Japanese encephalitis: an updated review
Pharmacological Reports (2022)
-
Natural products and their derivatives as anti-flavivirus drug candidates
Medicinal Chemistry Research (2021)
-
[1,2,4]Triazolo[1,5-a]pyrimidine derivative (Mol-5) is a new NS5-RdRp inhibitor of DENV2 proliferation and DENV2-induced inflammation
Acta Pharmacologica Sinica (2020)
