
Key Points
-
The filoviruses, Ebola virus and Marburg virus, cause lethal haemorrhagic fever in humans and non-human primates (NHPs), and there are currently no licensed vaccines or treatments available for human use.
-
Death occurs in 90% or more of patients infected with Zaire ebolavirus (EBOV) if the viral load exceeds 6 log genome copies per millilitre in untreated patients, so antiviral interventions must be initiated early in the disease course.
-
Among the post-exposure treatments and therapies developed to date against filoviruses, the monoclonal antibody-based approaches targeting filovirus glycoproteins appear to demonstrate the highest level of protective efficacy.
-
Antisense approaches including small interfering RNAs and phosphorodiamidate morpholino oligomers have shown efficacy in NHP models of filovirus infection, and the modular nature of these technologies offers flexibility should mutations in a filovirus occur or a new filovirus be identified.
-
Although preclinical data are sparse, the broad-spectrum antivirals favipiravir and GS-5734 have a number of advantages as interventions for filovirus infections, and their efficacy is not likely to be affected by mutations in a filovirus or the emergence of a previously unknown filovirus.
-
The importance of advanced supportive care, including electrolyte balance, ventilation and dialysis, was likely pivotal in the success of treating EBOV-infected individuals in developed countries during the 2013–2016 EBOV epidemic.
Abstract
The filoviruses — Ebola virus and Marburg virus — cause lethal haemorrhagic fever in humans and non-human primates (NHPs). Filoviruses present a global health threat both as naturally acquired diseases and as potential agents of bioterrorism. In the recent 2013–2016 outbreak of Ebola virus, the most promising therapies for post-exposure use with demonstrated efficacy in the gold-standard NHP models of filovirus disease were unable to show statistically significant protection in patients infected with Ebola virus. This Review briefly discusses these failures and what has been learned from these experiences, and summarizes the current status of post-exposure medical countermeasures in development, including antibodies, small interfering RNA and small molecules. We outline how our current knowledge could be applied to the identification of novel interventions and ways to use interventions more effectively.
Similar content being viewed by others

Introduction
Among viruses that cause haemorrhagic fever, members of the two genera of the family Filoviridae, Marburgvirus and Ebolavirus, are the most feared because of their dramatic clinical presentation, unusually high case fatality rates (up to 90%) and uncertain natural history. Bats are thought to have an important role in the maintenance of filoviruses in nature, but which bat species carry filoviruses and whether other species are involved are unknown1,2. Historical filovirus outbreaks originated in Central Africa and ranged in size from a few to more than 400 cases; these outbreaks were reasonably well controlled by quarantine and contact tracing. However, this changed in December 2013, when an unprecedented outbreak caused by the Zaire species of Ebolavirus began in the West African countries of Guinea, Liberia and Sierra Leone. The epidemic continued unabated for over 2 years, finally ending in January 2016 with 28,616 cases and 11,301 deaths3. In addition to concerns of natural outbreaks in regions of Central and West Africa, filoviruses are known to have been a focus of former biological weapons programmes and have the potential for deliberate misuse4. Currently, there are no preventive filovirus vaccines or post-exposure treatments approved for human use. Work with infectious filoviruses must be conducted under biosafety level 4 (BSL-4) containment, which has further slowed the development of effective interventions. For these reasons, members of the genera Ebolavirus and Marburgvirus have recently been included as two of only eleven human pathogens and two of only four viruses named on the US Department of Health and Human Services Tier 1 list of Category A select agents, defined as affecting humans and having the “potential to pose a severe threat to public health and safety” (Ref. 5). In addition to being important human pathogens, filoviruses have devastated great ape populations in the Congo basin, further harming an already endangered species6,7.
Although remarkable progress has been made over the past decade in developing and advancing vaccines that prevent filovirus infection, here we focus on interventions that could be deployed after exposure to a filovirus. The abundant in vitro data and data using rodent models to evaluate the efficacy of anti-filovirus interventions have not accurately predicted results in the more robust non-human primate (NHP) models8,9 (Box 1); therefore, this Review focuses on studies of post-exposure treatments and therapies performed in NHPs and on recent data obtained in human patients during the 2013–2016 Zaire ebolavirus epidemic. We also outline topics including phylogeny, clinical manifestations, preventive vaccines (Box 2), animal models, the US Food and Drug Administration (FDA) Animal Rule (Box 3), pathology, viral and host targets and cell-based antiviral screening assays (Box 4).
Infections caused by filoviruses
Filovirus taxonomy
Filovirus taxonomy has changed many times since the discovery of Marburg virus in 1967 and of two different species of Ebola virus simultaneously in 1976. This has complicated nomenclature and made it highly confusing and controversial even for those in the filovirus community. Currently, the family Filoviridae contains two genera of disease-causing viruses, Marburgvirus and Ebolavirus. The Marburgvirus genus contains a single species, Marburg marburgvirus, which contains two members, Marburg virus (MARV) and Ravn virus (RAVV). The Ebolavirus genus consists of five distinct species: Bundibugyo ebolavirus (BDBV), Reston ebolavirus (RESTV), Sudan ebolavirus (SUDV), Tai Forest ebolavirus (TAFV; previously termed 'Côte d'Ivoire ebolavirus' but more commonly known as 'Ivory Coast ebolavirus') and Zaire ebolavirus (EBOV). MARV, RAVV, BDBV, SUDV and EBOV are important human pathogens, with case fatality rates often up to 90% for MARV, RAVV and EBOV, and around 55% for SUDV8,10. On the basis of two small outbreaks in 2007 and 2012, BDBV seems to be the least pathogenic, with a case fatality rate of about 40–48%11,12. In 1994, TAFV was associated with a number of deaths in chimpanzees and a single symptomatic but non-lethal human infection in the Republic of Côte d'Ivoire13,14. RESTV is lethal for macaques but is not thought to cause disease in humans15. An outbreak of RESTV was reported in pigs in the Philippines in 2008; however, it remains uncertain whether the disease observed in the pigs was caused by RESTV or other agents reported to have co-infected the animals16.
Clinical manifestations
Clinical and laboratory signs of disease caused by filovirus infection are nonspecific and usually associated with an incubation period of 2–21 days (mean 4–10 days)8,17. Illness begins with an abrupt onset of fever, asthenia, myalgia and arthralgia, and can include a number of other nonspecific indications8,17. Hypovolaemic shock and multiple organ failure are associated with severe and fatal cases. Clinical pathological observations include leukopenia and lymphocytopenia with increased circulating numbers of neutrophils, as well as thrombocytopenia. Prolonged blood clotting times and increased circulating levels of D-dimers, tissue factor, thrombomodulin and von Willebrand factor have been reported with filovirus infections, and substantially abnormal values for some of these factors have been associated with a fatal outcome18,19,20,21,22,23. Complications collectively referred to as 'post-Ebola syndrome', including musculoskeletal pain, headache and ocular problems, have been noted in a number of survivors of filovirus infection24,25,26,27,28,29. In rare cases, encephalitis has also been noted30,31,32,33. Filoviruses have also been detected by PCR and/or virus isolation in the breast milk34,35 and semen36,37,38,39 of survivors. The persistence of filoviruses in semen is particularly concerning, with detection in one individual occurring 565 days after discharge from an EBOV treatment centre in Liberia38.
Pathology and tissue tropism
Humans and NHPs are naturally exposed to filoviruses through mucosal surfaces or small abrasions and/or breaks in the skin8,17. Filoviruses have a broad cell tropism and productively replicate in numerous cell types. Electron microscopic examination of tissues from experimentally infected NHPs and from fatal human cases has demonstrated that monocytes, macrophages, dendritic cells, hepatocytes, adrenal cortical cells, endothelial cells, fibroblasts and several types of epithelial cell can all support replication of filoviruses40,41,42,43,44,45,46,47,48,49,50,51,52,53. Systematic studies in experimentally infected NHPs suggest that monocytes, macrophages and dendritic cells are the early replication sites for EBOV and MARV41,48,51. Filovirus infection of mononuclear phagocytes is thought to trigger a series of events that includes the production and release of the procoagulant protein tissue factor54,55 and an assortment of pro-inflammatory cytokines, chemokines and free radical species in NHPs and humans48,51,55,56,57,58,59,60,61,62,63,64,65,66,67,68. This dysregulated host response likely plays a greater role in the development of the observed pathology than any structural damage caused by viral replication in host cells and/or tissues.
During filovirus infection of humans and NHPs, lymphoid depletion, necrosis and apoptosis are frequently seen in the spleen, thymus and lymph nodes19,40,41,42,45,46,47,48,51,52,53,64,69,70,71. Although the lymphoid tissues are the primary sites of filovirus replication, inflammatory responses by infiltrating immune cells in these or other infected tissues are typically sparse. Interestingly, lymphocytes themselves do not support the production of progeny virus despite the large die-off of lymphocytes during the course of infection72.
Coagulation disorders are perhaps the most common feature of filovirus infection, although loss of blood is not usually the cause of death. Results from many studies have shown biochemical and histological evidence of disseminated intravascular coagulation in both experimentally infected NHPs and humans19,20,22,23,40,44,45,46,49,50,51,53,54,70,73,74,75,76,77,78,79. The mechanisms that cause these coagulation disorders are not fully understood, but the expression or release of tissue factor from filovirus-infected monocytes and macrophages seems to play a role54,55. However, the coagulation irregularities noted during filovirus haemorrhagic fever could be caused by other factors, particularly at the end stage of disease.
Targeting biology and replication
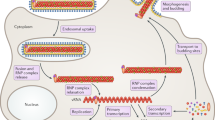
The development of medical countermeasures to combat filovirus infection has been built on the rapid advancement in the understanding of viral entry, replication and egress. The filovirus replication cycle is outlined in Fig. 1, and many of the pharmacological targets focus on the disruption of this cycle.

Infectious viral particles bind to host attachment factors and signaling molecules, including T cell immunoglobulin mucin receptor 1 (TIM1; also known as hepatitis A virus cellular receptor 1 (HAVCR1)), dendritic cell-specific intercellular adhesion molecule 3-grabbing non-integrin (DC-SIGN) and Toll-like receptor 4 (TLR4). Viral particles are internalized largely via macropinocytosis and subsequently bind to Niemann–Pick C1 protein (NPC1) to initiate virus–host membrane fusion at the endosomal membrane. The replication complex is composed of the nucleoprotein (NP), viral protein 35 (VP35), VP30, and the viral RNA-dependent RNA polymerase L (L), which initiate viral genome transcription and replication in viral factories known as inclusion bodies. Concurrently, several filoviral proteins (VP24, VP35 and VP40) block viral detection by direct interaction with host proteins involved in the antiviral response. A phenomenon unique to Ebolaviruses involves a transcriptional editing event that ultimately results in the production of several types of viral glycoproteins (GPs), which include both secreted dimeric forms (soluble GP (sGP) and small soluble GP (ssGP)) and the structural trimeric form GP that is subsequently cleaved in the Golgi compartment by host proteases into GP1 and GP2 and presented at the cell surface. All filovirus glycoproteins follow a classical secretory pathway for secretion outside of the infected cell; in addition, infected cells have been shown to increase the expression of a surface enzyme capable of cleaving and thus releasing a portion of the surface-bound structural form GP (known as shed GP) before it is incorporated into virions. These scenarios are thought to contribute to pathogenesis by providing an immunological decoy of sorts that is capable of consuming circulating filovirus GP-specific antibodies, as antibodies to GP isolated from human survivors of Ebolavirus infection also bind the soluble forms99. The production of the different glycoproteins from the same gene is dependent on the insertion of one or two additional uridine (U) residues in the GP gene editing site such that the non-edited mRNA expresses sGP and the 8Uand 9U mRNA species produce the structural form GP and soluble form ssGP, respectively. Marburgviruses do not contain this editing site. Interestingly, excessive in vitro passage of Ebolaviruses in Vero cells (interferon-deficient) favours the generation of genomes with an 8U genome, presumably because of a lack of host innate immune pressures. Of note, viral seed stocks with a predominance of 8U genomes have been shown to have a slower disease course in non-human primates and a shift from uniform lethality, which may have direct implications for the evaluation of medical countermeasures300. The viral genome structure as shown in the inset. dsRNA, double-stranded RNA; EBOV, Zaire ebolavirus; ER, endoplasmic reticulum; IFNAR1, IFNα/β receptor 1; JAK, Janus kinase; KPNA1, karyopherin subunit-α1 (also known as importin subunit α5); MARV, Marburg virus; MDA5, melanoma differentiation-associated protein 5; NP, nucleoprotein; PACT, protein activator of the interferon-induced protein kinase (also known as PRKRA); PKR, protein kinase R (also known as EIF2AK2); RIG-I, retinoic acid-inducible gene I protein; siRNA, small interfering RNA; STAT, signal transducer and activator of transcription; TACE, TNFα-converting enzyme (also known as ADAM17).
Entry
The filovirus glycoprotein (GP) is largely accepted to be the only viral protein on the viral surface and therefore the most immunologically available viral target. GP is a type I transmembrane protein. The binding of filoviruses to host cells has been associated with several attachment factors80,81,82,83,84,85, but entry and deposition of replication machinery for both Ebolaviruses and Marburgviruses have been directly linked to intravesicular cleavage of GP by host proteases, such as cathepsins86, and subsequent fusion of viral GP with the host protein Niemann–Pick C1 (NPC1)87,88.
Replication
Replication is immediately initiated upon release of the viral nucleocapsid, thereby freeing the encapsidated viral genome and replication machinery for subsequent cytoplasmic assembly of replication complexes for production of progeny viral genomes and proteins. At approximately 19 kb in length, filovirus genomes contain seven genes that encode an equal number of structural proteins, including nucleoprotein (NP), viral protein 35 (VP35), VP40, GP, VP30, VP24 and the RNA-dependent RNA polymerase L (also known as large structural protein)8. Filovirus RNA synthesis is reliant on NP, VP35, VP30 and L working in a complex89,90; however, during the process, VP24 has been shown to be closely associated with intracellular centres of virus replication, known as inclusion bodies, and has been shown to be required for nucleocapsid and genome packaging91,92,93. VP40 directly interacts with cellular endosomal trafficking elements and is key for viral budding94. GP requires post-translational modifications in the endoplasmic reticulum and Golgi processes and hence follows a different trafficking pathway to the plasma membrane than do the other viral proteins95,96,97. Unique to members of the Ebolavirus genus, an artefact of transcriptional editing of the GP gene allows for the production of several nonstructural GP derivatives, each of which is thought to contribute to pathogenesis in a different way, some of which may have direct relevance to interventions specific to Ebolavirus infections97,98,99,100,101.
Immune evasion
Fatal filovirus infections are accompanied by extremely high infectious viral burdens in the host, which contribute to rapid pathogenesis and possibly to communicability. This unbridled replication is thought to stem from the multiple means by which filovirus proteins can subvert the host immune system. In addition to serving structural and replication roles, both Ebolavirus and Marburgvirus proteins exploit specific intracellular and/or extracellular immune-mediated antiviral pathways8,102,103.
One of the best studied and likely most versatile of these proteins is VP35, which suppresses the cytosolic detection of viral products, thus blunting traditional intracellular antiviral responses. VP35 binds to double-stranded RNA, a by-product of viral replication, which serves two purposes: avoidance of RNA-dependent antiviral sensing pathways — including retinoic acid-inducible gene I protein (RIG-I; also known as probable ATP-dependent RNA helicase DDX58), melanoma differentiation-associated protein 5 (MDA5; also known as interferon-induced helicase C domain-containing protein 1 (IFIH1)) and protein kinase R (PKR; also known as interferon-induced, double-stranded RNA-activated protein kinase (EIF2AK2)) — and protection against endogenous host RNAses104,105,106,107. This protein has also been shown to interrupt interferon regulatory factor 3 (IRF3) signalling directly and IRF7 signalling indirectly, thereby decreasing the expression of interferons. Further, direct binding of VP35 to protein activator of the interferon-induced protein kinase (PACT; also known as interferon-inducible double-stranded RNA-dependent protein kinase activator A (PRKRA)), a multifunctional cellular protein important for activation of PKR signalling, abrogates this antiviral response. Some of the first cells to be infected are immune surveillance cells, including dendritic cells, monocytes and macrophages, in which the infection-mediated blockade of immune activation is particularly powerful as it reduces the potential for these cells to become activated early in the course of infection. The contribution of VP35 to immune evasion is best exhibited in vivo: Ebolavirus with mutant VP35 is non-lethal in animal models including mice108 and guinea pigs109.
Both Ebolaviruses and Marburgviruses abrogate the response to exogenous interferon by directly altering the downstream signalling cascade. EBOV VP24 disrupts signal transducer and activator of transcription 1 (STAT1) translocation to the nucleus to prevent transcription of interferon response genes110,111. MARV VP40 directly blocks phosphorylation of Janus kinase 1 (JAK1), STAT1 and STAT2 (Ref. 112). Despite these means to blunt the response to interferon, infected cells still enact antiviral responses, albeit at a lower level.
Expression of the cellular protein tetherin (also known as bone marrow stromal antigen 2) at the plasma membrane serves to 'tether' the budding virion to the infected cell, thereby decreasing viral spread. Through an incompletely understood mechanism, filovirus GP is able to circumvent the binding ability of tetherin and hence allow for productive budding of infectious particles113,114.
Molecular mimicry has also been suggested to take place within filovirus genomes. mRNAs from filoviruses (those encoding VP35, VP30, VP24 and L) contain initiation sequences similar to those found in the host proteins that remain present after cellular stress, thus shifting host translation towards the production of viral mRNAs, a phenomenon that is supported by the lack of stress granule formation in EBOV-infected cells115,116,117.
Treatment and therapy options
Medical countermeasures should seek to exploit what is known about filovirus biology and replication. Particular focus should be put on crippling filovirus defences to enable recognition by the immune system as early as possible. Efforts to control virus replication are also paramount in order to afford as much time as possible for hosts to mount immune responses. Targeting multifunctional proteins such as VP35 or GP could simultaneously disarm multiple aspects of the viral arsenal and thus tip the balance back in favour of the host.
Herein, we use the term 'post-exposure' to indicate interventions given after virus exposure but before the onset of clinical signs or viraemia. The terms 'therapy' or 'therapeutic' indicate that an intervention was administered any time after the onset of clinical signs, including viraemia.
In the 2013–2016 outbreak of EBOV in West Africa, the most advanced post-exposure treatments — the monoclonal antibody (mAb) cocktail ZMapp and the small interfering RNA (siRNA) TKM-Ebola, which had efficacy in NHP models — did not provide statistically significant protection in clinical trials to patients infected with Ebola. Disconnects between preclinical NHP data and these human trials were likely caused by time constraints and poor trial design, which is understandable in such a massive epidemic in a resource-poor setting, with an agent associated with an extraordinarily high case fatality rate. In these clinical studies, the interventions were given to many patients with high viral loads who were near death; virtually no intervention under these conditions could have resulted in substantial patient survival. Notably, treatment of repatriated patients with these same drugs in medical facilities with advanced supportive care resulted in remarkable survival.
In the following sections, we discuss the different treatment modalities that have shown efficacy in preclinical trials (the discussion is limited to agents investigated in NHPs) and/or have been used in clinical trials in West Africa. Detailed information on drug dose, administration route, challenge dose and other aspects is provided in Tables 1, 2, 3, 4.
Antibodies targeting filovirus proteins
Convalescent blood. Blood-based antibody products have been used for treating numerous infectious diseases (including anthrax, botulism, cytomegalovirus, hepatitis B, rabies, tetanus and vaccinia). Indeed, 15 mAb products are currently licensed for use against infectious agents in the USA and Europe118. However, until recently, the use of antibody-based approaches to treat filovirus infections has been highly controversial. The initial application of passive antibody therapy to treat a filovirus infection occurred in November 1976, when a laboratory worker in the UK experienced an accidental needle-stick exposure119. As it was unclear whether the exposure was to EBOV or SUDV, the worker was treated with anti-EBOV convalescent serum on day 8 after exposure and with anti-SUDV convalescent serum on day 11 after exposure. The patient developed clinical disease and survived, but as the patient also received interferon and supportive care in a medical facility in a developed nation, the contribution of antibodies in the convalescent serum to the survival of the patient is unknown.
The next use of antibodies to treat human filovirus infections occurred during a large outbreak of EBOV in Kikwit in 1995 (Ref. 120). Eight patients were treated in a nonrandomized uncontrolled study with convalescent whole blood, and seven survived. However, the authors involved with the study concluded that a number of factors other than administration of convalescent blood could have contributed to the high survival observed among these patients.
Two preclinical studies in NHPs have attempted to replicate this study with the use of either convalescent NHP whole blood121 or convalescent NHP serum122 (Table 1). In an initial study, four rhesus monkeys were treated with homologous convalescent whole blood from rhesus macaque donors immediately after EBOV (Kikwit strain) challenge and again on day 3 or 4 after exposure121. Serum levels of anti-EBOV antibodies were comparable to levels associated with protective vaccination; however, all treated animals succumbed to the infection with no delay in death versus controls121. In a more recent study, four rhesus monkeys were treated with pooled homologous convalescent serum from rhesus macaque donors on days 3, 6, and 9 after EBOV (Makona strain) challenge122. As observed in the whole blood transfer study, none of the treated macaques survived, and there was no delay in the time to death versus controls. Finally, during the 2013–2016 EBOV outbreak in West Africa, a nonrandomized, comparative study was conducted using convalescent plasma123. Specifically, 99 patients of various ages with confirmed EBOV infection received two consecutive transfusions of 200–250 ml of convalescent plasma, each from a separate donor. The transfusions were initiated on the day of diagnosis or up to 2 days later. Treatment was not associated with a significant improvement in survival.
To date, the use of blood products obtained from convalescent donors has not convincingly demonstrated any strong survival benefit in either preclinical NHP studies or infected patients. Although convalescent blood products were used in repatriated patients with EBOV infection during the 2013–2016 outbreak, those patients also received other experimental therapies and advanced supportive care, thus confounding interpretation of therapeutic benefit124,125,126,127. Furthermore, host rejection is a risk — acute respiratory distress was reported in a health-care worker in Spain and was attributed to treatment with convalescent plasma126.
Equine hyperimmune antibodies. The first use of passive antibody therapy with anti-filovirus antibodies not obtained from convalescent donors was in a study conducted in a baboon model of EBOV haemorrhagic fever128. This study assessed the protective efficacy of an equine hyperimmune immunoglobulin G (IgG) preparation. In brief, baboons were challenged with low doses of EBOV (undefined 1976 strain, 10–30 times the median lethal dose (LD50)), and groups of animals were treated before or shortly after exposure. The equine IgG completely protected animals if administered before or up to 1 hour after EBOV challenge, but survival dropped to 29% if treatment was delayed until 2 hours after exposure. This product was subsequently evaluated in cynomolgus macaques using the gold-standard, high-dose 1,000 plaque-forming unit (PFU) challenge of EBOV (Kikwit strain). No protection was observed if the equine IgG was administered before or on the day of EBOV challenge, and partial protection (33%) was noted if animals were treated with the equine IgG shortly after exposure and again on day 5 (Refs 129,130). A repeat of a portion of this study in rhesus macaques showed no protection in four of four animals when a purportedly more potent equine IgG treatment was given shortly after EBOV (Kikwit strain) challenge and again on day 5 after exposure (T.W.G., unpublished observations).
Monoclonal and purified antibodies. During the late 1990s, after clinical failures of a number of murine mAbs for a variety of indications, it became apparent that species matching was important for the success of this technology131,132,133. These clinical experiences with murine mAbs fostered the development of human-like antibodies by chimerization of murine mAb variable regions with human constant regions, humanization of murine mAbs or by the use of fully human mAbs. In 1999, a potent neutralizing mAb, KZ52, was generated from a survivor of an EBOV (Kikwit strain) infection134. This mAb targets EBOV GP. On the basis of the success of KZ52 in protecting guinea pigs if administered at a dose of 25 mg per kg shortly after challenge with guinea pig-adapted EBOV (Mayinga strain)135, a study was conducted with KZ52 in rhesus monkeys136. Four rhesus monkeys were given KZ52 at 50 mg per kg 1 day before challenge with EBOV (Kikwit strain) and again on day 4 after virus challenge. Three of the four treated animals succumbed with no delay in death, whereas one animal had a delay in death and succumbed on day 28. The authors were not able to detect viral neutralization escape mutants, and it was evident that neutralization of the virus in the blood had little to no impact on the disease course or viral burden. These results were very discouraging and, along with the polyclonal equine IgG data in the macaque models, dampened enthusiasm for pursuing antibody-based approaches for treating filovirus disease.
The first successes with antibody-based therapies were reported in 2012 by three different groups. Dye et al.137 used polyclonal IgG purified from the convalescent serum of NHPs vaccinated with experimental EBOV or MARV vaccines that survived subsequent filovirus challenge. For EBOV, three rhesus monkeys were treated with total IgG administered beginning on day 2 and again on days 4 and 8 after homologous EBOV (Kikwit strain) challenge. All three treated animals survived. The effector concentration for half-maximum response (EC50) and 80% maximum response (EC80) values of the IgG preparation, determined by plaque reduction neutralization assay, were approximately 5 and 15 μg per ml, respectively. This is striking because the purified anti-EBOV IgG was not as potent in neutralizing EBOV as was KZ52, which failed to protect macaques against lethal disease136. This demonstrates that in vitro neutralization does not always correlate with in vivo protection. In the companion MARV arm of the study, three rhesus monkeys were treated with total IgG administered beginning on day 2 and again on days 4 and 8 after homologous MARV (Ci67 strain) challenge. All three treated animals survived.
Work also reported in 2012 by independent studies from two different groups showed partial to complete protection of NHPs. Each group used a different cocktail of three mouse mAbs targeting the EBOV GP, administered beginning either 1 day or 2 days after EBOV (Kikwit strain) challenge. Qiu et al.138 used a pool of three mouse mAbs (1H3, 2G4 and 4G7), designated as ZMAb, and demonstrated 50–100% protection (depending on dosing schedule) after EBOV infection of cynomolgus monkeys. Olinger et al.139 used a pool of three mouse–human chimeric mAbs (13C6, 13F6 and 6D8), designated MB-003, and demonstrated 67% protection (two of three animals) after EBOV infection of rhesus monkeys. Pettitt et al.140 also assessed the utility of MB-003 as a true therapeutic by administering the cocktail when EBOV (Kikwit strain)-infected rhesus monkeys were first determined to be febrile and PCR-positive for EBOV. In this study, animals were given MB-003 on days 4, 7 and 10 after EBOV infection; three of the seven treated animals survived. Interestingly, evaluation of the two surviving animals from the Olinger et al. study139 identified the emergence of escape mutants in the two animals that did not survive EBOV challenge141.
The seminal event in the advancement of mAbs for treating filovirus infections occurred as the result of a collaboration between the inventors of MB-003 and ZMAb. The team chimerized the ZMAb mAbs and used the guinea pig model of EBOV disease to identify the most protective combination of three mAbs from the two cocktails142. The best combination of anti-EBOV GP mAbs (13C6, 2G4 and 4G7), produced in tobacco plants, is now known as ZMapp. This cocktail was shown to completely protect rhesus monkeys if administered beginning as late as 5 days after EBOV (Kikwit strain) challenge, when the animals were at an advanced stage of disease. The mechanism of protection afforded by the mAbs included in the ZMapp formulation is not completely known. Work done by Davidson et al.143 showed that 13C6 binds to a residue at the tip of the glycan cap of GP, and the authors suggest that 13C6 likely neutralizes EBOV through the use of complement, antibody-dependent cell-mediated cytotoxicity or another crystallizable fragment (Fc)-mediated mechanism. 2G4 and 4G7 bind to epitopes in the base of GP and neutralize the virus through a structural mechanism143.
Further work in NHPs to optimize ZMapp was reported in 2016 by Qiu et al.144. In this study, the individual components of ZMapp were produced in Chinese hamster ovary (CHO) cells and compared with plant-derived components. The authors showed that a cocktail of two of the three chimeric (c) mAbs (c13C6 and c2G4), termed MIL77 and produced in CHO cells, completely protected rhesus monkeys (three of three) from 1,000 PFU EBOV (Makona strain) challenge at 50 mg per kg beginning 3 days after EBOV exposure (treatment administered on days 3, 6 and 9). Interestingly, the same cocktail of the two mAbs produced in plants protected two of three macaques from lethal infection, following the same treatment regimen at a dose of 50 mg per kg. In addition to ZMapp, other groups have recently explored the use of human anti-EBOV GP mAbs as treatments in NHPs. Specifically, Corti et al.145 showed that monotherapy with mAb114 completely protected rhesus monkeys from 1,000 PFU EBOV (Kikwit strain) challenge at 50 mg per kg beginning either 1 day (three of three; treatment regimen on days 1, 2 and 3) or 5 days (three of three; treatment regimen on days 5, 6 and 7)145 after EBOV exposure.
ZMapp gained notoriety in August 2014 when, on the basis of the strength of preclinical NHP studies, it was used compassionately to treat two patients repatriated to the USA124. ZMapp was subsequently used on at least four additional patients evacuated from West Africa to Europe and the USA146,147,148. ZMAb was also used in at least two cases32,149, as was MIL77 (Refs 32,150). The role of ZMapp, ZMAb or MIL77 in patient survival is impossible to discern as all the patients were repatriated to developed countries and received advanced supportive care; most also received other experimental therapies.
In 2015, a randomized controlled trial of ZMapp plus the available standard of care versus the available standard of care alone was conducted in patients diagnosed with EBOV haemorrhagic fever using PCR151. A total of 72 patients were enrolled at sites in Guinea, Liberia, Sierra Leone and the USA. Death occurred in 13 of 35 patients (37%) who received the current standard of care alone and in 8 of 36 patients (22%) who received the current standard of care plus ZMapp (50 mg per kg every third day for up to three doses). The observed posterior probability that ZMapp plus the current standard of care was superior was 91.2%, which fell short of the prespecified threshold of 97.5%. Several confounders likely precluded ZMapp from reaching the prespecified threshold. Notably, as the epidemic was dying down when the trial was initiated, the authors were not able to enrol their target of 100 participants per group. More importantly, seven of the eight deaths that occurred in ZMapp recipients happened after one of the three planned infusions. Therefore, if ZMapp protection requires two or three full doses, then these seven patients died before optimal dosing was achieved. In NHPs, a period of 5 days from the first therapeutic treatment was required to protect >90% of NHPs from lethal EBOV infection142; therefore, treatment was likely initiated too late in some patients in the trial. Despite missing statistical significance in this trial, ZMapp likely has benefit.
Although all anti-filovirus antibody uses in humans have been for EBOV infections, this technology has also been advanced in preclinical models of other filoviruses. Most notably, a fully human anti-Marburgvirus GP mAb known as MR191-N completely protected rhesus monkeys against MARV (Angola strain) infection if treatment was initiated on day 4 after exposure and protected 80% of animals if treatment was initiated on day 5 after MARV challenge152. Importantly, this same mAb completely protected rhesus macaques against RAVV infection when treatment was initiated on day 5 after exposure. Although there have been no preclinical studies in NHPs assessing mAbs for BDBV or SUDV, recent studies have identified fully human mAbs with exceptionally potent pan-Ebolavirus neutralizing activity and protective efficacy against three virulent Ebolaviruses in small animals153,154, suggesting that such interventions may soon be available.
Small interfering RNAs
In 2010, the first work demonstrating the potential for siRNA in treating filovirus infections in preclinical NHP models was reported155 (Table 2). This study assessed a pool of three siRNAs that were chemically modified not to induce a nonspecific innate immune response and packaged in a novel lipid nanoparticle (LNP) as the delivery vehicle. The pool included siRNAs targeting L, VP24 and VP35. Groups of rhesus monkeys were treated by intravenous injection 30 minutes after EBOV (Kikwit strain) challenge with pooled anti-EBOV siRNAs and again either on days 1, 3 and 5 after virus exposure or daily through day 6 after virus exposure. Complete protection from lethal infection was observed in macaques receiving seven doses of the siRNAs, whereas 66% protection was observed in macaques receiving four doses of the siRNAs.
Although no additional NHP work was reported on anti-filovirus siRNAs until 2014, the anti-EBOV formulation was modified by eliminating the anti-VP24 siRNA and modifying the LNP. This formulation, which targeted the Kikwit strain of EBOV, became known as TKM-Ebola (TKM-100802). During the early stages of the 2013–2016 EBOV epidemic in West Africa, the TKM-100802 formulation was modified by two nucleotide substitutions in the VP35 siRNA and a single nucleotide substitution in the L siRNA to ensure specificity to the West African Makona strain of EBOV156. This formulation was termed TKM-130803, and the siRNA component was referred to as siEbola-3. This formulation was further modified by changing the LNP to an LNP2 composition. This LNP2 formulation containing siEbola-3 was rapidly assessed in rhesus macaques79. Animals were challenged with EBOV (Makona strain) and treated intravenously with the LNP2 siEbola-3 formulation (0.5 mg per kg) beginning on day 3 after virus exposure and continuing daily for a total of seven treatments. All animals treated with LNP2 siEbola-3 were completely protected from EBOV challenge, whereas all untreated controls succumbed to the virus.
During the early part of the 2013–2016 EBOV epidemic, TKM-100802 was administered to five patients infected with EBOV who were medically evacuated to Europe and the USA and to one individual as post-exposure prophylaxis127,146,156,157. Because the patients were also treated with other experimental products and received advanced supportive care, it was not possible to assess the effectiveness or safety of TKM-100802 in this setting. TKM-100802 was also given to a physician who was medically evacuated to the USA from West Africa after potentially being exposed to EBOV157. However, the patient never became overtly infected or seroconverted to EBOV. Subsequently, the newer TKM-130803 formulation (administered by intravenous infusion in a single daily dose of 0.3 mg per kg for seven days, in addition to supportive care) was assessed in a small, single-arm, phase II trial conducted between March and June 2015 in Sierra Leone156. The trial had an unusual design developed early in the outbreak in 2014 with the aim of enabling the rapid generation of initial evidence about potential treatments. The primary outcome was survival up to 14 days after admission (excluding patients who died within 48 hours of admission). Analysis of data from 1,820 adult patients from the 2014–2015 outbreak with PCR-confirmed EBOV infection was used to pre-specify a futility boundary based on a target survival probability of p> 0.55, which was reached after 14 patients had been treated (2 treated patients were excluded from the analysis, as they died within 48 hours of admission). Of the 12 treated patients included in the final analysis, 9 died and 3 survived. Overall, TKM-130803 was well-tolerated but was not shown to improve survival as defined by the criteria of the trial.
However, the potential to draw meaningful conclusions about the possible effectiveness of TKM-130803 is heavily limited by the characteristics of the study. Most importantly, the TKM-130803 recipients had exceptionally severe disease, with mean pre-treatment viral loads of >1 × 109 RNA copies per millilitre of plasma prior to the first infusion (>2 orders of magnitude higher than those seen in the ZMapp study151). Therefore no knowledge was gained on the potential effectiveness of TKM-130803 in patients with lower viral loads and a more realistic chance of survival. Unfortunately, only patients who had very severe disease presented at the treatment centre during the study, but the trial design did not include a plan to stratify patients based on disease severity or a plan to allow compassionate use outside the trial for patients presenting with exceptionally high viral loads.
It is also unfortunate that only the LNP1 formulation was available for use in this trial, not the improved LNP2 formulation that protected EBOV-infected NHPs79. Finally, it is noteworthy that the dose used in the clinical trial (0.3 mg per kg) was based on the maximum tolerated dose in a healthy volunteer study, but was substantially lower than doses used to protect NHPs (0.5–2.0 mg per kg). Overall, no firm conclusion about the potential value of future clinical evaluation of TKM-130803 or related agents can be drawn from this study.
The siRNA technology has also been applied to filoviruses other than EBOV in preclinical NHP models. Thi et al.158 assessed the post-exposure and therapeutic potential of an siRNA directed against Marburgvirus NP encapsulated in LNP and termed NP-718m-LNP. Groups of rhesus monkeys (four per group) were challenged with MARV (Angola strain) and treated intravenously with NP-718m-LNP beginning 30–45 minutes, 1 day, 2 days or 3 days after virus exposure (the onset of clinical illness occurs at day 3). Animals in all treatment groups received a total of seven daily doses. The five control animals all succumbed to MARV challenge, whereas all 20 treated animals survived. More recently, the same group using the same experimental design showed that NP-718m-LNP treatment of rhesus monkeys, beginning at day 4 or 5 after MARV (Angola strain) challenge, protected 100% and 50% of animals, respectively, whereas treatment of RAVV-infected macaques beginning at either day 3 or surprisingly even day 6 after virus exposure resulted in 100% protection of all animals159. Thi et al.160 also assessed the post-exposure and therapeutic potential of LNP-encapsulated siRNAs directed against the SUDV VP24, VP35 or NP. Initial post-exposure studies in rhesus monkeys showed that the anti-SUDV VP35 siRNA was the most promising candidate. A therapeutic study was then performed in which groups of rhesus monkeys (four per group) were challenged with a uniformly lethal dose of SUDV (Gulu strain) and treated intravenously with the anti-SUDV VP35 siRNA beginning at either day 4 or 5 after virus exposure. Animals in all treatment groups received a total of seven daily doses. All macaques in the day-4 group and 50% of the animals in the day-5 group survived challenge. In both the MARV study and SUDV study, the presence of infectious filovirus or antigen was not detected in immune-privileged tissues of surviving animals, suggesting that siRNAs may be a good option for treating relapsed patients with complications. There are no published reports assessing the potential of siRNA technology for BDBV and RAVV in NHPs or human patients.
Phosphorodiamidate morpholino oligomers
Phosphorodiamidate morpholino oligomers (PMOs) are uncharged antisense agents that consist of moieties with six-sided morpholino bases linked by phosphorodiamidate linkages. PMOs inhibit viral replication by interfering with the translation of gene products by sterically obstructing mRNA. An initial report161 assessed the potential of this technology in a preclinical NHP study (Table 2). The authors treated one group of rhesus monkeys by intramuscular injection with a pool of PMOs targeting sequences of viral mRNAs for EBOV VP24, VP35 and L and a second group of animals with PMOs targeting only EBOV VP35, beginning 2 days before EBOV (Kikwit strain) challenge. The treated animals received a daily dose through day 9 after virus challenge. Whereas all of the control animals or those that received the anti-VP35 PMO died, 50% of the treated animals receiving the pooled PMOs survived. In a subsequent study162, the PMO technology was changed to the positively charged PMOplus. These new PMOs contain piperazine linkages within the molecular backbone and have a small number of positive charges (two to five)163 that are intended to enhance avidity to the negatively charged viral RNA target sequence and to enhance entry into cells. A pooled anti-EBOV VP24 and anti-EBOV VP35 PMOplus, termed AVI-6002, protected 63% of rhesus monkeys against homologous EBOV (Kikwit strain) challenge when AVI-6002 was given subcutaneously and intraperitoneally daily for 10–14 days beginning 30–60 minutes after virus challenge163. Further work164 assessed the importance of each component of AVI-6002. Groups of rhesus monkeys were challenged with EBOV (Kikwit strain) and treated intravenously with different PMOplus compounds beginning approximately 1 hour after virus challenge. Animals received a total of 14 daily PMOplus treatments. The anti-VP35 PMOplus resulted in no survival, the PMOplus directed against both VP24 and VP35 protected 63% of animals and the anti-VP24 PMOplus alone protected 75% of macaques. This study showed that the protective efficacy of AV-6002 was due to the anti-VP24 activity of the compound.
The PMO technology has also been used for MARV. A pool of PMOplus molecules specific to MARV VP24 and NP, AVI-6003, was assessed in MARV (Musoke strain)-infected rhesus monkeys162. Treatment was initiated 30–60 minutes after virus exposure and continued daily for 14 total treatments. All treated animals, regardless of the route of AVI-6003 administration (intraperitoneal, subcutaneous, intravenous or intravenous plus subcutaneous) survived. Furthermore, PMOplus containing only molecules specific to MARV NP, termed AVI-7288, was effective in a delayed treatment approach in NHPs165. In this study, the authors challenged groups of cynomolgus monkeys with MARV (Musoke strain) and treated animals once daily for 14 days intravenously with AVI-7288 beginning 1 hour, 1 day, 2 days or 4 days after virus exposure. All animals in the 2-day group survived MARV challenge, and 83% survival was observed in all other treated groups. There are no published reports assessing the potential of the PMO technology for BDBV, SUDV or RAVV in NHPs and no reports of this technology being employed in any patient exposed to or infected with a filovirus.
Small-molecule antiviral compounds
Favipiravir. Favipiravir, also known as T-705 or Avigan (6-fluoro-3-hydroxy-2-pyrazinecarboxamide), is a pyrazine derivative prodrug that has broad-spectrum antiviral activity against a number of RNA viruses166. The mechanism of action is thought to be due to selective inhibition of viral RNA-dependent RNA polymerase167, although it has also been suggested that favipiravir induces lethal RNA transversion mutations168. There have been no published preclinical studies in NHPs using favipiravir as a post-exposure treatment or therapeutic.
It is not completely clear why favipiravir was used during the 2013–2016 EBOV epidemic to treat infected patients. However, it was the only promising drug available at the time with a fairly well-known safety profile that could be repurposed and delivered orally, and it had been shown to have antiviral activity against EBOV in vitro and in mice169,170. Similar to ZMapp and TKM-100802, favipiravir was used to treat a number of patients evacuated from West Africa to Europe126,146,149,171, and the role of favipiravir in patient survival is impossible to discern given that all patients received advanced supportive care and most also received other experimental therapies.
A historically controlled, single-arm, proof-of-concept trial of favipiravir, known as the JIKI trial, was conducted during the 2013–2016 EBOV epidemic in Guinea172. The trial enrolled 126 patients with EBOV infection, of whom 111 were analysed. All patients were given oral favipiravir on the day of enrolment and daily from days 1 through 9. Mortality was 20% in patients with lower viral loads and 91% in patients with high viral loads. Overall, there was no statistically significant difference in survival of patients receiving favipiravir versus historical controls. The authors concluded that monotherapy with favipiravir in patients with medium but not high EBOV viraemia merits further study. Subsequent analysis of data from the JIKI trial suggested that favipiravir plasma concentrations failed to achieve the target exposure defined before the trial173. Whether this had any impact on the outcome of the JIKI trial is unknown.
Favipiravir was also used in certain areas in Sierra Leone during the 2013–2016 EBOV epidemic as part of a WHO (World Health Organization)-recommended supportive therapy standard of care or in combination with another WHO-recommended therapy such as ZMapp151. A retrospective analysis of favipiravir therapy suggested some benefit in long-term survival, although the difference was not statistically significant174.
GS-5734. GS-5734 is a monophosphoramidate prodrug of an adenosine nucleoside analogue. In a study of GS-5734 in an NHP model of EBOV haemorrhagic fever175, groups of EBOV (Kikwit strain)-infected animals were treated intravenously with GS-5734 once daily for 12 days beginning at various times after virus exposure employing different dosing regimens (Table 2). At the highest doses, 100% protection was observed even if treatment was initiated at day 3 after virus exposure. Intriguingly, systemic viral RNA was detected in only 33% of treated animals if GS-5734 treatment was initiated at day 3, which is inconsistent with other EBOV studies in rhesus macaques similarly challenged, in which a very high percent of animals are PCR-positive by day 3 (Refs 79,142,144,176). This makes the day 3 results of this study particularly hard to compare with other NHP post-exposure treatment and therapeutic studies. Differences in the sensitivity of PCR assays or the viral stock used9 may also explain the discrepancy among studies. There are no published reports assessing the potential of GS-5734 for BDBV, SUDV, MARV or RAVV in NHPs.
GS-5734 has seen limited use in patients with EBOV infection. GS-5734 was used in a relapsed patient with meningoencephalitis who had been repatriated to Europe and had previously been treated with other experimental therapies32. This relapsed patient received GS-5734 and advanced supportive care, including high-dose corticosteroids, and recovered. One study reported the compassionate use of GS-5734 in combination with ZMapp and a convalescent buffy coat transfusion in a newborn baby who survived EBOV infection177. GS-5734 is currently being used in West Africa in a trial called PREVAIL for men with persistent EBOV RNA in semen178.
BCX4430. BCX4430 is a synthetic adenosine analogue that inhibits viral RNA polymerase function by acting as a non-obligate RNA chain terminator. Groups of cynomolgus monkeys were challenged with MARV (Musoke strain) and treated with BCX4430 intramuscularly twice daily for 14 days beginning 1 hour, 1 day or 2 days after virus exposure179 (Table 2). In this study, 100% of animals in the day 1 and 2 groups and 83% in the 1-hour group were protected. For EBOV, post-exposure treatment of cynomolgus monkeys with BCX4430 starting 2 days after EBOV (Kikwit strain) challenge resulted in no protection180. By contrast, in a rhesus monkey model of EBOV (Kikwit strain) challenge with a higher dose of BCX4430 and treatment being initiated at 30–60 minutes after virus challenge, the authors observed 100% protection of treated animals. There are no published reports assessing the potential of BCX4430 against BDBV, SUDV or RAVV in NHPs and no reports of this technology being used in a patient exposed to or infected with a filovirus.
Brincidofovir. Brincidofovir is an orally available ether–lipid-conjugated prodrug of cidofovir that has antiviral activity against a number of double-stranded DNA viruses, most notably smallpox181. Cidofovir is an acyclic nucleoside phosphonate analogue of deoxycytidine monophosphate and blocks viral DNA polymerase by serving as an alternative substrate, thereby inhibiting viral DNA synthesis. There have been no preclinical reports assessing the use of brincidofovir in any animal model of filovirus infection. Brincidofovir was shown to have some in vitro activity against EBOV182 through unknown mechanisms (EBOV is an RNA virus), albeit relatively weak activity versus other EBOV antivirals. Nonetheless, brincidofovir was used most famously to treat an individual with EBOV haemorrhagic fever in Dallas, Texas, who did not survive183. Brincidofovir was also used in other repatriated patients in the USA and Europe who also received other experimental drugs and advanced supportive care32,125,148. A single-arm phase II trial of brincidofovir was initiated in Liberia in January 2015 (Ref. 184). The trial was stopped by the manufacturer after all four patients treated with brincidofovir died of EBOV haemorrhagic fever. Although the sample size was small, these patients had mid-level viral loads. Brincidofovir is unlikely to be an effective monotherapy for disease caused by EBOV infection.
Amiodarone. Amiodarone is a multi-ion channel inhibitor and adrenoreceptor agonist that is used as an antiarrhythmic drug185. No preclinical reports have assessed amiodarone in an animal model of filovirus infection. Amiodarone has been reported to inhibit filovirus cell entry in vitro186 and interfere with fusion of the EBOV viral envelope with the endosomal membrane187.
Amiodarone was used to treat at least two individuals infected with EBOV during the 2013–2016 epidemic148,188. In the patient for which details are available, a Ugandan physician became infected with EBOV and self-initiated amiodarone therapy188. This patient was then medically evacuated to Germany, where amiodarone treatment was discontinued because of its potential cardiac side effects and unclear antiviral effectiveness in vivo188. The patient received advanced supportive care and other experimental therapies and survived. During this same period of time, an Italian nongovernmental aid relief organization proposed a trial of amiodarone189, but it was not recommended by the WHO because of documented adverse effects of amiodarone that could exacerbate EBOV haemorrhagic fever190. Amiodarone does not seem to have much utility for treating filovirus infections given the high associated risks and lack of any supportive preclinical data in NHPs.
Vaccine vectors as post-exposure treatments
Similar to the rabies vaccine used, in combination with hyperimmune globulin to treat humans exposed to rabies virus, filovirus vaccines have been used to treat NHPs experimentally infected with filoviruses as well as patients potentially exposed to filoviruses. All these vaccines utilize viral vectors that express a filovirus GP. In the initial proof-of-concept work191, treatment of rhesus monkeys with a recombinant vesicular stomatitis virus (rVSV)-based MARV (Musoke strain) vaccine shortly after a homologous MARV challenge resulted in complete protection of all animals from disease and death (Table 3). Subsequent studies by the same group showed that rVSV–EBOV (Mayinga strain) protected 50% of rhesus monkeys against EBOV (Kikwit strain) challenge192 and that an rVSV–SUDV (Boniface strain) vaccine protected 100% of rhesus macaques against homologous SUDV challenge193. As the vaccines in all of these initial studies were administered 20–30 minutes after filovirus challenge, questions were raised about the real-world utility of this approach. In follow-up studies, treatment of rhesus monkeys with rVSV–MARV (Musoke strain), initiated 1 day after homologous MARV challenge, resulted in survival of five of six monkeys, and two of six macaques were protected when the rVSV–MARV vaccine was administered 2 days after virus exposure194. None of the control animals, which were treated with nonspecific rVSV filovirus vaccines, survived filovirus challenge in any of these studies, suggesting that protection requires the development of a humoral or cellular immune response against the specific pathogen.
More recently, the rVSV–EBOV vaccine was used against the new Makona strain of EBOV195. The authors treated groups of rhesus monkeys with one dose of rVSV–EBOV either as a single injection of vaccine at 1 hour or 1 day after homologous EBOV exposure or as two injections (half of the dose at each time point). A control group of macaques was given the same dose of an rVSV–MARV vaccine at both time points, and another cohort of animals was not treated with any vaccine. All untreated animals succumbed to EBOV infection, whereas 33–67% of the macaques in each treatment group survived, including the group treated with the nonspecific rVSV–MARV vaccine. In contrast to previous studies, the authors suggest that protection from post-exposure vaccination is antigen-nonspecific and rather due to an early activation of the innate immune system. The small numbers of control animals treated with nonspecific rVSV filovirus vaccines (seven collectively in the previous studies and three in this study) make it difficult to definitively say whether post-exposure protection by the rVSV filovirus vaccines is mediated by early activation of the innate immune system or by specific antigen responses, or whether the differing results are due to individual animal variability, as the case fatality rate for the Makona strain of EBOV in untreated rhesus macaque controls is not well established. As noted previously, there are occasional (although infrequent) untreated control rhesus monkeys that survive exposure to the Kikwit strain of EBOV9,139. Collectively, studies to date suggest that an early innate response is required to slow down virus replication and buy time for the host to mount a protective adaptive immune response.
The rVSV–EBOV vaccine was used to treat accidental laboratory exposures and repatriated patients during the 2013–2016 epidemic. The initial use in humans was to treat a laboratory exposure to EBOV about 40 hours after the incident196. The individual survived with no evidence of overt clinical illness; however, whether the patient was actually exposed to EBOV remains uncertain. Subsequently, there were at least six cases of medical staff working in West Africa during the 2013–2016 epidemic who were treated with a single dose of the rVSV–EBOV vaccine at times ranging from approximately 24 to 43 hours after the potential exposure to EBOV157,197,198. All these individuals survived with mild vaccine-related clinical symptoms but no overt clinical illness, and it is again unknown whether any of these patients were actually exposed to EBOV.
In addition to the rVSV–EBOV vaccine, adenovirus serotype 5 (Ad5) vaccine vectors expressing either the EBOV GP (Ad5–EBOV) or an Ad5 vector adjuvanted with IFNα (Ad5–IFNα) have been used in post-exposure treatment studies in NHPs176,199. This work is discussed in the combination treatment section below.
Interferons
There have been several attempts to boost the host innate immune response during filovirus infection through the use of interferons in preclinical NHP models (Table 4). However, results have been discouraging. Treatment of cynomolgus monkeys with recombinant human interferon-α2b (IFNα2b)130 or treatment of rhesus monkeys with recombinant human IFNβ200 after exposure to EBOV (Kikwit strain) offered no survival benefit. A small pilot study of IFNβ given 1 hour after exposure to MARV (Musoke strain) resulted in survival of one of three animals200.
A small, historically controlled, single-arm, proof-of-concept trial of IFNβ1 was conducted in 2015 (Ref. 201). In this trial, IFNβ1a was given subcutaneously daily for up to 17 days. Six of the nine patients survived, which was not statistically different from historical controls; however, the different viral loads between these two groups complicates the comparison and interpretation of the results. On the basis of the available preclinical NHP data and the limited EBOV clinical trial data, it does not appear that interferons have much promise as a monotherapy for filovirus infection.
Modulation of the coagulation system
Coagulation disorders are a common pathological feature of filovirus infections in humans and NHPs and contribute to hypotension and multiple organ failure in haemorrhagic fever. There have been several studies investigating the use of compounds that modulate blood coagulation disorders as a non-antiviral approach to treating filovirus disease. Recombinant nematode anticoagulant protein c2 (rNAPc2) blocks the activity of a complex comprising coagulation factor VIIa and tissue factor and was administered shortly after challenge (beginning immediately or on day 1) to groups of rhesus monkeys exposed to EBOV (Kikwit strain)55; 33% of the animals survived (Table 4). In rhesus monkeys challenged with MARV (Angola strain), rNAPc2 treatment resulted in the survival of only one of six treated animals50.
Recombinant human activated protein C (Drotrecogin alfa; Xigris) has also been considered as a treatment option for filovirus infection in NHPs. The mechanism of action of activated protein C is not completely known. However, it is thought to exert antithrombotic effects by inhibiting coagulation factors Va and VIIIa and may have indirect profibrinolytic activity by inhibiting plasminogen activator inhibitor 1 (PAI1; also known as SERPINE1). Activated protein C also has anti-inflammatory effects. A single study in NHPs assessed the efficacy of activated protein C by exposing rhesus monkeys to EBOV (Kikwit strain) and treating them with a continuous intravenous infusion of recombinant human activated protein C for 7 days78. Two of the eleven animals survived EBOV infection, and there was a significant delay in death in treated animals.
In 1975, two patients infected with MARV (Ozolin strain) in South Africa were given aggressive supportive care and prophylactic heparin19. Both patients survived infection, although the role of heparin treatment in survival could not be discerned. In 1976, during the original EBOV (Mayinga strain) outbreak, a single patient was also treated with heparin but did not survive202. Heparin was used in two instances during the 2013–2016 EBOV epidemic32,149. Both patients in these reports survived, but they also received antiviral drugs and advanced supportive care; therefore, the role of heparin is unknown.
During the 2013–2016 EBOV epidemic, a fibrin-derived peptide, FX06, was used to treat vascular leakage in a repatriated doctor in Germany188. On the 11th day after the onset of symptoms and demonstration of biophysical evidence of vascular leak syndrome, the patient received intravenous FX06. The patient also received advanced supportive care, including mechanical ventilation and renal replacement therapy, and survived.
Collectively, data assessing compounds that modulate the coagulation system during filovirus infection are sparse but suggest that these strategies may have utility as part of an overall supportive care regimen in combination with interventions that directly control viral replication.
Immunomodulatory approaches
A fatal outcome in human and NHP filovirus infection is associated with dysregulation of normal host immune responses, characterized by a cytokine storm, with unbridled production of many pro-inflammatory mediators. There was a strong push for the use of statins and angiotensin receptor blockers during the recent EBOV epidemic as a way to modulate the host response and to maintain and restore endothelial barrier integrity. Statins are best known for their capacity to inhibit cholesterol synthesis, but they also have anti-inflammatory effects by, for example, inhibiting leukocyte–endothelial adhesion, thereby muting the production of pro-inflammatory cytokines and modulating regulatory T cell activity203,204. Anecdotal stories suggest that local physicians in Sierra Leone successfully used two statins, atorvastatin and irbesartan205. However, the methods and data from these studies have not been made available for adequate review.
A concern with the use of any immunomodulatory agent to treat filovirus disease is that, without strong preclinical data in NHP models, it is hard to predict whether such interventions will have beneficial or unintended deleterious effects. As an example, EBOV (Kikwit strain)-infected rhesus monkeys that were given several doses of a mAb to IFNα/β receptor 1 (IFNAR1) not only all succumbed to infection but did so with significant decrease in the mean time to death versus untreated controls200. As another example, an antibody against integrin-β1 resulted in a worse outcome in EBOV (Makona strain)-infected rhesus monkeys, with a significant decrease in survival and time to death versus untreated controls (T.W.G., unpublished data). Overall, there is currently little evidence that immunomodulatory approaches have any benefit as monotherapies for the treatment of filovirus infections, but they may have some utility as part of an overall supportive care regimen or, as mentioned above, as modulators of coagulation.
Combination approaches
There have been several attempts to assess the benefit of combining several different interventions (Table 4). ZMAb was combined with Ad5–IFNα to treat cynomolgus or rhesus monkeys beginning 3 days after exposure to EBOV (Kikwit strain)176. Notably, 75% (three of four) and 100% (four of four) of cynomolgus and rhesus macaques, respectively, survived challenge.
A combination of the Ad5–EBOV vector, the Ad5–IFNα vector and universal type I interferon with or without rNAPc2 has also been investigated199. Treatment of EBOV (Kikwit strain)-infected cynomolgus monkeys with different doses of Ad5–EBOV and Ad5–IFNα initiated 30 minutes after exposure combined with universal interferon given on days 5–21 protected 67% (six of nine) of the animals from death. If treatment was delayed until 1 day after virus exposure, 25% (one of four) of cynomolgus monkeys and 33% (one of three) of rhesus monkeys survived the lethal challenge. In a study that used Ad5–EBOV and Ad5–IFNα vectors 30 minutes after exposure, followed by universal interferon and rNAPc2 (days 5–21), 67% protection (two of three animals) was observed199.
Conclusion and outlook
The 2013–2016 EBOV epidemic triggered an enormous interest in filoviruses. Substantial efforts have been directed towards the discovery and development of medical countermeasures, including preventive vaccines and post-exposure treatments. The field has made the most progress on preventive vaccines, with several clinical trials conducted during the 2013–2016 EBOV epidemic and continuing today (Box 2). Therapeutic interventions have lagged behind, and some of the most promising approaches have been used to treat repatriated patients. However, clinical trials of post-exposure treatments have been limited and most of them were poorly designed. There is not a lack of filovirus animal models in general, but viral challenges with wild-type filovirus isolates are largely limited to immunocompromised rodents and NHPs (Box 1). Model refinement may be needed in the future to study viral persistence and post-Ebola syndrome as well as to address treatment of those conditions. Well-characterized animal models, particularly NHPs, will be needed for licensure of promising medical countermeasures under the FDA Animal Rule (Box 3). The development of cell-based screening systems based on reverse genetic approaches that can be applied to high-throughput evaluations under lower-level biocontainment has provided the necessary tools for future discoveries (Box 4).
The well-publicized success of the rVSV–EBOV preventive vaccine from the ring vaccination, cluster-randomized clinical trial in Guinea206,207 (Box 2), and likely impending licensure based on this strong data, should pave the way for other medical countermeasures. Importantly, the rVSV vaccine work in Guinea shows how a clinical trial can be done effectively during an outbreak, in contrast to clinical trials for ZMapp151, favipiravir172 and TKM-130803 (Ref. 156), which were important for compassionate use but were not well-conducted clinical studies. However, these trials do provide valuable information that can be used to improve future clinical validation of experimental therapies, particularly reiteration of the association between high viral load and reduced survival208,209,210. A number of studies have correlated nonsurvival at 90% or more if the viral load exceeds 6 log copies per millilitre210,211,212,213,214 in untreated patients. In a study of 84 patients infected with EBOV in West Africa, all patients with a viral load >7.71 log copies per millilitre of blood died, whereas all those with counts below this threshold survived215. Thus, for example, in the case of the TKM-130803 EBOV trial, the patients who survived would almost certainly have died without at least receiving supportive care in addition to TKM-130803, but whether treatment with TKM-130803 had any contribution was not possible to determine with confidence owing to the issues with the trial discussed earlier. Clearly, this shows the importance of rapid and reliable diagnostics, including viral load determination, for conducting appropriate clinical trials.
Among the post-exposure treatments and therapies developed to date against filoviruses, the mAb-based approaches that target filovirus GPs seem to demonstrate the highest level of protective efficacy. These mAbs have strong preclinical efficacy in clinically ill NHPs as true therapeutics, and human mAbs are generally safe. The main disadvantage of mAbs is that they rely on cross reactivity to conserved epitopes. Small changes in the GP of known filoviruses or the presence of a new filovirus could be problematic. Pan-filovirus mAb approaches may help153,154, and the field should continue to move in this direction. Cost and production time are important, and transitioning mAb production from plant-based systems (as was used for the original ZMapp formulation for EBOV, as well as for MR-191-N for MARV) to mammalian-based systems should, in part, reduce these concerns. The requirement for multiple intravenous administrations of mAbs is not ideal in resource-poor settings. Studies should be conducted in NHPs to determine whether fewer treatments and/or less invasive routes can still be effective.
siRNAs have been efficacious in NHPs as true therapeutics, nearly equal to mAbs, and are safe in humans156. The modular nature of the siRNA technology offers flexibility should mutations in a filovirus occur or a new filovirus be identified. Because siRNA recognition is sequence-dependent, adjustments for viral nucleotide changes can be made rapidly as long as sequence data are made available in real time. This emphasizes the importance of on-site sequence capabilities in future outbreaks as demonstrated during the West African epidemic216. In addition, siRNAs may benefit relapsed patients in whom the virus is present in immune-privileged tissues that mAbs may have difficulty accessing. Similar to mAbs, siRNAs are administered intravenously, and administration of siRNAs requires seven daily injections. PMOs have most of the same advantages and disadvantages of siRNAs, although PMOs are not as far along in terms of demonstrated preclinical efficacy in NHPs. Both siRNA and PMO technologies have promise for future development, but delivery technology and treatment regimens need optimization.
The broad-spectrum antivirals favipiravir and GS-5734 have a number of advantages as interventions for filovirus infections. Their effectiveness is less likely to be affected by mutations in a filovirus or by the emergence of a previously unknown filovirus. For favipiravir, oral administration is a major benefit in resource-poor settings. Clinical trials of favipiravir in West Africa suggested some benefit in patients with lower viral loads when treatment was initiated172. Favipiravir was safe in phase I trials as well as in a clinical trial in patients with EBOV infection172. Favipiravir may also have some benefit in treating relapsed patients in whom the virus is present in immune-privileged tissues. However, there are currently no strong data in NHPs or in phase II/III clinical trials. By contrast, although GS-5734 requires intravenous delivery, the initial preclinical work in NHPs against EBOV appears strong, and future work on GS-5734 should focus on showing efficacy in NHPs against other filoviruses as well as optimizing delivery.
Post-exposure vaccines may be useful if treatment occurs very soon after exposure. Data in NHPs show that therapeutic vaccination after a certain time following filovirus exposure will not work well194. The rVSV–EBOV vaccine is safe and effective206,207 and available to respond rapidly to future EBOV outbreaks.
The importance of advanced supportive care, including electrolyte balance, ventilation and dialysis, was likely pivotal in the successful treatment of individuals infected with EBOV in developed countries during the recent EBOV epidemic32,125,126,127,148,149,188,217,218. Only two of the twelve patients with EBOV (17%) treated in the USA during this epidemic (two imported cases, two infections identified in the USA and eight medically evacuated patients) died. One patient who died within 48 hours of arriving in the USA was at a very advanced stage of disease on arrival217. From published articles and news reports, there were also likely 15 patients infected with EBOV in Europe during the epidemic (two imported cases, one infection identified in Spain and twelve medically evacuated patients), of whom three died (20%). By comparison, mortality for EBOV reported in treatment centres in West Africa was 37–74%211,219,220,221,222. Advanced supportive care clearly improved patient survival. However, most patients with EBOV infection who were medically evacuated to the USA or Europe also received experimental antiviral therapies, sometimes in combination (one patient was given ZMapp, TKM-Ebola and favipiravir146). The combination of experimental antiviral interventions and advanced supportive care likely contributed to the improved outcome. Future studies in preclinical NHP models will need to assess whether there are any adverse effects or additive or synergistic benefits associated with mixtures of different countermeasures. Indeed, there may be enhanced efficacy from combining drugs that target different parts of the virus or different pathways important for viral replication. This should be a major focus of future research.
Numerous survivors of the 2013–2016 EBOV epidemic now have post-Ebola syndrome. Although this syndrome was noted in survivors of past filovirus outbreaks223,224,225, the magnitude of the 2013–2016 epidemic resulted in a large number of survivors. This syndrome draws attention to the importance of identifying, developing and optimizing drugs that can reach immune-privileged tissues such as the central nervous system and reproductive organs.
Until recently, the development of intervention therapies almost completely lacked therapeutic combination approaches. Future efforts should include the combination of certain promising monotherapies by studying synergistic and/or additive effects of drug compounds. For example, mAb treatment can be combined with siRNA or PMO approaches, as those have distinct mechanisms and should therefore not interfere with each other, so they are likely to be additive in improving outcomes. In addition, supportive care has to be optimized and integrated into any treatment strategy. One approach could be the inclusion of strategies that target host defence mechanisms, such as anticoagulants and immunomodulators, which have shown little utility as monotherapies but may be beneficial if added to supportive or virus-specific therapy.
In summary, the best options for treating small numbers of cases of filovirus infections at present seem to be ZMapp, MR-191-N and other human mAbs under development. For relapsed patients or convalescent patients who are shedding filoviruses, favipiravir, siRNAs and/or GS-5734 may be more appropriate. The potential for widespread filovirus outbreaks has increased, as demonstrated by the recent West African epidemic. A post-exposure ring vaccination approach may prove useful in future outbreaks, given its recent success in clinical trials, both as a post-exposure treatment and as a means for targeted vaccine deployment in an emergency setting. Looking to the future, continued development of broad-spectrum treatments that are active against multiple strains and species of filoviruses, as well as drugs that can be given orally or by intranasal spray, may be the best strategy.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
20 April 2018
The discussion of the phase II trial of TKM-130803 reported in reference 156 has been revised to correct inaccuracies in the original text.
References
Leroy, E. M. et al. Fruit bats as reservoirs of Ebola virus. Nature 438, 575–576 (2005).
Towner, J. S. et al. Marburg virus infection detected in a common African bat. PLoS ONE 2, e764 (2007).
World Health Organization. Ebola data and statistics. Situation summary. WHO http://apps.who.int/gho/data/view.ebola-sitrep.ebola-summary-latest?lang=enhttp://apps.who.int/gho/data/view.ebola-sitrep.ebola-summary-latest?lang=en (2016).
Borio, L. et al. Hemorrhagic fever viruses as biological weapons: medical and public health management. JAMA 287, 2391–2405 (2002).
Centers for Disease Control and Prevention. Possession, use, and transfer of select agents and toxins; biennial review of the list of select agents and toxins and enhanced biosafety requirements. Final rule. Fed. Regist 82, 6278–6294 (2017).
Walsh, P. D. et al. Catastrophic ape decline in western equatorial Africa. Nature 422, 611–614 (2003).
Leroy, E. M. et al. Multiple Ebola virus transmission events and rapid decline of central African wildlife. Science 303, 387–390 (2004).
Feldmann, H., Sanchez, A. & Geisbert, T. W. in Fields Virology 6th edn (eds Knipe, D. M. & Howley, P. M.) 923–956. (Lippincott Williams & Wilkins, 2013).
Geisbert, T. W., Strong, J. E. & Feldmann, H. Considerations in the use of nonhuman primate models of ebola virus and marburg virus infection. J. Infect. Dis. 212 (Suppl. 2), S91–S97 (2015). This review article provides a description of the NHP models used for the evaluation of post-exposure treatments and therapies against filoviruses.
Rougeron, V., Feldmann, H., Grard, G., Becker, S. & Leroy, E. M. Ebola and Marburg haemorrhagic fever. J. Clin. Virol. 64, 111–119 (2015).
Towner, J. S. et al. Newly discovered ebola virus associated with hemorrhagic fever outbreak in Uganda. PLoS Pathog. 4, e1000212 (2008).
Kratz, T. et al. Ebola virus disease outbreak in Isiro, Democratic Republic of the Congo, 2012: signs and symptoms, management and outcomes. PLoS ONE 10, e0129333 (2015).
Le Guenno, B. et al. Isolation and partial characterisation of a new strain of Ebola virus. Lancet 345, 1271–1274 (1995).
Formenty, P. et al. Human infection due to Ebola virus, subtype Cote d'Ivoire: clinical and biologic presentation. J. Infect. Dis. 179 (Suppl. 1), S48–S53 (1999).
Jahrling, P. B. et al. Preliminary report: isolation of Ebola virus from monkeys imported to USA. Lancet 335, 502–505 (1990).
Barrette, R. W. et al. Discovery of swine as a host for the Reston ebolavirus. Science 325, 204–206 (2009).
Baseler, L., Chertow, D. S., Johnson, K. M., Feldmann, H. & Morens, D. M. The pathogenesis of Ebola virus disease. Annu. Rev. Pathol. 12, 387–418 (2017).
World Health Organization. Ebola haemorrhagic fever in Zaire, 1976. Bull. World Health Organ. 56, 271–293 (1978).
Gear, J. S. et al. Outbreak of Marburg virus disease in Johannesburg. Br. Med. J. 4, 489–493 (1975).
Rollin, P. E., Bausch, D. G. & Sanchez, A. Blood chemistry measurements and D-dimer levels associated with fatal and nonfatal outcomes in humans infected with Sudan Ebola virus. J. Infect. Dis. 196 (Suppl. 2), S364–S371 (2007).
McElroy, A. K. et al. Von Willebrand factor is elevated in individuals infected with Sudan virus and is associated with adverse clinical outcomes. Viral Immunol. 28, 71–73 (2015).
McElroy, A. K. et al. Ebola hemorrhagic fever: novel biomarker correlates of clinical outcome. J. Infect. Dis. 210, 558–566 (2014).
Kash, J. C. et al. Longitudinal peripheral blood transcriptional analysis of a patient with severe Ebola virus disease. Sci. Transl Med. 9, eaai9321 (2017).
Varkey, J. B. et al. Persistence of Ebola virus in ocular fluid during convalescence. N. Engl. J. Med. 372, 2423–2427 (2015).
Mattia, J. G. et al. Early clinical sequelae of Ebola virus disease in Sierra Leone: a cross-sectional study. Lancet Infect. Dis. 16, 331–338 (2016).
Scott, J. T. et al. Post-Ebola syndrome, Sierra Leone. Emerg. Infect. Dis. 22, 641–646 (2016).
Tiffany, A. et al. Ebola virus disease complications as experienced by survivors in Sierra Leone. Clin. Infect. Dis. 62, 1360–1366 (2016).
Hereth-Hebert, E. et al. Ocular complications in survivors of the Ebola outbreak in Guinea. Am. J. Ophthalmol. 175, 114–121 (2017).
Shantha, J. G., Yeh, S. & Nguyen, Q. D. Ebola virus disease and the eye. Curr. Opin. Ophthalmol. 27, 538–544 (2017).
Sagui, E. et al. Severe Ebola virus infection with encephalopathy: evidence for direct virus involvement. Clin. Infect. Dis. 61, 1627–1628 (2015).
de Greslan, T. et al. Ebola virus-related encephalitis. Clin. Infect. Dis. 63, 1076–1078 (2016).
Jacobs, M. et al. Late Ebola virus relapse causing meningoencephalitis: a case report. Lancet 388, 498–503 (2016).
Billioux, B. J., Smith, B. & Nath, A. Neurological complications in Ebola virus infection. Neurotherapeutics 13, 461–470 (2016).
Nordenstedt, H. et al. Ebola virus in breast milk in an Ebola virus-positive mother with twin babies, Guinea, 2015. Emerg. Infect. Dis. 22, 759–760 (2015).
Sissoko, D. et al. Ebola virus persistence in breast milk after no reported illness: a likely source of virus transmission from mother to child. Clin. Infect. Dis. 64, 513–516 (2017).
Deen, G. F. et al. Ebola RNA persistence in semen of ebola virus disease survivors — preliminary report. N. Engl. J. Med. 377, 1428–1437 (2015).
Mate, S. E. et al. Molecular evidence of sexual transmission of Ebola virus. N. Engl. J. Med. 373, 2448–2454 (2015).
Soka, M. J. et al. Prevention of sexual transmission of Ebola in Liberia through a national semen testing and counselling programme for survivors: an analysis of Ebola virus RNA results and behavioural data. Lancet Glob. Health 4, e736–e743 (2016).
Sow, M. S. et al. New evidence of long-lasting persistence of Ebola virus genetic material in semen of survivors. J. Infect. Dis. 214, 1475–1476 (2016).
Jaax, N. K. et al. Lethal experimental infection of rhesus monkeys with Ebola-Zaire (Mayinga) virus by the oral and conjunctival route of exposure. Arch. Pathol. Lab. Med. 120, 140–155 (1996).
Twenhafel, N. A. et al. Pathology of experimental aerosol Zaire ebolavirus infection in rhesus macaques. Vet. Pathol. 50, 514–529 (2013).
Murphy, F. A., Simpson, D. I., Whitfield, S. G., Zlotnik, I. & Carter, G. B. Marburg virus infection in monkeys. Ultrastructural studies. Lab. Invest. 24, 279–291 (1971).
Baskerville, A., Fisher-Hoch, S. P., Neild, G. H. & Dowsett, A. B. Ultrastructural pathology of experimental Ebola haemorrhagic fever virus infection. J. Pathol. 147, 199–209 (1985).
Geisbert, T. W., Jahrling, P. B., Hanes, M. A. & Zack, P. M. Association of Ebola-related Reston virus particles and antigen with tissue lesions of monkeys imported to the United States. J. Comp. Pathol. 106, 137–152 (1992).
Geisbert, T. W. & Jaax, N. K. Marburg hemorrhagic fever: report of a case studied by immunohistochemistry and electron microscopy. Ultrastruct. Pathol. 22, 3–17 (1998).
Ryabchikova, E. I., Kolesnikova, L. V. & Luchko, S. V. An analysis of features of pathogenesis in two animal models of Ebola virus infection. J. Infect. Dis. 179 (Suppl. 1), S199–S202 (1999).
Zaki, S. R. & Goldsmith, C. S. Pathologic features of filovirus infections in humans. Curr. Top. Microbiol. Immunol. 235, 97–116 (1999).
Geisbert, T. W. et al. Pathogenesis of Ebola hemorrhagic fever in cynomolgus macaques: evidence that dendritic cells are early and sustained targets of infection. Am. J. Pathol. 163, 2347–2370 (2003).
Geisbert, T. W. et al. Pathogenesis of Ebola hemorrhagic fever in primate models: evidence that hemorrhage is not a direct effect of virus-induced cytolysis of endothelial cells. Am. J. Pathol. 163, 2371–2382 (2003).
Geisbert, T. W. et al. Marburg virus Angola infection of rhesus macaques: pathogenesis and treatment with recombinant nematode anticoagulant protein c2. J. Infect. Dis. 196 (Suppl. 2), S372–S381 (2007).
Hensley, L. E. et al. Pathogenesis of Marburg hemorrhagic fever in cynomolgus macaques. J. Infect. Dis. 204 (Suppl. 3), S1021–S1031 (2011).
Martines, R. B., Ng, D. L., Greer, P. W., Rollin, P. E. & Zaki, S. R. Tissue and cellular tropism, pathology and pathogenesis of Ebola and Marburg viruses. J. Pathol. 235, 153–174 (2015).
Davis, K. J. et al. Pathology of experimental Ebola virus infection in African green monkeys. Involvement of fibroblastic reticular cells. Arch. Pathol. Lab. Med. 121, 805–819 (1997).
Geisbert, T. W. et al. Mechanisms underlying coagulation abnormalities in ebola hemorrhagic fever: overexpression of tissue factor in primate monocytes/macrophages is a key event. J. Infect. Dis. 188, 1618–1629 (2003).
Geisbert, T. W. et al. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet 362, 1953–1958 (2003). This paper suggests modulation of blood coagulation pathways as an intervention for Ebola infection.
Villinger, F. et al. Markedly elevated levels of interferon (IFN)-γ, IFN-α, interleukin (IL)-2, IL-10, and tumor necrosis factor-α associated with fatal Ebola virus infection. J. Infect. Dis. 179 (Suppl. 1), S188–S191 (1999).
Hutchinson, K. L. et al. Multiplex analysis of cytokines in the blood of cynomolgus macaques naturally infected with Ebola virus (Reston serotype). J. Med. Virol. 65, 561–566 (2001).
Baize, S. et al. Inflammatory responses in Ebola virus-infected patients. Clin. Exp. Immunol. 128, 163–168 (2002).
Hensley, L. E., Young, H. A., Jahrling, P. B. & Geisbert, T. W. Proinflammatory response during Ebola virus infection of primate models: possible involvement of the tumor necrosis factor receptor superfamily. Immunol. Lett. 80, 169–179 (2002).
Sanchez, A. et al. Analysis of human peripheral blood samples from fatal and nonfatal cases of Ebola (Sudan) hemorrhagic fever: cellular responses, virus load, and nitric oxide levels. J. Virol. 78, 10370–10377 (2004).
Wauquier, N., Becquart, P., Padilla, C., Baize, S. & Leroy, E. M. Human fatal zaire ebola virus infection is associated with an aberrant innate immunity and with massive lymphocyte apoptosis. PLoS Negl. Trop. Dis. 4, e837 (2010).
Ebihara, H. et al. Host response dynamics following lethal infection of rhesus macaques with Zaire ebolavirus. J. Infect. Dis. 204 (Suppl. 3), S991–S999 (2011).
Fernando, L. et al. Immune response to Marburg virus Angola infection in nonhuman primates. J. Infect. Dis. 212 (Suppl. 2), S234–S241 (2015).
Lin, K. L. et al. Temporal characterization of Marburg virus Angola infection following aerosol challenge in rhesus macaques. J. Virol. 89, 9875–9885 (2015).
Marzi, A. et al. Delayed disease progression in cynomolgus macaques infected with Ebola virus Makona strain. Emerg. Infect. Dis. 21, 1777–1783 (2015).
Ewers, E. C. et al. Natural history of aerosol exposure with Marburg virus in rhesus macaques. Viruses 8, 87 (2016).
McElroy, A. K. et al. Kinetic analysis of biomarkers in a cohort of US patients With Ebola virus disease. Clin. Infect. Dis. 63, 460–467 (2016).
Jiang, T. et al. Features of Ebola virus disease at the late outbreak stage in Sierra Leone: clinical, virological, immunological, and evolutionary analyses. J. Infect. Dis. 215, 1107–1110 (2017).
Gedigk, P., Bechtelsheimer, H. & Korb, G. Pathological anatomy of the “Marburg virus” disease (the so-called “Marburg monkey disease”) [German]. Dtsch. Med. Wochenschr. 93, 590–601 (1968).
Zlotnik, I. Marburg agent disease: pathology. Trans. R. Soc. Trop. Med. Hyg. 63, 310–327 (1969).
Alves, D. A. et al. Aerosol exposure to the angola strain of Marburg virus causes lethal viral hemorrhagic fever in cynomolgus macaques. Vet. Pathol. 47, 831–851 (2010).
Geisbert, T. W. et al. Apoptosis induced in vitro and in vivo during infection by Ebola and Marburg viruses. Lab. Invest. 80, 171–186 (2000).
Baskerville, A., Bowen, E. T., Platt, G. S., McArdell, L. B. & Simpson, D. I. The pathology of experimental Ebola virus infection in monkeys. J. Pathol. 125, 131–138 (1978).
Fisher-Hoch, S. P. et al. Haematological and biochemical monitoring of Ebola infection in rhesus monkeys: implications for patient management. Lancet 2, 1055–1058 (1983).
Fisher-Hoch, S. P. et al. Pathophysiology of shock and hemorrhage in a fulminating viral infection (Ebola). J. Infect. Dis. 152, 887–894 (1985).
Fisher-Hoch, S. P. et al. Pathogenic potential of filoviruses: role of geographic origin of primate host and virus strain. J. Infect. Dis. 166, 753–763 (1992).
Bray, M., Hatfill, S., Hensley, L. & Huggins, J. W. Haematological, biochemical and coagulation changes in mice, guinea-pigs and monkeys infected with a mouse-adapted variant of Ebola Zaire virus. J. Comp. Pathol. 125, 243–253 (2001).
Hensley, L. E. et al. Recombinant human activated protein C for the postexposure treatment of Ebola hemorrhagic fever. J. Infect. Dis. 196 (Suppl. 2), S390–S399 (2007).
Thi, E. P. et al. Lipid nanoparticle siRNA treatment of Ebola-virus-Makona-infected nonhuman primates. Nature 521, 362–365 (2015).
Becker, S., Spiess, M. & Klenk, H. D. The asialo-glycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol. 76, 393–399 (1995).
Takada, A. et al. Downregulation of beta1 integrins by Ebola virus glycoprotein: implication for virus entry. Virology 278, 20–26 (2000).
Chan, S. Y. et al. Folate receptor-α is a cofactor for cellular entry by Marburg and Ebola viruses. Cell 106, 117–126 (2001).
Alvarez, C. P. et al. C-Type lectins DC-SIGN and L-SIGN mediate cellular entry by Ebola virus in cis and in trans. J. Virol. 76, 6841–6844 (2002).
Simmons, G. et al. DC-SIGN and DC-SIGNR bind ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology 305, 115–123 (2003).
Kondratowicz, A. S. et al. T-Cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl Acad. Sci. USA 108, 8426–8431 (2011).
Brecher, M. et al. Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J. Virol. 86, 364–372 (2012).
Cote, M. et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 477, 344–348 (2011).
Carette, J. E. et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477, 340–343 (2011).
Muhlberger, E. Filovirus replication and transcription. Future Virol. 2, 205–215 (2007).
Martin, B., Canard, B. & Decroly, E. Filovirus proteins for antiviral drug discovery: structure/function bases of the replication cycle. Antiviral Res. 141, 48–61 (2017).
Hoenen, T. et al. Inclusion bodies are a site of ebolavirus replication. J. Virol. 86, 11779–11788 (2012).
Nanbo, A., Watanabe, S., Halfmann, P. & Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci. Rep. 3, 1206 (2013).
Banadyga, L. et al. Ebola virus VP24 interacts with NP to facilitate nucleocapsid assembly and genome packaging. Sci. Rep. 7, 7698 (2017).
Harty, R. N., Brown, M. E., Wang, G., Huibregtse, J. & Hayes, F. P. A. PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: implications for filovirus budding. Proc. Natl Acad. Sci. USA 97, 13871–13876 (2000).
Feldmann, H., Will, C., Schikore, M., Slenczka, W. & Klenk, H. D. Glycosylation and oligomerization of the spike protein of Marburg virus. Virology 182, 353–356 (1991).
Feldmann, H., Nichol, S. T., Klenk, H. D., Peters, C. J. & Sanchez, A. Characterization of filoviruses based on differences in structure and antigenicity of the virion glycoprotein. Virology 199, 469–473 (1994).
Volchkov, V. E. Processing of the Ebola virus glycoprotein. Curr. Top. Microbiol. Immunol. 235, 35–47 (1999).
Volchkov, V. E. et al. GP mRNA of Ebola virus is edited by the Ebola virus polymerase and by T7 and vaccinia virus polymerases. Virology 214, 421–430 (1995).
Sanchez, A., Trappier, S. G., Mahy, B. W., Peters, C. J. & Nichol, S. T. The virion glycoproteins of Ebola viruses are encoded in two reading frames and are expressed through transcriptional editing. Proc. Natl Acad. Sci. USA 93, 3602–3607 (1996).
Volchkova, V. A., Klenk, H. D. & Volchkov, V. E. Delta-peptide is the carboxy-terminal cleavage fragment of the nonstructural small glycoprotein sGP of Ebola virus. Virology 265, 164–171 (1999).
Mehedi, M. et al. A new Ebola virus nonstructural glycoprotein expressed through RNA editing. J. Virol. 85, 5406–5414 (2011).
Messaoudi, I., Amarasinghe, G. K. & Basler, C. F. Filovirus pathogenesis and immune evasion: insights from Ebola virus and Marburg virus. Nat. Rev. Microbiol. 13, 663–676 (2015).
Prescott, J. B. et al. Immunobiology of Ebola and Lassa virus infections. Nat. Rev. Immunol. 17, 195–207 (2017).
Basler, C. F. et al. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc. Natl Acad. Sci. USA 97, 12289–12294 (2000).
Basler, C. F. et al. The Ebola virus VP35 protein inhibits activation of interferon regulatory factor 3. J. Virol. 77, 7945–7956 (2003).
Leung, D. W., Prins, K. C., Basler, C. F. & Amarasinghe, G. K. Ebolavirus VP35 is a multi-functional virulence factor. Virulence 1, 526–531 (2010).
Edwards, M. R. et al. Differential Regulation of Interferon Responses by Ebola and Marburg Virus VP35 Proteins. Cell Rep. 14, 1632–1640 (2016).
Hartman, A. L. et al. Inhibition of IRF-3 activation by VP35 is critical for the high level of virulence of ebola virus. J. Virol. 82, 2699–2704 (2008).
Prins, K. C. et al. Mutations abrogating VP35 interaction with double-stranded RNA render Ebola virus avirulent in guinea pigs. J. Virol. 84, 3004–3015 (2010).
Reid, S. P. et al. Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. J. Virol. 80, 5156–5167 (2006).
Reid, S. P., Valmas, C., Martinez, O., Sanchez, F. M. & Basler, C. F. Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J. Virol. 81, 13469–13477 (2007).
Valmas, C. et al. Marburg virus evades interferon responses by a mechanism distinct from ebola virus. PLoS Pathog. 6, e1000721 (2010).
Kaletsky, R. L., Francica, J. R., Agrawal-Gamse, C. & Bates, P. Tetherin-mediated restriction of filovirus budding is antagonized by the Ebola glycoprotein. Proc. Natl Acad. Sci. USA 106, 2886–2891 (2009).
Lopez, L. A. et al. Ebola virus glycoprotein counteracts BST-2/Tetherin restriction in a sequence-independent manner that does not require tetherin surface removal. J. Virol. 84, 7243–7255 (2010).
Nelson, E. V. et al. Ebola virus does not induce stress granule formation during infection and sequesters stress granule proteins within viral inclusions. J. Virol. 90, 7268–7284 (2016).
Le Sage, V. et al. Ebola virus VP35 blocks stress granule assembly. Virology 502, 73–83 (2017).
Shabman, R. S. et al. An upstream open reading frame modulates ebola virus polymerase translation and virus replication. PLoS Pathog. 9, e1003147 (2013).
Sparrow, E., Friede, M., Sheikh, M. & Torvaldsen, S. Therapeutic antibodies for infectious diseases. Bull. World Health Organ. 95, 235–237 (2017).
Emond, R. T., Evans, B., Bowen, E. T. & Lloyd, G. A case of Ebola virus infection. Br. Med. J. 2, 541–544 (1977).
Mupapa, K. et al. Treatment of Ebola hemorrhagic fever with blood transfusions from convalescent patients. J. Infect. Dis. 179 (Suppl. 1), S18–S23 (1999).
Jahrling, P. B., Geisbert, J. B., Swearengen, J. R., Larsen, T. & Geisbert, T. W. Ebola hemorrhagic fever: evaluation of passive immunotherapy in nonhuman primates. J. Infect. Dis. 196 (Suppl. 2), S400–S403 (2007).
Mire, C. E. et al. Passive immunotherapy: assessment of convalescent serum against Ebola virus Makona infection in nonhuman primates. J. Infect. Dis. 214, S367–S374 (2016).
van Griensven, J. et al. Evaluation of convalescent plasma for Ebola virus disease in Guinea. N. Engl. J. Med. 374, 33–42 (2016). This article reports that the treatment of patients during the 2013–2016 Ebola epidemic with convalescent plasma did not improve survival.
Lyon, G. M. et al. Clinical care of two patients with Ebola virus disease in the United States. N. Engl. J. Med. 371, 2402–2409 (2014).
Florescu, D. F. et al. Administration of brincidofovir and convalescent plasma in a patient with Ebola virus disease. Clin. Infect. Dis. 61, 969–973 (2015).
Mora-Rillo, M. et al. Acute respiratory distress syndrome after convalescent plasma use: treatment of a patient with Ebola virus disease contracted in Madrid, Spain. Lancet Respir. Med. 3, 554–562 (2015).
Kraft, C. S. et al. The Use of TKM-100802 and convalescent plasma in 2 patients with Ebola virus disease in the United States. Clin. Infect. Dis. 61, 496–502 (2015).
Kudoyarova-Zubavichene, N. M., Sergeyev, N. N., Chepurnov, A. A. & Netesov, S. V. Preparation and use of hyperimmune serum for prophylaxis and therapy of Ebola virus infections. J. Infect. Dis. 179 (Suppl. 1), S218–S223 (1999).
Jahrling, P. B. et al. Passive immunization of Ebola virus-infected cynomolgus monkeys with immunoglobulin from hyperimmune horses. Arch. Virol. Suppl. 11, 135–140 (1996).
Jahrling, P. B. et al. Evaluation of immune globulin and recombinant interferon-α2b for treatment of experimental Ebola virus infections. J. Infect. Dis. 179 (Suppl. 1), S224–S234 (1999).
Drewe, E. & Powell, R. J. Clinically useful monoclonal antibodies in treatment. J. Clin. Pathol. 55, 81–85 (2002).
Zucca, E. Therapeutic use of monoclonal antibodies: a new era? Ann. Oncol. 9, 683–685 (1998).
Dimitrov, D. S. Therapeutic antibodies, vaccines and antibodyomes. mAbs 2, 347–356 (2010).
Maruyama, T. et al. Ebola virus can be effectively neutralized by antibody produced in natural human infection. J. Virol. 73, 6024–6030 (1999).
Parren, P. W., Geisbert, T. W., Maruyama, T., Jahrling, P. B. & Burton, D. R. Pre- and postexposure prophylaxis of Ebola virus infection in an animal model by passive transfer of a neutralizing human antibody. J. Virol. 76, 6408–6412 (2002).
Oswald, W. B. et al. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLoS Pathog. 3, e9 (2007).
Dye, J. M. et al. Postexposure antibody prophylaxis protects nonhuman primates from filovirus disease. Proc. Natl Acad. Sci. USA 109, 5034–5039 (2012).
Qiu, X. et al. Successful treatment of ebola virus-infected cynomolgus macaques with monoclonal antibodies. Sci. Transl Med. 4, 138ra81 (2012). References 137 and 138 are the first studies to report the successful treatment of NHPs with antibody-based interventions.
Olinger, G. G. Jr et al. Delayed treatment of Ebola virus infection with plant-derived monoclonal antibodies provides protection in rhesus macaques. Proc. Natl Acad. Sci. USA 109, 18030–18035 (2012).
Pettitt, J. et al. Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB-003 monoclonal antibody cocktail. Sci. Transl Med. 5, 199ra113 (2013).
Kugelman, J. R. et al. Emergence of Ebola virus escape variants in infected nonhuman primates treated with the MB-003 antibody cocktail. Cell Rep. 12, 2111–2120 (2015).
Qiu, X. et al. Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 514, 47–53 (2014). This seminal paper, reporting the complete protection of NHPs against Ebola when ZMapp was administered at advanced stages of disease, laid the groundwork for the use of ZMapp to treat repatriated patients during the 2013–2016 Ebola epidemic.
Davidson, E. et al. Mechanism of binding to ebola virus glycoprotein by the ZMapp, ZMAb, and MB-003 cocktail antibodies. J. Virol. 89, 10982–10992 (2015).
Qiu, X. et al. Two-mAb cocktail protects macaques against the Makona variant of Ebola virus. Sci. Transl Med. 8, 329ra33 (2016).
Corti, D. et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 351, 1339–1342 (2016).
The Nordic Page. Ebola-infected Norwegian doctor fully recovers. The Nordic Page https://www.tnp.no/norway/panorama/4660-ebola-infected-norwegian-doctor-fully-recovers-oslo-norwayhttps://www.tnp.no/norway/panorama/4660-ebola-infected-norwegian-doctor-fully-recovers-oslo-norway (2014).
Pooley, W. Ebola: perspectives from a nurse and patient. Am. J. Trop. Med. Hyg. 92, 223–224 (2015).
Uyeki, T. M. et al. Clinical management of Ebola virus disease in the United States and Europe. N. Engl. J. Med. 374, 636–646 (2016). This article describes the highly successful aggressive supportive care given to 27 Ebola patients medically evacuated from West Africa in 2014–2015 and treated in hospitals in the USA and Europe.
Schibler, M. et al. Clinical features and viral kinetics in a rapidly cured patient with Ebola virus disease: a case report. Lancet Infect. Dis. 15, 1034–1040 (2015).
Mundasad, S. British medic declared free of Ebola. BBC News http://www.bbc.com/news/health-32088310 (2015).
Davey, R. T. Jr et al. A randomized, controlled trial of ZMapp for Ebola virus infection. N. Engl. J. Med. 375, 1448–1456 (2016). This paper reports the results of a randomized controlled trial of ZMapp conducted in West Africa and the USA during the 2013–2016 Ebola epidemic.
Mire, C. E. et al. Therapeutic treatment of Marburg and Ravn virus infection in nonhuman primates with a human monoclonal antibody. Sci. Transl Med. 9, eaai8711 (2017). This article reports the development of a human mAb that completely protects NHPs against Marburg and Ravn viruses when administered beginning at advanced stages of disease.
Wec, A. Z. et al. Antibodies from a human survivor define sites of vulnerability for broad protection against ebolaviruses. Cell 169, 878–890.e15 (2017).
Zhao, X. et al. Immunization-elicited broadly protective antibody reveals ebolavirus fusion loop as a site of vulnerability. Cell 169, 891–904.e15 (2017).
Geisbert, T. W. et al. Postexposure protection of non-human primates against a lethal Ebola virus challenge with RNA interference: a proof-of-concept study. Lancet 375, 1896–1905 (2010). This study using siRNAs is the first to show complete post-exposure protection of NHPs against Ebola virus.
Dunning, J. et al. Experimental treatment of Ebola virus disease with TKM-130803: a single-arm Phase 2 clinical trial. PLoS Med. 13, e1001997 (2016).
Wong, K. K. et al. Use of postexposure prophylaxis after occupational exposure to Zaire ebolavirus. Clin. Infect. Dis. 63, 376–379 (2016).
Thi, E. P. et al. Marburg virus infection in nonhuman primates: therapeutic treatment by lipid-encapsulated siRNA. Sci. Transl Med. 6, 250ra116 (2014).
Thi, E. P. et al. siRNA rescues nonhuman primates from advanced Marburg and Ravn virus disease. J. Clin. Invest. 10.1172/JCI96185 (2017). This article reports complete post-exposure protection of NHPs against Marburg virus and Ravn virus with the use of siRNAs if treatment is initiated beginning at advanced stages of disease.
Thi, E. P. et al. Rescue of non-human primates from advanced Sudan ebolavirus infection with lipid encapsulated siRNA. Nat. Microbiol. 1, 16142 (2016).
Warfield, K. L. et al. Gene-specific countermeasures against Ebola virus based on antisense phosphorodiamidate morpholino oligomers. PLoS Pathog. 2, e1 (2006).
Warren, T. K. et al. Advanced antisense therapies for postexposure protection against lethal filovirus infections. Nat. Med. 16, 991–994 (2010). This paper describes the development of morpholino oligomers and demonstrates post-exposure protection against Ebola and Marburg viruses in NHPs.
Swenson, D. L. et al. Chemical modifications of antisense morpholino oligomers enhance their efficacy against Ebola virus infection. Antimicrob. Agents Chemother. 53, 2089–2099 (2009).
Warren, T. K. et al. A single phosphorodiamidate morpholino oligomer targeting VP24 protects rhesus monkeys against lethal Ebola virus infection. mBio 6, e02344–14 (2015).
Warren, T. K. et al. Delayed time-to-treatment of an antisense morpholino oligomer is effective against lethal Marburg virus infection in cynomolgus macaques. PLoS Negl. Trop. Dis. 10, e0004456 (2016).
Furuta, Y. et al. Favipiravir (T-705), a novel viral RNA poly-merase inhibitor. Antiviral Res. 100, 446–454 (2013).
Jin, Z., Smith, L. K., Rajwanshi, V. K., Kim, B. & Deval, J. The ambiguous base-pairing and high substrate efficiency of T-705 (favipiravir) ribofuranosyl 5′-triphosphate towards influenza A virus polymerase. PLoS ONE 8, e68347 (2013).
Baranovich, T. et al. T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 87, 3741–3751 (2013).
Oestereich, L. et al. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antiviral Res. 105, 17–21 (2014).
Smither, S. J. et al. Post-exposure efficacy of oral T-705 (Favipiravir) against inhalational Ebola virus infection in a mouse model. Antiviral Res. 104, 153–155 (2014).
Agrati, C. et al. Longitudinal characterization of dysfunctional T cell-activation during human acute Ebola infection. Cell Death Dis. 7, e2164 (2016).
Sissoko, D. et al. Experimental treatment with favipiravir for Ebola virus disease (the JIKI Trial): a historically controlled, single-arm proof-of-concept trial in Guinea. PLoS Med. 13, e1001967 (2016). This paper reports the results of a historically controlled trial of favipiravir conducted in Guinea during the 2013–2016 Ebola epidemic.
Nguyen, T. H. et al. Favipiravir pharmacokinetics in Ebola-Infected patients of the JIKI trial reveals concentrations lower than targeted. PLoS Negl. Trop. Dis. 11, e0005389 (2017).
Bai, C. Q. et al. Clinical and virological characteristics of Ebola virus disease patients treated with favipiravir (T-705)-Sierra Leone, 2014. Clin. Infect. Dis. 63, 1288–1294 (2016).
Warren, T. K. et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 531, 381–385 (2016). This paper describes the development of GS-5734 and demonstrates post-exposure protection against Ebola virus in NHPs.
Qiu, X. et al. mAbs and Ad-vectored IFN-α therapy rescue Ebola-infected nonhuman primates when administered after the detection of viremia and symptoms. Sci. Transl Med. 5, 207ra143 (2013).
Dornemann, J. et al. First newborn baby to receive experimental therapies survives Ebola virus disease. J. Infect. Dis. 215, 171–174 (2013).
Siegel, D. et al. Discovery and Synthesis of a phosphoramidate prodrug of a pyrrolo[2,1-f][triazin-4-amino] adenine C-nucleoside (GS-5734) for the treatment of Ebola and emerging viruses. J. Med. Chem. 60, 1648–1661 (2017).
Warren, T. K. et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508, 402–405 (2014). This article describes the development of BCX4430 and demonstrates post-exposure protection against Marburg virus in NHPs.
Taylor, R. et al. BCX4430 — a broad-spectrum antiviral adenosine nucleoside analog under development for the treatment of Ebola virus disease. J. Infect. Public Health 9, 220–226 (2016).
Olson, V. A. et al. In vitro efficacy of brincidofovir against variola virus. Antimicrob. Agents Chemother. 58, 5570–5571 (2014).
McMullan, L. K. et al. The lipid moiety of brincidofovir is required for in vitro antiviral activity against Ebola virus. Antiviral Res. 125, 71–78 (2016).
Kroll, D. in Chimerix's brincidofovir given to Dallas, Nebraska Ebola patients. Forbes https://www.forbes.com/sites/davidkroll/2014/10/07/chimerixs-brincidofovir-given-to-dallas-nebraska-ebola-patients/#236eedff274e (2014).
Dunning, J. et al. Experimental treatment of Ebola virus disease with brincidofovir. PLoS ONE 11, e0162199 (2016). This paper reports the use of brincidofovir to treat four cases of Ebola infection in Liberia in 2015.
Vamos, M. & Hohnloser, S. H. Amiodarone and dronedarone: an update. Trends Cardiovasc. Med. 26, 597–602 (2016).
Gehring, G. et al. The clinically approved drugs amiodarone, dronedarone and verapamil inhibit filovirus cell entry. J. Antimicrob. Chemother. 69, 2123–2131 (2014).
Salata, C. et al. Amiodarone and metabolite MDEA inhibit Ebola virus infection by interfering with the viral entry process. Pathog. Dis. 73, ftv032 (2015).
Wolf, T. et al. Severe Ebola virus disease with vascular leakage and multiorgan failure: treatment of a patient in intensive care. Lancet 385, 1428–1435 (2015). This article reports the use of a fibrin-derived peptide, FX06, to treat vascular leakage in a repatriated doctor in Germany during the 2013–2016 Ebola epidemic.
Turone, F. Doctors trial amiodarone for Ebola in Sierra Leone. BMJ 349, g7198 (2014).
Gupta-Wright, A., Lavers, J. & Irvine, S. Concerns about the off-licence use of amiodarone for Ebola. BMJ 350, h272 (2015).
Daddario-DiCaprio, K. M. et al. Postexposure protection against Marburg haemorrhagic fever with recombinant vesicular stomatitis virus vectors in non-human primates: an efficacy assessment. Lancet 367, 1399–1404 (2006). This work is the first to show complete post-exposure protection of NHPs against Marburg virus with the use of an rVSV-based vaccine.
Feldmann, H. et al. Effective post-exposure treatment of Ebola infection. PLoS Pathog. 3, e2 (2007). This article reports the use of an rVSV-based vaccine as a post-exposure treatment for Ebola virus infection in NHPs.
Geisbert, T. W. et al. Recombinant vesicular stomatitis virus vector mediates postexposure protection against Sudan Ebola hemorrhagic fever in nonhuman primates. J. Virol. 82, 5664–5668 (2008).
Geisbert, T. W. et al. Postexposure treatment of Marburg virus infection. Emerg. Infect. Dis. 16, 1119–1122 (2010).
Marzi, A. et al. Efficacy of vesicular stomatitis virus-Ebola virus postexposure treatment in rhesus macaques infected with Ebola virus Makona. J. Infect. Dis. 214, S360–S366 (2016).
Gunther, S. et al. Management of accidental exposure to Ebola virus in the biosafety level 4 laboratory, Hamburg, Germany. J. Infect. Dis. 204 (Suppl. 3), S785–S790 (2011).
Cnops, L. et al. Risk of misinterpretation of Ebola virus PCR results after rVSV ZEBOV-GP vaccination. Clin. Infect. Dis. 60, 1725–1726 (2015).
Lai, L. et al. Emergency postexposure vaccination with vesicular stomatitis virus-vectored Ebola vaccine after needlestick. JAMA 313, 1249–1255 (2015).
Wong, G. et al. Adenovirus-vectored vaccine provides postexposure protection to Ebola virus-infected vonhuman primates. J. Infect. Dis. 212 (Suppl. 2), S379–S383 (2015).
Smith, L. M. et al. Interferon-β therapy prolongs survival in rhesus macaque models of Ebola and Marburg hemorrhagic fever. J. Infect. Dis. 208, 310–318 (2013).
Konde, M. K. et al. Interferon β-1a for the treatment of Ebola virus disease: a historically controlled, single-arm proof-of-concept trial. PLoS ONE 12, e0169255 (2017). This paper reports the results of a historically controlled trial of IFNβ1a conducted in Guinea during the 2013–2016 Ebola epidemic.
Isaacson, M., Sureau, P. H., Courteille, G. & Pattyn, S. R. in Ebola Virus Haemorrhagic Fever (ed. Pattyn, S. R.) 22–26 (Elsevier, 1978).
Abeles, A. M. & Pillinger, M. H. Statins as antiinflammatory and immunomodulatory agents: a future in rheumatologic therapy? Arthritis Rheum. 54, 393–407 (2006).
Forero-Pena, D. A. & Gutierrez, F. R. Statins as modulators of regulatory T-cell biology. Mediators Inflamm. 2013, 167086 (2013).
Fedson, D. S. & Rordam, O. M. Treating Ebola patients: a 'bottom up' approach using generic statins and angiotensin receptor blockers. Int. J. Infect. Dis. 36, 80–84 (2015).
Henao-Restrepo, A. M. et al. Efficacy and effectiveness of an rVSV-vectored vaccine expressing Ebola surface glycoprotein: interim results from the Guinea ring vaccination cluster-randomised trial. Lancet 386, 857–866 (2015).
Henao-Restrepo, A. M. et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ca Suffit!). Lancet 389, 505–518 (2017).
Crowe, S. J. et al. Prognostic indicators for Ebola patient survival. Emerg. Infect. Dis. 22, 217–223 (2016).
Hartley, M. A. et al. Predicting Ebola severity: a clinical prioritization score for Ebola virus disease. PLoS Negl. Trop. Dis. 11, e0005265 (2017).
Fitzpatrick, G. et al. The contribution of Ebola viral load at admission and other patient characteristics to mortality in a Medecins Sans Frontieres Ebola case management centre, Kailahun, Sierra Leone, June–October 2014. J. Infect. Dis. 212, 1752–1758 (2015).
Schieffelin, J. S. et al. Clinical illness and outcomes in patients with Ebola in Sierra Leone. N. Engl. J. Med. 371, 2092–2100 (2014).
de La Vega, M. A. et al. Ebola viral load at diagnosis associates with patient outcome and outbreak evolution. J. Clin. Invest. 125, 4421–4428.
Zhang, X. et al. Prognostic analysis of patients with Ebola virus disease. PLoS Negl. Trop. Dis. 9, e0004113 (2015).
Vernet, M. A. et al. Clinical, virological, and biological parameters associated with outcomes of Ebola virus infection in Macenta, Guinea. JCI Insight 2, e88864 (2017).
Lanini, S. et al. Blood kinetics of Ebola virus in survivors and nonsurvivors. J. Clin. Invest. 125, 4692–4698 (2015).
Quick, J. et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 530, 228–232 (2016).
Sueblinvong, V. et al. Critical care for multiple organ failure secondary to Ebola virus disease in the United States. Crit. Care Med. 43, 2066–2075 (2015).
Connor, M. J. Jr et al. Successful delivery of RRT in Ebola virus disease. J. Am. Soc. Nephrol. 26, 31–37 (2015).
Bah, E. I. et al. Clinical presentation of patients with Ebola virus disease in Conakry. Guinea. N. Engl. J. Med. 372, 40–47 (2015).
Qin, E. et al. Clinical features of patients with Ebola virus disease in Sierra Leone. Clin. Infect. Dis. 61, 491–495 (2015).
Hunt, L. et al. Clinical presentation, biochemical, and haematological parameters and their association with outcome in patients with Ebola virus disease: an observational cohort study. Lancet Infect. Dis. 15, 1292–1299 (2015).
Ji, Y. J. et al. Clinical presentations and outcomes of patients with Ebola virus disease in Freetown, Sierra Leone. Infect. Dis. Poverty 5, 101 (2016).
Rowe, A. K. et al. Clinical, virologic, and immunologic follow-up of convalescent Ebola hemorrhagic fever patients and their household contacts, Kikwit, Democratic Republic of the Congo. Commission Lutte Contre Epidemies Kikwit. J. Infect. Dis. 179 (Suppl. 1), S28–S35 (1999).
Okware, S. I. et al. An outbreak of Ebola in Uganda. Trop. Med. Int. Health 7, 1068–1075 (2002).
Clark, D. V. et al. Long-term sequelae after Ebola virus disease in Bundibugyo, Uganda: a retrospective cohort study. Lancet Infect. Dis. 15, 905–912 (2015).
Cross, R. W. et al. Comparison of the pathogenesis of the Angola and Ravn strains of Marburg virus in the outbred guinea pig model. J. Infect. Dis. 212 (Suppl. 2), S258–S270 (2015).
Qiu, X. et al. Establishment and characterization of a lethal mouse model for the Angola strain of Marburg virus. J. Virol. 88, 12703–12714 (2014).
Bray, M., Davis, K., Geisbert, T., Schmaljohn, C. & Huggins, J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J. Infect. Dis. 178, 651–661 (1998).
Connolly, B. M. et al. Pathogenesis of experimental Ebola virus infection in guinea pigs. J. Infect. Dis. 179 (Suppl. 1), S203–S217 (1999).
Warfield, K. L. et al. Development and characterization of a mouse model for Marburg hemorrhagic fever. J. Virol. 83, 6404–6415 (2009).
Ebihara, H. et al. A Syrian golden hamster model recapitulating ebola hemorrhagic fever. J. Infect. Dis. 207, 306–318 (2013).
Cross, R. W., Fenton, K. A., Geisbert, J. B., Mire, C. E. & Geisbert, T. W. Modeling the disease course of Zaire ebolavirus infection in the outbred guinea pig. J. Infect. Dis. 212 (Suppl. 2), S305–S315 (2015).
Wong, G. et al. Development and characterization of a guinea pig-adapted Sudan virus. J. Virol. 90, 392–399 (2015).
Marzi, A. et al. A hamster model for Marburg virus infection accurately recapitulates Marburg hemorrhagic fever. Sci. Rep. 6, 39214 (2017).
Ebihara, H. et al. Molecular determinants of Ebola virus virulence in mice. PLoS Pathog. 2, e73 (2006).
Geisbert, T. W. et al. Evaluation in nonhuman primates of vaccines against Ebola virus. Emerg. Infect. Dis. 8, 503–507 (2002).
Cross, R. W. et al. The domestic ferret (Mustela putorius furo) as a lethal infection model for 3 species of ebolavirus. J. Infect. Dis. 214, 565–569 (2016).
Bird, B. H. et al. Humanized mouse model of Ebola virus disease mimics the immune responses in human disease. J. Infect. Dis. 213, 703–711 (2016).
Mire, C. E. et al. Oral and conjunctival exposure of nonhuman primates to low doses of Ebola Makona virus. J. Infect. Dis. 214, S263–S267 (2016).
Towner, J. S. et al. Marburgvirus genomics and association with a large hemorrhagic fever outbreak in Angola. J. Virol. 80, 6497–6516 (2006).
Hevey, M., Negley, D., Pushko, P., Smith, J. & Schmaljohn, A. Marburg virus vaccines based upon alphavirus replicons protect guinea pigs and nonhuman primates. Virology 251, 28–37 (1998).
Herbert, A. S. et al. Venezuelan equine encephalitis virus replicon particle vaccine protects nonhuman primates from intramuscular and aerosol challenge with ebolavirus. J. Virol. 87, 4952–4964 (2013).
Sullivan, N. J. et al. Accelerated vaccination for Ebola virus haemorrhagic fever in non-human primates. Nature 424, 681–684 (2003).
Geisbert, T. W. et al. Vector choice determines immunogenicity and potency of genetic vaccines against Angola Marburg virus in nonhuman primates. J. Virol. 84, 10386–10394 (2010).
Geisbert, T. W. et al. Recombinant adenovirus serotype 26 (Ad26) and Ad35 vaccine vectors bypass immunity to Ad5 and protect nonhuman primates against ebolavirus challenge. J. Virol. 85, 4222–4233 (2011).
Swenson, D. L. et al. Vaccine to confer to nonhuman primates complete protection against multistrain Ebola and Marburg virus infections. Clin. Vaccine Immunol. 15, 460–467 (2008).
Wu, S. et al. An adenovirus vaccine expressing Ebola virus variant Makona glycoprotein is efficacious in guinea pigs and nonhuman primates. J. Infect. Dis. 214, S326–S332 (2016).
Stanley, D. A. et al. Chimpanzee adenovirus vaccine generates acute and durable protective immunity against ebolavirus challenge. Nat. Med. 20, 1126–1129 (2014).
Bukreyev, A. et al. Successful topical respiratory tract immunization of primates against Ebola virus. J. Virol. 81, 6379–6388 (2007).
Meyer, M. et al. Aerosolized Ebola vaccine protects primates and elicits lung-resident T cell responses. J. Clin. Invest. 125, 3241–3255 (2015).
Johnson, R. F. et al. An inactivated Rabies virus-based Ebola vaccine, FILORAB1, adjuvanted with glucopyranosyl lipid a in stable emulsion confers complete protection in nonhuman primate challenge models. J. Infect. Dis. 214, S342–S354 (2016).
Blaney, J. E. et al. Antibody quality and protection from lethal Ebola virus challenge in nonhuman primates immunized with rabies virus based bivalent vaccine. PLoS Pathog. 9, e1003389 (2013).
Jones, S. M. et al. Live attenuated recombinant vaccine protects nonhuman primates against Ebola and Marburg viruses. Nat. Med. 11, 786–790 (2005).
Daddario-DiCaprio, K. M. et al. Cross-protection against Marburg virus strains by using a live, attenuated recombinant vaccine. J. Virol. 80, 9659–9666 (2006).
Geisbert, T. W. et al. Single-injection vaccine protects nonhuman primates against infection with Marburg virus and three species of Ebola virus. J. Virol. 83, 7296–7304 (2009).
Mire, C. E. et al. Vesicular stomatitis virus-based vaccines protect nonhuman primates against Bundibugyo ebolavirus. PLoS Negl Trop. Dis. 7, e2600 (2013).
Marzi, A. et al. VSV-EBOV rapidly protects macaques against infection with the 2014/15 Ebola virus outbreak strain. Science 349, 739–742 (2015).
Mire, C. E. et al. Single-dose attenuated Vesiculovax vaccines protect primates against Ebola Makona virus. Nature 520, 688–691 (2015).
Warfield, K. L. et al. Ebola virus-like particle-based vaccine protects nonhuman primates against lethal Ebola virus challenge. J. Infect. Dis. 196 (Suppl. 2), S430–S437 (2007).
Warfield, K. L. et al. Homologous and heterologous protection of nonhuman primates by Ebola and Sudan virus-like particles. PLoS ONE 10, e0118881 (2015).
Swenson, D. L. et al. Monovalent virus-like particle vaccine protects guinea pigs and nonhuman primates against infection with multiple Marburg viruses. Expert Rev. Vaccines 7, 417–429 (2008).
Marzi, A. et al. An Ebola whole-virus vaccine is protective in nonhuman primates. Science 348, 439–442 (2015).
Sullivan, N. J., Sanchez, A., Rollin, P. E., Yang, Z. Y. & Nabel, G. J. Development of a preventive vaccine for Ebola virus infection in primates. Nature 408, 605–609 (2000).
Falzarano, D. et al. Single immunization with a monovalent vesicular stomatitis virus-based vaccine protects nonhuman primates against heterologous challenge with Bundibugyo ebolavirus. J. Infect. Dis. 204 (Suppl. 3), S1082–S1089 (2011).
Hensley, L. E. et al. Demonstration of cross-protective vaccine immunity against an emerging pathogenic ebolavirus species. PLoS Pathog. 6, e1000904 (2010).
Choi, J. H. et al. A single dose respiratory recombinant adenovirus-based vaccine provides long-term protection for non-human primates from lethal Ebola infection. Mol. Pharm. 12, 2712–2731 (2015).
Mire, C. E. et al. Durability of a vesicular stomatitis virus-based Marburg virus vaccine in nonhuman primates. PLoS ONE 9, e94355 (2014).
Huttner, A. et al. The effect of dose on the safety and immunogenicity of the VSV Ebola candidate vaccine: a randomised double-blind, placebo-controlled phase 1/2 trial. Lancet Infect. Dis. 15, 1156–1166 (2015).
Huttner, A. et al. A dose-dependent plasma signature of the safety and immunogenicity of the rVSV-Ebola vaccine in Europe and Africa. Sci. Transl Med. 9, eaaj1701 (2017).
Agnandji, S. T. et al. Phase 1 trials of rVSV Ebola vaccine in Africa and Europe. N. Engl. J. Med. 374, 1647–1660 (2016).
Regules, J. A. et al. A recombinant vesicular stomatitis virus Ebola vaccine. N. Engl. J. Med. 376, 330–341 (2017).
ElSherif, M. S. et al. Assessing the safety and immunogenicity of recombinant vesicular stomatitis virus Ebola vaccine in healthy adults: a randomized clinical trial. CMAJ 189, E819–E827 (2017).
Zhu, F. C. et al. Safety and immunogenicity of a novel recombinant adenovirus type-5 vector-based Ebola vaccine in healthy adults in China: preliminary report of a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet 385, 2272–2279 (2015).
Zhu, F. C. et al. Safety and immunogenicity of a recombinant adenovirus type-5 vector-based Ebola vaccine in healthy adults in Sierra Leone: a single-centre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 389, 621–628 (2017).
Li, J. X. et al. Immunity duration of a recombinant adenovirus type-5 vector-based Ebola vaccine and a homologous prime-boost immunisation in healthy adults in China: final report of a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Glob. Health 5, e324–e334 (2017).
Dolzhikova, I. V. et al. Safety and immunogenicity of GamEvac-Combi, a heterologous VSV- and Ad5-vectored Ebola vaccine: An open phase I/II trial in healthy adults in Russia. Hum. Vaccin Immunother. 13, 613–620 (2017).
De Santis, O. et al. Safety and immunogenicity of a chimpanzee adenovirus-vectored Ebola vaccine in healthy adults: a randomised, double-blind, placebo-controlled, dose-finding, phase 1/2a study. Lancet Infect. Dis. 16, 311–320 (2016).
Ewer, K. et al. A monovalent chimpanzee adenovirus Ebola vaccine boosted with MVA. N. Engl. J. Med. 374, 1635–1646 (2016).
Tapia, M. D. et al. Use of ChAd3-EBO-Z Ebola virus vaccine in Malian and US adults, and boosting of Malian adults with MVA-BN-Filo: a phase 1, single-blind, randomised trial, a phase 1b, open-label and double-blind, dose-escalation trial, and a nested, randomised, double-blind, placebo-controlled trial. Lancet Infect. Dis. 16, 31–42 (2016).
Ledgerwood, J. E. et al. Chimpanzee adenovirus vector Ebola vaccine. N. Engl. J. Med. 376, 928–938 (2017).
Milligan, I. D. et al. Safety and immunogenicity of novel adenovirus Type 26- and modified vaccinia ankara-vectored Ebola vaccines: a randomized clinical trial. JAMA 315, 1610–1623 (2016).
Kibuuka, H. et al. Safety and immunogenicity of Ebola virus and Marburg virus glycoprotein DNA vaccines assessed separately and concomitantly in healthy Ugandan adults: a phase 1b, randomised, double-blind, placebo-controlled clinical trial. Lancet 385, 1545–1554 (2015).
Sarwar, U. N. et al. Safety and immunogenicity of DNA vaccines encoding Ebolavirus and Marburgvirus wild-type glycoproteins in a phase I clinical trial. J. Infect. Dis. 211, 549–557 (2015).
Snoy, P. J. Establishing efficacy of human products using animals: the US food and drug administration's “animal rule”. Vet. Pathol. 47, 774–778 (2010).
Geisbert, T. W. et al. Vesicular stomatitis virus-based ebola vaccine is well-tolerated and protects immunocompromised nonhuman primates. PLoS Pathog. 4, e1000225 (2008).
Mire, C. E. et al. Recombinant vesicular stomatitis virus vaccine vectors expressing filovirus glycoproteins lack neurovirulence in nonhuman primates. PLoS Negl. Trop. Dis. 6, e1567 (2012).
Ebihara, H. et al. In vitro and in vivo characterization of recombinant Ebola viruses expressing enhanced green fluorescent protein. J. Infect. Dis. 196 (Suppl. 2), S313–S322 (2007).
Albarino, C. G. et al. Development of a reverse genetics system to generate recombinant Marburg virus derived from a bat isolate. Virology 446, 230–237 (2013).
Albarino, C. G., Wiggleton Guerrero, L., Lo, M. K., Nichol, S. T. & Towner, J. S. Development of a reverse genetics system to generate a recombinant Ebola virus Makona expressing a green fluorescent protein. Virology 484, 259–264 (2015).
Koehler, A. et al. A single amino acid change in the Marburg virus matrix protein VP40 provides a replicative advantage in a species-specific manner. J. Virol. 90, 1444–1454 (2015).
Hoenen, T., Groseth, A., de Kok-Mercado, F., Kuhn, J. H. & Wahl-Jensen, V. Minigenomes, transcription and replication competent virus-like particles and beyond: reverse genetics systems for filoviruses and other negative stranded hemorrhagic fever viruses. Antiviral Res. 91, 195–208 (2011).
Muhlberger, E., Lotfering, B., Klenk, H. D. & Becker, S. Three of the four nucleocapsid proteins of Marburg virus, NP, VP35, and L, are sufficient to mediate replication and transcription of Marburg virus-specific monocistronic minigenomes. J. Virol. 72, 8756–8764 (1998).
Muhlberger, E., Weik, M., Volchkov, V. E., Klenk, H. D. & Becker, S. Comparison of the transcription and replication strategies of marburg virus and Ebola virus by using artificial replication systems. J. Virol. 73, 2333–2342 (1999).
Watanabe, S. et al. Production of novel ebola virus-like particles from cDNAs: an alternative to ebola virus generation by reverse genetics. J. Virol. 78, 999–1005 (2004).
Hoenen, T. et al. Infection of naive target cells with virus-like particles: implications for the function of ebola virus VP24. J. Virol. 80, 7260–7264 (2006).
Watt, A. et al. A novel life cycle modeling system for Ebola virus shows a genome length-dependent role of VP24 in virus infectivity. J. Virol. 88, 10511–10524 (2014).
Hoenen, T., Watt, A., Mora, A. & Feldmann, H. Modeling the lifecycle of Ebola virus under biosafety level 2 conditions with virus-like particles containing tetracistronic minigenomes. J. Vis. Exp. 91, 52381 (2014).
Hoenen, T. & Feldmann, H. Reverse genetics systems as tools for the development of novel therapies against filoviruses. Expert Rev. Anti Infect. Ther. 12, 1253–1263 (2014).
Wenigenrath, J., Kolesnikova, L., Hoenen, T., Mittler, E. & Becker, S. Establishment and application of an infectious virus-like particle system for Marburg virus. J. Gen. Virol. 91, 1325–1334 (2010).
Geisbert, T. W., Strong, J. E. & Feldmann, H. Considerations in the use of nonhuman primate models of ebola virus and marburg virus infection. J. Infect. Dis. 212 (Suppl. 2), S91–S97 (2015).
Acknowledgements
The authors thank many colleagues in the field for helpful discussions as well as the intramural and extramural divisions of the National Institutes of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH) for financial support of their work over the past decade. This work was supported in part by the US Department of Health and Human Services, NIH grant U19AI109711 to T.W.G. The opinions, interpretations, conclusions and recommendations are those of the authors and are not necessarily endorsed by the University of Texas Medical Branch at Galveston or the NIH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
T.W.G. and H.F. are listed on several patents and/or patent applications for a number of vaccines and treatments for filoviruses. These patents were related to work performed as official duties when T.W.G. was employed by the US Department of Defense and when H.F. was employed by the Public Health Agency of Canada. T.W.G. and H.F. have not received any royalties related to any filovirus vaccine or treatment patents. T.W.G. receives grants related to filovirus research from the US National Institutes of Health (HHSN261200800001E, R01AI09894301, R01AI09881701, U19AI109711, U19AI109945 and R01AI119185) and the US Department of Defense (W911QY-14-1-001, HDTRA1-12-1-0051 and HDTRA1-17-C-0009).
Glossary
- Biosafety level 4 (BSL-4) containment
-
The highest level of biocontainment. BSL-4 precautions are required for work with agents that can be aerosolized and cause fatal disease.
- Asthenia
-
Muscular weakness.
- Myalgia
-
Muscular pain.
- Arthralgia
-
Joint pain.
- Leukopenia
-
A condition of having a low number of circulating leukocytes, including neutrophils, eosinophils, basophils, lymphocytes and monocytes.
- Lymphocytopenia
-
A condition of having a low number of circulating leukocytes, including natural killer cells, T cells and B cells.
- Thrombocytopenia
-
A condition of having a low number of circulating thrombocytes, also known as platelets.
- D-dimers
-
Products of clot degradation. D-dimers consist of two crosslinked D fragments of fibrin.
- Encephalitis
-
Inflammation of the brain.
- Cell tropism
-
A situation in which viruses replicate in one cell type but not another. This tropism likely results from the differential expression of host surface proteins.
- Disseminated intravascular coagulation
-
A process in which the blood clots in numerous small vessels.
- Type I transmembrane protein
-
An integral single-pass membrane protein that has an extracellular N-terminus and a cytoplasmic C-terminus.
- Stress granule
-
A structure found in stressed cells that contains proteins and RNA stored in stalled translational complexes.
- Viraemia
-
The presence of virus particles in the blood.
- Convalescent serum
-
Blood serum obtained from individuals who have recovered from a disease.
- Avidity
-
The strength of binding between two molecules.
- Prodrug
-
A compound that, once processed in the body, releases a therapeutic drug.
- Nucleoside analogue
-
A compound that interferes with nucleic acid elongation owing to structural similarities with nucleosides. Nucleoside analogues are commonly used as antiviral agents.
- Meningoencephalitis
-
A condition in which both the brain and the meninges are inflamed.
- Buffy coat
-
A blood fraction that is particularly high in leukocytes and platelets.
- Vascular leak syndrome
-
A condition characterized by the escape of plasma through capillary walls.
- Cytokine storm
-
A potentially fatal condition characterized by the excessive release of cytokines and associated inflammation. Ebola virus and Marburg virus cause lethal haemorrhagic fever in humans and non-human primates \(NHPs\). Filoviruses present a global health threat both as naturally acquired diseases and as potential agents of bioterrorism.
Rights and permissions
About this article

Cite this article
Cross, R., Mire, C., Feldmann, H. et al. Post-exposure treatments for Ebola and Marburg virus infections. Nat Rev Drug Discov 17, 413–434 (2018). https://doi.org/10.1038/nrd.2017.251
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd.2017.251
This article is cited by
-
Integrative role of small non-coding RNAs in viral immune response: a systematic review
Molecular Biology Reports (2024)
-
Strong anti-viral nano biocide based on Ag/ZnO modified by amodiaquine as an antibacterial and antiviral composite
Scientific Reports (2022)
-
A cryptic pocket in Ebola VP35 allosterically controls RNA binding
Nature Communications (2022)
-
MSC-derived exosomes carrying a cocktail of exogenous interfering RNAs an unprecedented therapy in era of COVID-19 outbreak
Journal of Translational Medicine (2021)
-
Combination therapy protects macaques against advanced Marburg virus disease
Nature Communications (2021)
