
Key Points
-
Severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) are examples of emerging zoonotic coronavirus infections capable of person-to-person transmission that result in large-scale epidemics with substantial effects on patient health and socioeconomic factors. Unlike patients with mild illnesses that are caused by other human-pathogenic coronaviruses, patients with SARS or MERS coronavirus infections may develop severe acute respiratory disease with multi-organ failure. The case–fatality rates of SARS and MERS are approximately 10% and 35%, respectively.
-
Both SARS and MERS pose major clinical management challenges because there is no specific antiviral treatment that has been proven to be effective in randomized clinical trials for either infection. Substantial efforts are underway to discover new therapeutic agents for coronavirus infections.
-
Virus-based therapies include monoclonal antibodies and antiviral peptides that target the viral spike glycoprotein, viral enzyme inhibitors, viral nucleic acid synthesis inhibitors and inhibitors of other viral structural and accessory proteins.
-
Host-based therapies include agents that potentiate the interferon response or affect either host signalling pathways involved in viral replication or host factors utilized by coronaviruses for viral replication.
-
The major challenges in the clinical development of novel anti-coronavirus drugs include the limited number of suitable animal models for the evaluation of potential treatments for SARS and MERS, the current absence of new SARS cases, the limited number of MERS cases — which are also predominantly geographically confined to the Middle East — as well as the lack of industrial incentives to develop antivirals for mild infections caused by other, less pathogenic coronaviruses.
-
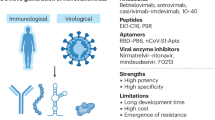
The continuing threat of MERS-CoV to global health 3 years after its discovery presents a golden opportunity to tackle current obstacles in the development of new anti-coronavirus drugs. A well-organized, multidisciplinary, international collaborative network consisting of clinicians, virologists and drug developers, coupled to political commitment, should be formed to carry out clinical trials using anti-coronavirus drugs that have already been shown to be safe and effective in vitro and/or in animal models, particularly lopinavir–ritonavir, interferon beta-1b and monoclonal antibodies and antiviral peptides targeting the viral spike glycoprotein.
Abstract
In humans, infections with the human coronavirus (HCoV) strains HCoV-229E, HCoV-OC43, HCoV-NL63 and HCoV-HKU1 usually result in mild, self-limiting upper respiratory tract infections, such as the common cold. By contrast, the CoVs responsible for severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS), which were discovered in Hong Kong, China, in 2003, and in Saudi Arabia in 2012, respectively, have received global attention over the past 12 years owing to their ability to cause community and health-care-associated outbreaks of severe infections in human populations. These two viruses pose major challenges to clinical management because there are no specific antiviral drugs available. In this Review, we summarize the epidemiology, virology, clinical features and current treatment strategies of SARS and MERS, and discuss the discovery and development of new virus-based and host-based therapeutic options for CoV infections.
Similar content being viewed by others

Main
Coronaviruses (CoVs; subfamily Coronavirinae, family Coronaviridae, order Nidovirales) are a group of highly diverse, enveloped, positive-sense, single-stranded RNA viruses that cause respiratory, enteric, hepatic and neurological diseases of varying severity in a broad range of animal species, including humans1,2,3. CoVs are subdivided into four genera: Alphacoronavirus, Betacoronavirus (βCoV), Gammacoronavirus and Deltacoronavirus2,3,4,5,6,7. Over the past 12 years, two novel βCoVs, severe acute respiratory syndrome CoV (SARS-CoV) and Middle East respiratory syndrome CoV (MERS-CoV), have emerged, and these viruses can cause severe human diseases8,9. The lack of effective drug treatment and associated high morbidity and mortality rates of these two CoVs as well as their potential to cause epidemics highlight the need for novel drug discovery for the treatment of CoV infections.
Epidemiology of SARS and MERS
SARS. SARS-CoV emerged first in southern China and rapidly spread around the globe in 2002–2003 (Refs 8,10,11). In November 2002, an unusual epidemic of atypical pneumonia with a high rate of nosocomial transmission to health-care workers occurred in Foshan, Guangdong, China12,13. In March 2003, a novel CoV was confirmed to be the causative agent for SARS, and was thus named SARS-CoV14,15,16,17. A 64-year-old nephrologist who travelled from southern China to Hong Kong on 21 February 2003 became the index case of subsequent large community and health-care-associated outbreaks of SARS in Hong Kong and other regions10,11,18,19,20,21. The high infectivity of SARS was highlighted by the super-spreading event at a major teaching hospital in Hong Kong in which 138 people, including many previously healthy health-care workers, were infected within 2 weeks of exposure to an index patient who was being managed in a general medical ward for community-acquired pneumonia10,22. Through international air travel, SARS-CoV was spread to 29 countries and regions with a total of 8,098 cases and 774 fatalities (9.6% of cases) by the end of the epidemic in July 2003 (Ref. 23) (see Supplementary information S1 (figure, parts a,b)).
A retrospective serological survey suggested that cross-species transmission of SARS-CoV or its variants from animal species to humans might have occurred frequently in the wet market, where high seroprevalence was detected among asymptomatic animal handlers24. A close variant of SARS-CoV was isolated in palm civets in Dongmen market, Shenzhen, China, in 2003 (Ref. 25). During the small-scale SARS outbreaks in late 2003 and early 2004, three of the four patients had direct or indirect contact with palm civets26,27. However, viral genetic sequence analysis demonstrated that the SARS-CoV-like virus had not been circulating among masked palm civets in markets for a long time, and a serological study showed that only caged market civets and not wild civets were infected with the SARS-CoV-like virus28. CoVs that are highly similar to SARS-CoV have been isolated from Chinese horseshoe bats since 2005 (Refs 29,30,31,32). These SARS-like CoVs from bats share 88–95% sequence homology with human or civet CoV isolates, which suggests that bats were probably the natural reservoir of a close ancestor of SARS-CoV4,33,34.
MERS. The isolation of a novel βCoV from a patient in Jeddah, Saudi Arabia, who died of severe pneumonia and multi-organ failure in June 2012, was first reported in September 2012 (Ref. 35). Initially named 'human coronavirus Erasmus Medical Center', the virus was later renamed MERS-CoV by international consensus, and the disease was called Middle East respiratory syndrome (MERS)36. Retrospective analysis of a cluster of nosocomial cases in April 2012 in Jordan confirmed that MERS-CoV was also responsible for that outbreak37. Over the past 3 years, MERS-CoV has continued to spread within and beyond the Middle East, and there are ongoing reports of sporadic cases and community and health-care-associated clusters of infected individuals in the Middle East, especially in Saudi Arabia and the United Arab Emirates9,38. Travel-related cases and clusters have also been increasingly reported on other continents9. As of 9 October 2015, 1,593 laboratory-confirmed cases of MERS, including 568 deaths, have been reported to the World Health Organization39 (see Supplementary information S1 (figure, parts c,d)).
MERS-CoV is considered primarily to be a zoonotic virus that has the capability of non-sustained person-to-person spread9. Serological and virological studies have shown that camels and bats are the most likely animal reservoirs of MERS-CoV9,40,41,42,43,44,45,46,47. Although not all primary cases of MERS were individuals who had direct contact with camels, such exposure is considered to be an important factor for the spread of MERS-CoV, as evidenced by the substantially increased seroprevalence of anti-MERS-CoV antibodies among individuals with occupational exposure to camels, such as camel shepherds and slaughterhouse workers, relative to the general population in Saudi Arabia48,49. Person-to-person transmission of MERS-CoV has occurred in health-care facilities and family clusters50,51,52,53. The recent, large health-care-associated outbreaks in Jeddah and South Korea have been attributed to poor compliance with infection control measures54,55. Further studies are needed to fully understand the exact mode of transmission and other potential sources of MERS-CoV for optimization of treatment and prevention strategies for MERS56,57.
Clinical features of SARS and MERS. Patients with SARS or MERS present with various clinical features, ranging from asymptomatic or mild respiratory illness to fulminant severe acute respiratory disease with extrapulmonary manifestations8,9. Both diseases have predominantly respiratory manifestations, but extrapulmonary features may occur in severe cases56 (see Supplementary information S2 (table)). Notably, early treatment is especially important for patients with severe MERS because this disease progresses to respiratory distress, renal failure and death much more rapidly than SARS does. The three- to four-fold higher case-fatality rate of MERS relative to SARS may be related to the higher median age and prevalence of comorbidities in patients with MERS as well as the different pathogenesis of the two diseases9,58,59,60,61. Comorbidities associated with severe MERS include obesity, diabetes mellitus, systemic immunocompromising conditions and chronic cardiac and pulmonary diseases9,60,62,63. Although the rate of secondary transmission among household contacts of index MERS patients (which is approximately 4%) and the estimated pandemic potential of MERS-CoV are lower than those of SARS-CoV, the rapidly progressive clinical course and high fatality of MERS continues to pose a major threat to at-risk populations64,65,66,67,68,69,70,71 (see Supplementary information S2 (table)).
Current management strategies for SARS and MERS. Supportive care — including organ support and prevention of complications, especially acute respiratory distress syndrome, organ failure and secondary nosocomial infections — remains the most important management strategy for SARS and MERS, as there is currently no specific antiviral treatment that has been proven to be effective in randomized controlled trials8,9,56,72,73,74,75. Numerous compounds have been found to inhibit the entry and/or replication of SARS-CoV and MERS-CoV in cell culture or in animal models, but activity in vitro and even in animal experiments does not necessarily translate into efficacy in humans8,9. Owing to the high morbidity and mortality rates of SARS and MERS, some of these antiviral drugs and immunomodulators have been used empirically or evaluated in uncontrolled trials8,10,21,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90 (Table 1). Substantial efforts are underway to discover new therapeutic agents for CoV infections and these investigations are based on our understanding of the basic virology of CoVs. Importantly, treatment with these investigational therapies requires application of standard research treatment protocols and systematic clinical and virological data collection in controlled research trials, with the approval of the local ethics committee.
Development of anti-CoV therapeutics
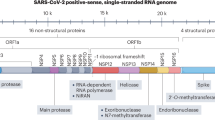
Key CoV targets for new drug development. Despite their high species diversity, CoVs share key genomic elements that are essential for the design of therapeutic targets (Fig. 1). The large replicase polyprotein 1a (pp1a) and pp1ab, which are encoded by the 5′-terminal open reading frame 1a/b (ORF1a/b), are cleaved by two viral proteases, the papain-like protease (PLpro) and the 3C-like protease (3CLpro), to produce non-structural proteins (NSPs) such as RNA-dependent RNA polymerase (RdRp) and helicase, which are involved in the transcription and replication of the virus9,91 (Fig. 2). Numerous enzyme inhibitors targeting these proteins have shown anti-CoV activity in vitro.

The typical coronavirus (CoV) genome is a single-stranded, non-segmented RNA genome, which is approximately 26–32 kb. It contains 5′-methylated caps and 3′-polyadenylated tails and is arranged in the order of 5′, replicase genes, genes encoding structural proteins (spike glycoprotein (S), envelope protein (E), membrane protein (M) and nucleocapsid protein (N)), polyadenylated tail and then the 3′ end. The partially overlapping 5′-terminal open reading frame 1a/b (ORF1a/b) is within the 5′ two-thirds of the CoV genome and encodes the large replicase polyprotein 1a (pp1a) and pp1ab. These polyproteins are cleaved by papain-like cysteine protease (PLpro) and 3C-like serine protease (3CLpro) to produce non-structural proteins, including RNA-dependent RNA polymerase (RdRp) and helicase (Hel), which are important enzymes involved in the transcription and replication of CoVs. The 3′ one-third of the CoV genome encodes the structural proteins (S, E, M and N), which are essential for virus–cell-receptor binding and virion assembly, and other non-structural proteins and accessory proteins that may have immunomodulatory effects297. Particle image from Ref. 296, Nature Publishing Group. MERS, Middle East respiratory syndrome; SARS, severe acute respiratory syndrome.

Binding between the receptor-binding domain on the S1 subunit of spike glycoprotein (S) and the host receptor triggers conformational changes in the S2 subunit of S. This leads to fusion of the viral and cell membranes. Coronaviruses (CoVs) enter the host cell using the endosomal pathway and/or the cell surface non-endosomal pathway. Endosomal cell entry of CoVs is facilitated by low pH and the pH-dependent endosomal cysteine protease cathepsins. S is activated and cleaved into the S1 and S2 subunits by other host proteases, such as transmembrane protease serine 2 (TMPRSS2) and TMPRSS11D, which enables cell surface non-endosomal virus entry at the plasma membrane. Middle East respiratory syndrome (MERS)-CoV S is additionally activated by the serine endoprotease furin. CoVs then dissemble intracellularly to release the nucleocapsid and viral RNA into the cytoplasm for the translation of ORF1a/b into the large replicase polyprotein 1a (pp1a) and pp1ab and for the replication of genomic RNA. pp1a and pp1ab are cleaved by papain-like protease (PLpro) and 3C-like protease (3CLpro) to produce non-structural proteins (NSPs), including RNA-dependent RNA polymerase (RdRp) and helicase, which are involved in the transcription and replication of the virus. The NSPs produced by the cleavage of pp1a and pp1ab form the replication–transcription complex. Attachment of the hydrophobic domains of the CoV replication–transcription complex to the limiting membrane derived from the endoplasmic reticulum (ER) produces typical CoV replication structures including double-membrane vesicles and convoluted membranes. The full-length positive-strand genomic RNA is transcribed to form a full-length negative-strand template for synthesis of new genomic RNAs and overlapping subgenomic negative-strand templates. Subgenomic mRNAs are then synthesized and translated to produce the structural and accessory proteins. The helical nucleocapsid formed by the assembly of nucleocapsid protein (N) and genomic RNA interacts with the other structural proteins to form the assembled virion, which is then released by exocytosis into the extracellular compartment. Virus- and host-based treatment options are highlighted in red and blue, respectively. +, positive-strand RNA; −, negative-strand RNA; AP, accessory protein; CYP, cyclophilin; dec-RVKR-CMK, decanoyl-Arg-Val-Lys-Arg-chloromethylketone; DRACO, double-stranded RNA-activated caspase oligomerizer; E, envelope protein; ER, endoplasmic reticulum; ERGIC, endoplasmic reticulum Golgi intermediate compartment; ERK, extracellular signal-regulated kinase; M, membrane; mAb, monoclonal antibody; MAPK, mitogen-activated protein kinase; MPA, mycophenolic acid; mTOR, mammalian target of rapamycin; N, nucleocapsid protein; NAAE, N-(2-aminoethyl)-1-aziridine-ethanamine; NFAT, nuclear factor of activated T cells; ORF, open reading frame; PI3K, phosphoinositide 3-kinase; poly(I:C), polyinosinic:polycytidylic acid; RdRp, RNA-dependent RNA polymerase; S, spike glycoprotein; SARS-CoV, severe acute respiratory syndrome coronavirus; siRNA, small interfering RNA. *Only siRNAs that have been evaluated in published reports are included. siRNAs directed against other parts of the CoV genome would also be expected to diminish the accumulation or translation of genomic and all upstream subgenomic RNAs. Adapted with permission from Ref. 9, American Society for Microbiology.
The surface structural spike glycoprotein (S) is of particular interest for antiviral development because of its critical role in the virus–cell receptor interaction. S is composed of the amino-terminal receptor-binding S1 and carboxy-terminal membrane fusion S2 subunits. Cleavage at the protease site at the S1–S2 junction is required to activate membrane fusion, virus entry and syncytium formation9. Binding of the S1 subunit receptor-binding domain (RBD) to the host receptor triggers conformational changes in the S2 subunit (the stalk region of S) to bring the viral and cell membranes into close proximity and enable fusion92. Monoclonal antibodies (mAbs) against the S1 subunit RBD and fusion inhibitors targeting the S2 subunit have potent anti-CoV activity in vitro and/or in vivo92,93,94,95,96,97,98,99,100. The key functional host receptors utilized by human pathogenic CoVs include angiotensin-converting enzyme 2 (ACE2; used by SARS-CoV and human CoV (HCoV)-NL63), dipeptidyl peptidase 4 (DPP4; used by MERS-CoV), aminopeptidase N (used by HCoV-229E), and O-acetylated sialic acid (used by HCoV-OC43 and HCoV-HKU1)101,102,103,104,105,106. The host receptor is important in determining the pathogenicity, tissue tropism and host range of the virus. mAbs or agents that target the host receptor are potential anti-CoV agents so long as they do not induce immunopathological effects in animal models.
CoVs enter the host cell using the endosomal pathway and/or the cell surface non-endosomal pathway9 (Fig. 2). Low pH and the pH-dependent endosomal cysteine protease cathepsins help to overcome the energetically unfavourable membrane fusion reaction and facilitate endosomal cell entry of CoVs107,108. Other host proteases, such as transmembrane protease serine 2 (TMPRSS2) and TMPRSS11D (also known as airway trypsin-like protease), cleave S into the S1 and S2 subunits to activate S for cell surface non-endosomal virus entry at the plasma membrane109. Inhibitors of these proteases can abrogate this proteolytic cleavage and partially block cell entry109. MERS-CoV S is also activated by furin, a serine endoprotease that has been implicated in the processing of fusion proteins and cell entry of other RNA viruses, including HIV, avian influenza A/H5N1 virus, Ebola virus, Marburg virus and flaviviruses110. Furin is also involved in MERS-CoV S1/S2 cleavage during egress from the infected cell110. Monotherapy and/or combinatorial treatment with inhibitors of host proteases involved in the various cell entry pathways have potent anti-CoV activity in vitro and should be further evaluated in animal studies109,111.
CoVs disassemble inside the host cell and release the nucleocapsid and viral RNA into the cytoplasm, after which ORF1a/b is translated into pp1a and pp1ab, and the genomic RNA is replicated91. The numerous NSPs produced by the cleavage of pp1a and pp1ab form the replication–transcription complex. Attachment of the hydrophobic domains of the CoV replication–transcription complex to the limiting membrane derived from the endoplasmic reticulum produces typical CoV replication structures including double-membrane vesicles (DMVs) and convoluted membranes112,113. Novel agents, such as K22, that target membrane-bound CoV RNA synthesis inhibit DMV formation of a broad range of human and animal CoVs112. The full-length positive strand of genomic RNA is transcribed to form a full-length negative-strand template for the synthesis of new genomic RNAs and overlapping subgenomic negative-strand templates9,91. Subgenomic mRNAs are then synthesized and translated to produce the structural and accessory proteins9,91. The helical nucleocapsid, formed by the assembly of nucleocapsid protein (N) and genomic RNA, then interacts with S, envelope protein (E), and membrane protein (M) to form the assembled virion9. The virion is released into the extracellular compartment by exocytosis and the viral replication cycle is repeated9. Small interfering RNAs (siRNAs) targeting these structural genes could be useful in the treatment of CoV infections, and further optimization of the in vivo delivery of siRNAs may enable their clinical use.
Approaches to anti-CoV drug screening. The only two human-pathogenic CoVs known before the SARS epidemic were HCoV-229E and HCoV-OC43, which usually cause self-limiting upper respiratory tract infections2. Therefore, researchers and research facilities, especially those involved in antiviral development, were underprepared when SARS-CoV suddenly emerged in 2003. Subsequently, three general approaches were used to discover potential anti-CoV treatment options for human-pathogenic CoVs — especially SARS-CoV and the emerging MERS-CoV — that are associated with more severe disease than the other HCoVs are9,114,115.
The first approach to drug discovery is to test existing broad-spectrum antiviral drugs that have been used to treat other viral infections by using standard assays that measure the effects of these drugs on the cytopathicity, virus yield and plaque formation of live and/or pseudotyped CoVs. Examples of drugs identified using this approach include interferon alfa, interferon beta, interferon gamma, ribavirin and inhibitors of cyclophilin8,74,116,117,118,119,120,121,122. These drugs have the obvious advantage of being readily available with known pharmacokinetic and pharmacodynamic properties, side effects and dosing regimens. However, they do not have specific anti-CoV effects and may be associated with severe adverse effects.
The second anti-CoV drug discovery approach involves the screening of chemical libraries comprising large numbers of existing compounds or databases that contain information on transcriptional signatures in different cell lines122,123,124,125,126,127. This approach provides rapid, high-throughput screening of many readily available compounds that can then be further evaluated by antiviral assays. Various classes of drugs have been identified in these drug repurposing programmes, including many that have important physiological and/or immunological effects such as those that affect neurotransmitter regulation, the oestrogen receptor, kinase signalling, lipid or sterol metabolism, protein processing and DNA synthesis or repair122,123,124,125,126,127. The major disadvantage of this approach is that although many of the identified drugs exhibit anti-CoV activities in vitro, most are not clinically useful because they are either associated with immunosuppressive effects or they have anti-CoV half-maximal effective concentration (EC50) values that markedly exceed the peak serum concentration (Cmax) levels that are achievable at therapeutic dosages. A notable exception, which was found to be effective in a non-human primate model and in non-randomized clinical trials, is the anti-HIV protease inhibitor lopinavir–ritonavir76,77,128 (Table 1).
The third approach for anti-CoV drug discovery involves the de novo development of novel, specific agents based on the genomic and biophysical understanding of the individual CoVs. Examples include siRNA molecules or inhibitors that target specific viral enzymes involved in the viral replication cycle, mAbs that target the host receptor, inhibitors of host cellular proteases, inhibitors of virus endocytosis by the host cell, human or humanized mAbs that target the S1 subunit RBD and antiviral peptides that target the S2 subunit (Fig. 2). Although most of these drugs have potent in vitro and/or in vivo anti-CoV activity, their pharmacokinetic and pharmacodynamic properties and side-effect profiles have yet to be evaluated in animal and human trials. Furthermore, the development of these candidate drugs into clinically useful therapeutic options with reliable delivery modes for patients usually takes years.
Overall, these three drug discovery approaches are often used together during emerging CoV outbreaks to identify candidate drug compounds that can be broadly classified into virus-based and host-based treatment options.
Virus-based anti-CoV treatment options
Viral nucleosides, nucleotides and nucleic acids. Nucleosides and nucleotides are the building blocks of viral nucleic acids (Fig. 2). Drugs that target nucleosides or nucleotides and/or viral nucleic acids generally have broad-spectrum activity against a wide range of CoVs and other viruses (Table 2). Mycophenolate mofetil is an anti-rejection drug that inhibits inosine monophosphate dehydrogenase and the synthesis of guanine monophosphate122. The active compound, mycophenolic acid, exhibits antiviral activity in vitro against various viruses, including hepatitis B virus (HBV), hepatitis C virus (HCV) and arboviruses122. Mycophenolic acid was identified as a potential anti-MERS-CoV drug using high-throughput screening and has potent anti-MERS-CoV activity in vitro122. However, a subsequent study in a non-human primate model showed that MERS-CoV-infected common marmosets treated with mycophenolate mofetil had a worse outcome with more severe disease and higher viral loads in necropsied lung and extrapulmonary tissues than untreated animals did128. Renal transplant recipients who were on maintenance mycophenolate mofetil therapy also developed severe or fatal MERS129,130. Thus, the usual dosage of mycophenolate mofetil monotherapy is unlikely to be useful for prophylaxis or treatment of CoV infections.
Ribozymes (also known as catalytic RNA or RNA enzymes) are RNA molecules that catalyse specific biochemical reactions. A chimeric DNA–RNA hammerhead ribozyme that specifically recognizes the base sequence GUC, which is present in the loop region of SARS-CoV mRNA, substantially reduces the expression of recombinant SARS-CoV RNA in vitro131. However, ribozymes are rapidly degraded in vivo and delivery methods would have to be optimized in humans before ribozymes could become clinically useful.
Agents targeting the specific host cell membrane-bound CoV replication complex have also been investigated. One such compound, K22, inhibits membrane-bound CoV RNA synthesis and is active against a broad range of CoVs in vitro112. In cell culture, K22 exerts potent anti-CoV activity during an early step of the viral replication cycle and impairs formation of DMVs112. HCoV-229E escape mutants that are resistant to K22 have substitutions in the potential membrane-spanning domains in nsp6, a membrane-spanning integral component of the CoV replication complex that is involved in DMV formation, including nsp6H121L and nsp6M159V (Ref. 112). The emergence of K22 resistance should be monitored in subsequent in vivo studies.
Recently, a new class of broad-spectrum antivirals that targets long viral double-stranded RNA (dsRNA) has been reported. For example, dsRNA-activated caspase oligomerizer (DRACO) is a chimeric protein with a viral dsRNA-binding domain and a pro-apoptotic domain that selectively induces apoptosis in cells that contain viral dsRNA but spares uninfected host cells132. DRACO is active against many RNA viruses in vitro and/or in vivo132. If an effective mode of DRACO delivery can be achieved, a broad-spectrum anti-CoV drug that targets highly conserved CoV RNA sequences might become a reality.
Viral enzymes. All of the major enzymes and proteins of CoVs that are involved in viral replication are potentially druggable targets (Table 2). The SARS-CoV and MERS-CoV PLpro enzymes exhibit proteolytic, deubiquitylating and deISGylating activities133,134,135. Crystallography has facilitated the characterization of these PLpro enzymes and the identification of PLpro inhibitors136. Numerous SARS-CoV PLpro inhibitors belonging to different classes have been identified, including small-molecule inhibitors, thiopurine compounds, natural products, zinc ion and zinc conjugate inhibitors and naphthalene inhibitors137. However, some of these drugs only inhibit the enzymatic activities of PLpro without inhibiting viral replication, or vice versa137,138,139. None has been validated in animal or human studies137,138. Furthermore, most PLpro inhibitors have narrow-spectrum activity because of the structural differences among the PLpro enzymes of different CoVs140,141. For example, most SARS-CoV PLpro inhibitors are inactive against MERS-CoV because of the structurally different, flexible blocking loop 2 (BL2) domains in the PLpro enzymes of SARS-CoV and MERS-CoV140.
3CLpro is the other major CoV protease that cleaves the large replicase polyproteins during viral replication. SARS-CoV 3CLpro can be targeted by numerous classes of protease inhibitors, including zinc or mercury conjugates, C2-symmetric diols, peptidomimetic-α,β-unsaturated esters, anilides, benzotriazole, N-phenyl-2-acetamide, biphenyl sulfone, glutamic acid and glutamine peptides with a trifluoromethylketone group, pyrimidinone and pyrazole analogues142. Some of these 3CLpro inhibitors demonstrate broad-spectrum in vitro activities against CoVs with highly similar key residues for substrate recognition at their 3CLpro enzymes143,144. Among these 3CLpro inhibitors, the most readily available one is lopinavir, a protease inhibitor used to treat HIV infections that is usually marketed as a ritonavir-boosted form (lopinavir–ritonavir). Lopinavir and/or lopinavir–ritonavir have anti-CoV activity in vitro, as well as in MERS-CoV-infected non-human primates and in non-randomized trials of SARS patients76,77,123,128,145. It is postulated that the 3CLpro-inhibiting activity of lopinavir–ritonavir contributes at least partially to its anti-CoV effects146. It remains to be seen if resistance emerges, as it has in patients with HIV infection, when lopinavir–ritonavir is routinely used to treat CoV infections.
RdRp is an essential part of the CoV replication–transcription complex and is involved in the production of genomic and subgenomic RNAs. Ribavirin is a guanosine analogue with broad-spectrum antiviral activity and has been used in the treatment of severe respiratory syncytial virus infection, HCV infection and viral haemorrhagic fevers. Its exact mechanism of action is undetermined, but inhibition of mRNA capping and induction of mutations in RNA-dependent viral replication are considered to be important for RNA viruses, including CoVs147. High-dose ribavirin has been used to treat SARS patients, but the benefits were unclear8,10,21,72,74,75,117. It exhibits moderate anti-MERS-CoV activity at high doses in vitro and in MERS-CoV-infected rhesus macaques, but there was no obvious survival benefit in small cohorts of MERS patients86,87,88,89,121,148. Moreover, the severe side effects associated with the use of high-dose ribavirin limit its clinical application in patients with severe CoV infections8,74. Recently, a novel adenosine analogue, BCX4430 (Immucillin-A), was developed149. It acts as a non-obligate RNA chain terminator to inhibit viral RNA polymerases of a wide range of RNA viruses, including CoVs such as SARS-CoV and MERS-CoV as well as filoviruses such as Ebola and Marburg viruses149. Its development for human use has been fast-tracked to increase the number of treatment options for the recent Ebola virus epidemic in West Africa. Existing nucleoside analogues, such as acyclovir, could be modified by incorporating fleximers, which have increased binding affinity and can overcome resistance caused by point mutations in biologically important binding sites150. These acyclic fleximer nucleoside analogues inhibit MERS-CoV and HCoV-NL63 in vitro at micromolar concentrations150. Notably, resistance to nucleoside analogues due to mutations in RdRp has been reported for other RNA viruses, and should be monitored when these agents are used to treat CoV infections. In addition to nucleoside analogues, siRNA molecules targeting SARS-CoV RdRp have been used to inhibit SARS-CoV in vitro151,152.
Helicase catalyses the unwinding of duplex oligonucleotides into single strands in an ATP-dependent reaction during the CoV replication cycle. Helicase inhibitors are attractive anti-CoV treatment options because the helicases of different CoVs are highly homologous. Based on their mechanisms of action, CoV helicase inhibitors can be broadly categorized into two groups. The first group includes bananins and 5-hydroxychromone derivatives, which inhibit the unwinding and ATPase activity of SARS-CoV helicase, resulting in inhibition of viral replication in vitro153,154. However, the toxicity resulting from the inhibition of cellular ATPases or kinases by these compounds has limited their development for human use. The second group of CoV helicase inhibitors includes compounds that selectively inhibit the unwinding activity but not the ATPase activity of CoV helicase. An example is SSYA10-001, a triazole that inhibits a broad range of CoVs, including SARS-CoV, MERS-CoV and mouse hepatitis virus155,156. The toxicity of SSYA10-001 should be evaluated in animal models.
Viral spike glycoprotein. The membrane-anchored glycoprotein, S, is a major immunogenic antigen and is essential for the interaction between the virus and the host cell receptor (Fig. 2). Adoptive transfer of sera containing anti-MERS-CoV-S antibodies blocked virus attachment and accelerated viral clearance from the lungs of MERS-CoV infected BALB/c mice that were recently transduced by adenoviral vectors expressing human DPP4 (Ref. 157) (Table 2). Small cohorts of SARS patients who received convalescent-phase plasma containing neutralizing antibodies that probably targeted CoV S had significantly higher discharge rates by 3 weeks after symptom onset and a lower mortality rate83,84. However, the use of convalescent-phase plasma therapy during emerging CoV outbreaks is limited by the good will of convalescent patients with high serum neutralizing antibody titres. Disease worsening associated with immune enhancement that results from treatment with products containing low antibody titres has been reported in cell line and animal studies158,159. To overcome these problems, mAbs targeting different regions of SARS-CoV S have been generated by immunization of human immunoglobulin transgenic mice, cloning of small chain variable regions from naive and convalescent patients as well as from immortalization of convalescent S-specific B cells160. Most of these mAbs target specific epitopes on the S1 subunit RBD to inhibit virus–cell receptor binding, whereas others bind to the S2 subunit to interrupt virus–cell fusion160. Regardless of their binding sites and mechanisms, these mAbs exhibit neutralizing activities and reduced viral titres in vitro and/or in small animal models. Similarly, several mAbs targeting different epitopes on the S1 subunit RBD of MERS-CoV S have been developed94,95,96,97,100. These monoclonal antibodies bind to the RBD with 10-fold to >450-fold higher affinity than does human DPP4, resulting in broader and higher neutralizing activity in vitro. Importantly, combination therapy with two or more synergistically acting humanized or human mAbs targeting non-cross-resistant epitopes or different regions of S may help to reduce the frequency with which viruses mutate to escape antibody-mediated neutralization94. Treatment with these mAbs showed protective effects in MERS-CoV-infected human DPP4-transgenic mice and mice transduced by adenoviral vectors expressing human DPP4 (Refs 100,161,162). Their safety profiles and treatment effects in patients should be further evaluated.
Antiviral peptides targeting different regions of S are another promising therapeutic strategy. The S2 subunits or stalk regions of both SARS-CoV and MERS-CoV S are class I viral fusion proteins that each contain an N-terminal fusion peptide, heptad repeat 1 (HR1) and HR2 domains, a transmembrane domain and a cytoplasmic domain92. Antiviral peptides analogous to the N terminus, pre-transmembrane domain or the loop region separating the HR1 and HR2 domains of SARS-CoV inhibited virus plaque formation by 40–80% at micromolar concentrations163,164. Similarly, antiviral peptides spanning the HR2 domain of MERS-CoV inhibit S-mediated cell–cell fusion and viral entry into cells in vitro92,93. A peptide called HP2P-M2 that is derived from the HR2 domain, if administered intranasally before or after viral challenge, protected C57BL/6 mice and mice deficient for V(D)J recombination-activating protein 1 (RAG1) that were recently transduced by adenoviral vectors expressing human DPP4 from MERS-CoV infection with 10-fold to >1,000-fold reduction in viral titres in the lung; this protection was enhanced by combining this peptide with interferon beta99. Combining antiviral peptides targeting different regions of the S2 subunit may be synergistic in vitro and overcome the theoretical risk of drug resistance165. Importantly, an analogous fusion inhibitor, enfuvirtide, which binds to glycoprotein 41 of HIV to block membrane fusion and HIV cell entry, has been successfully marketed for treatment of HIV-1 infection166. Primary resistance to enfuvirtide is rare and can be overcome by modifying the drug such that it contains secondary compensatory mutations167,168. This example of successful drug development includes measures to counteract drug resistance and therefore favours antiviral peptides over anti-CoV S siRNAs for further evaluation in vivo; siRNAs have remained in preclinical development despite their reported antiviral activities in vitro and in SARS-CoV-infected rhesus macaques owing to the lack of reliable drug delivery methods in humans169,170,171,172.
Another class of anti-CoV agents that target S to inhibit CoV entry is the carbohydrate-binding agents. Griffithsin is an antiviral protein originally isolated from the red alga Griffithsia spp.173. It binds specifically to oligosaccharides on viral surface glycoproteins such as S and HIV glycoprotein 120. It inhibits a broad range of CoVs, including SARS-CoV, HCoV-229E, HCoV-OC43 and HCoV-NL63 in vitro and in SARS-CoV-infected mice173,174. The optimal delivery modes and safety profiles of these agents in humans should be further evaluated.
Viral envelope, membrane, nucleocapsid and accessory proteins. E, M and N and some of the accessory proteins are not only essential for virion assembly but may also have additional functions that suppress the host immune response to facilitate viral replication. For example, the accessory proteins 4a and 4b, and possibly also M and accessory protein 5 of MERS-CoV, exhibit interferon antagonist activities, and SARS-CoV N acts as a viral suppressor of RNA silencing and suppresses RNA interference triggered by either short hairpin RNAs or siRNAs175,176,177,178 (Table 2). siRNAs targeting E, M, N, ORF3a, ORF7a or ORF7b of SARS-CoV inhibited viral replication in vitro179,180. However, similar to anti-CoV S siRNAs, none of these siRNAs is ready for human use until better delivery methods become available.
Alternatively, an increasing number of agents that target specific binding sites or functions of these proteins are being generated through crystallography and functional assays. Examples include the viroporin inhibitor hexamethylene amiloride, which reduces the ion channel activity of E in SARS-CoV and HCoV-229E, and PJ34, which binds to a distinct ribonucleotide-binding pocket at the N-terminal domain of N in HCoV-OC43 (Refs 181,182,183). However, these agents are likely to be narrow-spectrum as the binding sites and functions of these proteins are unique to individual CoVs.
Novel lipophilic thiazolidine derivatives, such as LJ001 and JL103, are membrane-binding photosensitizers that produce singlet oxygen molecules to induce changes in the properties of lipid membranes and prevent fusion between viral and target cell membranes. They exhibit broad-spectrum activities against numerous enveloped viruses and may be active against CoVs184,185,186,187.
Host-based anti-CoV treatment options
Broad-spectrum host innate immune response. The host innate interferon response is crucial for the control of viral replication after infection188. Although CoVs are able to suppress the interferon response for immune evasion, they remain susceptible to interferon treatment in vitro189,190. The interferon response can be augmented by the administration of recombinant interferons or interferon inducers (Table 3). Recombinant interferon alfa and interferon beta inhibit the replication of both SARS-CoV and MERS-CoV in vitro and in animal models8,99,116,121,122,128,148,191,192. Various combinations of interferon alfa or interferon beta with other antivirals such as ribavirin and/or lopinavir–ritonavir have been used to treat patients with SARS or MERS. Overall, combination treatments consisting of interferons and ribavirin did not consistently improve outcomes8,9,74,86,87,89. The apparent discrepancy between in vitro findings and in vivo outcomes may be related to the high EC50/Cmax ratios of these drugs and the delay between symptom onset and drug administration8,121,122. This delay is especially relevant for MERS patients, as they have a much shorter median time interval between symptom onset and death than do SARS patients9,58. The use of recombinant interferon beta-1b, which has the lowest EC50/Cmax ratio against MERS-CoV among tested preparations of recombinant interferons, should be evaluated in combination with other effective antivirals in clinical trials at early stages of the infection122,128.
Polyinosinic:polycytidylic acid (poly(I:C)) is a synthetic analogue of dsRNA that strongly induces type I interferons. It substantially reduced the MERS-CoV load in BALB/c mice that were transduced by adenoviral vectors expressing human DPP4 shortly before poly(I:C) administration, although its effects in standard cell culture protection assays are not published157. Intramuscular injection of poly(I:C) stabilized with poly-L-lysine and carboxymethylcellulose seemed to be well tolerated by patients with malignant gliomas in Phase II clinical trials193,194. Nitazoxanide is another potent type I interferon inducer that has been used in humans for parasitic infections195. It is a synthetic nitrothiazolyl–salicylamide derivative that exhibits broad-spectrum antiviral activities against both RNA and DNA viruses including canine CoV, influenza viruses, HBV, HCV, HIV, rotavirus, norovirus and flaviviruses195. It has been evaluated in Phase II and Phase III clinical trials for the treatment of HCV infection and influenza and has a good safety profile195,196,197. Other innate immunomodulators that have anti-SARS-CoV effects in animal models include the antimicrobial peptide rhesus θ-defensin 1 and protein cage nanoparticles that elicit a host immune response in inducible bronchus-associated lymphoid tissue198,199. The combined use of interferon inducers and innate immunomodulators with effective antiviral agents may be synergistic and should be evaluated in animal models.
Other host signalling pathways involved in viral replication. In addition to direct potentiation of the interferon response, other cell signalling pathways have been identified as potential anti-CoV treatment targets (Table 3). Cyclophilins interact with SARS-CoV nsp1 to modulate the calcineurin pathway, which is important in the T cell-mediated adaptive immune response120. The calcineurin inhibitor cyclosporine inhibits a broad range of CoVs in vitro118,119,120. However, its clinical application is limited by its immunosuppressive effects and high EC50/Cmax ratio at standard therapeutic dosages. The antiviral activities of newer, non-immunosuppressive calcineurin inhibitors, which are active against HCoV-NL63, should be evaluated for SARS-CoV and MERS-CoV200. Similarly, agents that modulate other cellular signalling pathways, such as kinase signalling pathway inhibitors, also exhibit anti-CoV activities and are commercially available124,125. However, their toxicities may limit their use in patients with severe CoV infections.
Host factors utilized by CoVs for viral replication. CoVs utilize specific host factors for virus entry and replication (Fig. 2). The host receptor can be targeted by specific monoclonal or polyclonal antibodies, peptides or functional inhibitors (Table 3). For example, anti-DPP4 mAbs inhibit MERS-CoV cell entry in vitro201. YS110 is a recombinant humanized IgG1 anti-DPP4 mAb that seems to be well tolerated in patients with advanced solid tumours201. For the treatment of SARS-CoV, small-molecule entry inhibitors such as N-(2-aminoethyl)-1-aziridine-ethanamine (NAAE) inhibit the catalytic activity of ACE2 and SARS-CoV S-mediated cell–cell fusion in vitro202. Synthetic peptides analogous to critical segments of ACE2 also have anti-SARS-CoV activity at micromolar concentrations in vitro203. However, none of these receptor-directed compounds has yet been tested in patients with CoV infections. Their anti-CoV activity is likely to be narrow-spectrum, as different CoVs utilize different host cell receptors. Furthermore, the risks of immunopathology must be assessed, especially given the multiple essential biological and immunological functions of these receptors.
The entry of CoVs into host cells via the endosomal and/or cell surface pathways is facilitated by host proteases that cleave and activate S. Cathepsins are cysteine proteases that are involved in the endosomal pathway and can be inhibited by cathepsin inhibitors such as K11777 and its related vinylsulfone analogues111. These compounds seem to be safe and effective against various parasitic infections in animal models, and have broad-spectrum activities against enveloped RNA viruses such as CoVs (SARS-CoV, MERS-CoV, HCoV-229E and HCoV-NL63), filoviruses (Ebola and Marburg viruses) and paramyxoviruses111,204,205,206. TMPRSS2 is a serine protease that mediates the cell surface entry pathway; camostat mesylate is a synthetic low-molecular-weight serine protease inhibitor that has been used to treat chronic pancreatitis in humans with minimal side effects207,208. This molecule inhibits SARS-CoV and MERS-CoV in vitro and improves survival of SARS-CoV-infected mice109,111. Furin, another ubiquitously expressed host protease, is also important in MERS-CoV S-mediated entry. Blocking furin with decanoyl-Arg-Val-Lys-Arg-chloromethylketone inhibits MERS-CoV entry and cell–cell fusion in vitro110.
Another group of candidate anti-CoV drugs target the endocytosis of CoV during cell entry. Chlorpromazine is an antipsychotic drug used in the treatment of schizophrenia that also affects the assembly of clathrin-coated pits at the plasma membrane. It is active against HCV, alphaviruses and numerous CoVs, including SARS-CoV and MERS-CoV, in vitro123. Cardiotonic steroids that bind sodium/potassium-transporting ATPase subunit α1, such as ouabain and bufalin, also inhibit clathrin-mediated endocytosis of MERS-CoV at nanomolar concentrations209. However, the use of these clathrin-mediated endocytosis inhibitors in patients with CoV infections is limited by either very high EC50/Cmax ratios or toxicity. Alternatively, endocytosis can also be suppressed by a high pH. Chloroquine is an anti-malarial drug that sequesters protons into lysosomes to increase the intracellular pH. It has broad-spectrum antiviral activities against numerous CoVs (SARS-CoV, MERS-CoV, HCoV-229E and HCoV-OC43) and other RNA viruses in vitro123,210,211,212,213,214. However, it did not substantially reduce viral replication in SARS-CoV-infected mice, possibly because the cell surface pathway was not simultaneously blocked215. The anti-CoV effects, pharmacokinetic and pharmacodynamic profiles and toxicity of the combinations of different protease and endocytosis inhibitors that target these different cell entry pathways should be further evaluated in vivo.
Development of MERS-CoV vaccines
Rapid diagnostics and effective vaccines are often complementary to antiviral treatment in the control of epidemics caused by emerging viruses (Box 1). Although there has not been any new human SARS case for over 10 years, sporadic cases and clusters of MERS continue to occur in the Middle East owing to the persistence of zoonotic sources in endemic areas, and these cases spread to other regions. Effective MERS-CoV vaccines are essential for interrupting the chain of transmission from animal reservoirs and infected humans to susceptible hosts. Live-attenuated vaccines, which have been previously evaluated in animal models for SARS, might be associated with disseminated infection in immunocompromised patients. Inactivated vaccines might be associated with immunopathology during animal challenge. These are unfavourable approaches for MERS vaccine development because a substantial proportion of patients with severe MERS have comorbidities or systemic immunocompromising conditions. Other vaccination strategies for MERS that use DNA plasmids, viral vectors, nanoparticles, virus-like particles and recombinant protein subunits are in development and some have been evaluated in animal models157,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233 (Table 4). The availability of a safe and effective MERS-CoV vaccine for dromedary camels and non-immune individuals at high risk of camel contact in endemic regions such as the Middle East and the greater Horn of Africa would be an important measure for controlling the ongoing epidemic.
Outlook and challenges
Animal models for testing anti-CoV drugs. Suitable animal models are especially important for testing anti-CoV drugs because most of these drugs have not been used in humans. In contrast to the limited number of animal models established for the mild infections caused by HCoV-229E, HCoV-OC43, HCoV-NL63 and HCoV-HKU1, various small animal and non-human primate models have been evaluated for studies of the pathogenesis and treatment of SARS and MERS234,235,236,237. The identification of ACE2 and DPP4 as the functional receptors for SARS-CoV and MERS-CoV, respectively, was essential to the development of animal models that are representative of severe human disease101,102. A number of different non-human primates were found to be permissive to SARS-CoV, but none of them consistently reproduced characteristics of the severe human disease, and mortality was not observed237. These models were predominantly useful to fulfil Koch's postulates238. Small animals — including young and aged BALB/c and C57BL/6 mice, knockout mice with deficiencies in T, B and/or NK cells, golden Syrian hamsters and ferrets — could be productively infected with SARS-CoV, but few of them developed clinically apparent disease237. The best available small animal models for SARS utilize transgenic mice that express human ACE2 and/or mouse-adapted SARS-CoV strains that are capable of causing lethal disease in mice239,240,241. The limited availability of these ACE2-transgenic mice and mouse-adapted virus strains remains a major obstacle to testing anti-SARS-CoV drugs.
Similar to SARS, non-human primate models were also used to fulfil Koch's postulates and investigate the pathogenesis of MERS. Rhesus macaques developed only a mild, self-limiting disease and were not optimal for the evaluation of treatments for MERS148,242,243. By contrast, MERS-CoV-infected common marmosets developed a disseminated and fatal infection that closely resembled severe human disease128,244. However, the availability of common marmosets is limited and experiments on these small primates are technically demanding. Therefore, other small animal models for MERS were evaluated. Unlike with SARS-CoV, most small animals — including BALB/c mice, golden Syrian hamsters, ferrets and rabbits — were not susceptible to MERS-CoV infection245,246,247. Intranasal inoculation of adenoviral vectors expressing human DPP4 followed by MERS-CoV inoculation was a novel method that rapidly rendered mice susceptible to MERS-CoV infection, but the disease was relatively mild and confined to the respiratory tract157. Transgenic mice expressing human DPP4 develop severe pulmonary and disseminated infection and are currently the best available small animal model for MERS248. Potential anti-MERS-CoV treatment options identified in in vitro antiviral assays should be further evaluated in these transgenic mice.
Generic challenges in the clinical development of novel anti-CoV drugs. There are a number of virological and patient-associated factors that pose major challenges in the clinical development of novel anti-CoV drugs. First, CoVs are one of the most diverse and rapidly mutating groups of viruses, and novel CoVs emerge repeatedly at unpredictable times. Therefore, most anti-CoV drugs that specifically target the replication apparatus of an existing CoV may not be effective against another novel CoV. This is especially applicable to viral enzyme inhibitors, mAbs and antiviral peptides that target S, as well as agents that target the host cell receptor. Second, there are a limited number of animal models available for infections caused by HCoV-229E, HCoV-OC43, HCoV-NL63 and HCoV-HKU1. Even for SARS and MERS, experiments using suitable animal models such as mice with transgenes encoding ACE2 or DPP4, and non-human primates, are only available in a few designated research biosafety level 3 laboratories, and these experiments are technically demanding. Last and most important, the mild clinical severity of infections caused by HCoV-229E, HCoV-OC43, HCoV-NL63 and HCoV-HKU1, together with the absence of new SARS cases, have made recruitment of patients into clinical trials difficult and reduced the incentives for pharmaceutical companies to develop specific antiviral drugs for these CoV infections. The continuing threat of MERS-CoV to global health security 3 years after its first discovery presents a golden opportunity to tackle current obstacles in the development of new anti-CoV drugs. It is prudent that a well-organized, multidisciplinary, international collaborative network consisting of clinicians, virologists and drug developers, coupled to political commitment, is formed to carry out clinical trials using anti-CoV drugs that have already been shown to be safe and effective in vitro and/or in animal models.
Prioritization of virus-based and host-based treatment options for clinical development. Despite the report of a large number of virus-based and host-based treatment options with potent in vitro activities for SARS and MERS, only a few are likely to fulfil their potential in the clinical setting in the foreseeable future. Most drugs have one or more major limitations that prevent them from proceeding beyond the in vitro stage. First, many drugs have high EC50/Cmax ratios at clinically relevant dosages. Examples of such drugs include cyclosporine, chlorpromazine and interferon alfa. Second, some have severe side effects or cause immunosuppression. For example, the use of high-dose ribavirin may be associated with haemolytic anaemia, neutropenia, teratogenicity and cardiorespiratory distress. MERS-CoV-infected common marmosets treated with mycophenolate mofetil developed a fatal infection with even higher viral loads in their lungs and extrapulmonary tissues than untreated controls did128. Agents targeting host signalling pathways or receptors may induce immunopathology. Furthermore, the lack of a reliable drug delivery method in vivo is particularly problematic for siRNAs and other agents that have not been previously used in humans.
Looking ahead, the most feasible options that should be further evaluated in clinical trials for the ongoing MERS epidemic include monotherapy or combinational therapies that include lopinavir–ritonavir, interferon beta-1b and/or mAbs and antiviral peptides targeting MERS-CoV S. These agents have protective effects against MERS in non-human primate or mouse models. Moreover, they are either marketed drugs (in the case of lopinavir–ritonavir and interferon beta-1b) or they have been successfully developed for other infections (such as palivizumab, which is used for respiratory syncytial virus infection, and enfuvirtide, which is used for HIV infection). In the long term, the development of novel, broad-spectrum, pan-CoV antiviral drugs that are active against a wide range of CoVs may become the ultimate treatment strategy for circulating and emerging CoV infections.
References
Woo, P. C., Lau, S. K., Huang, Y. & Yuen, K. Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 234, 1117–1127 (2009).
Chan, J. F. et al. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 65, 477–489 (2012).
Chan, J. F., Lau, S. K. & Woo, P. C. The emerging novel Middle East respiratory syndrome coronavirus: the 'knowns' and 'unknowns'. J. Formos. Med. Assoc. 112, 372–381 (2013).
Chan, J. F., To, K. K., Tse, H., Jin, D. Y. & Yuen, K. Y. Interspecies transmission and emergence of novel viruses: lessons from bats and birds. Trends Microbiol. 21, 544–555 (2013).
Chan, J. F., To, K. K., Chen, H. & Yuen, K. Y. Cross-species transmission and emergence of novel viruses from birds. Curr. Opin. Virol. 10, 63–69 (2015).
Woo, P. C. et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 86, 3995–4008 (2012).
Lau, S. K. et al. Discovery of a novel coronavirus, China Rattus coronavirus HKU24, from Norway rats supports the murine origin of Betacoronavirus 1 and has implications for the ancestor of Betacoronavirus lineage A. J. Virol. 89, 3076–3092 (2015).
Cheng, V. C., Lau, S. K., Woo, P. C. & Yuen, K. Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 20, 660–694 (2007). This article provides a comprehensive review of SARS-CoV and SARS, including antiviral treatment.
Chan, J. F. et al. Middle East respiratory syndrome coronavirus: another zoonotic betacoronavirus causing SARS-like disease. Clin. Microbiol. Rev. 28, 465–522 (2015). This article provides a comprehensive review of MERS-CoV and MERS, including antiviral treatment.
Lee, N. et al. A major outbreak of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 348, 1986–1994 (2003). This article reports the first major outbreak of SARS.
Tsang, K. W. et al. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N. Engl. J. Med. 348, 1977–1985 (2003).
Zhao, Z. et al. Description and clinical treatment of an early outbreak of severe acute respiratory syndrome (SARS) in Guangzhou, PR China. J. Med. Microbiol. 52, 715–720 (2003).
Xu, R. H. et al. Epidemiologic clues to SARS origin in China. Emerg. Infect. Dis. 10, 1030–1037 (2004).
Peiris, J. S. et al. Coronavirus as a possible cause of severe acute respiratory syndrome. Lancet 361, 1319–1325 (2003). This is the first report SARS-CoV as the likely cause of SARS.
Kuiken, T. et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 362, 263–270 (2003).
Drosten, C. et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1967–1976 (2003).
Ksiazek, T. G. et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 348, 1953–1966 (2003).
Peiris, J. S. et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: a prospective study. Lancet 361, 1767–1772 (2003).
Woo, P. C. et al. Relative rates of non-pneumonic SARS coronavirus infection and SARS coronavirus pneumonia. Lancet 363, 841–845 (2004).
Hsu, L. Y. et al. Severe acute respiratory syndrome (SARS) in Singapore: clinical features of index patient and initial contacts. Emerg. Infect. Dis. 9, 713–717 (2003).
Booth, C. M. et al. Clinical features and short-term outcomes of 144 patients with SARS in the greater Toronto area. JAMA 289, 2801–2809 (2003).
Wong, R. S. & Hui, D. S. Index patient and SARS outbreak in Hong Kong. Emerg. Infect. Dis. 10, 339–341 (2004).
World Health Organization. Summary of probable SARS cases with onset of illness from 1 November 2002 to 31 July 2003. WHO emergencies preparedness, response [online].
Du, L. et al. Analysis on the characteristics of blood serum Ab-IgG detective result of severe acute respiratory syndrome patients in Guangzhou, China. Zhonghua Liu Xing Bing Xue Za Zhi 25, 925–928 (in Chinese) (2004).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276–278 (2003).
Wang, M. et al. SARS-CoV infection in a restaurant from palm civet. Emerg. Infect. Dis. 11, 1860–1865 (2005).
Song, H. D. et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl Acad. Sci. USA 102, 2430–2435 (2005).
Tu, C. et al. Antibodies to SARS coronavirus in civets. Emerg. Infect. Dis. 10, 2244–2248 (2004).
Lau, S. K. et al. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl Acad. Sci. USA 102, 14040–14045 (2005). This is the first report of a SARS-CoV-like bat coronavirus in Chinese horseshoe bats and the likely role of these bats as the natural animal reservoir of SARS-CoV.
Li, W. et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 310, 676–679 (2005).
Ge, X. Y. et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 503, 535–538 (2013).
He, B. et al. Identification of diverse alphacoronaviruses and genomic characterization of a novel severe acute respiratory syndrome-like coronavirus from bats in China. J. Virol. 88, 7070–7082 (2014).
Woo, P. C., Lau, S. K. & Yuen, K. Y. Infectious diseases emerging from Chinese wet-markets: zoonotic origins of severe respiratory viral infections. Curr. Opin. Infect. Dis. 19, 401–407 (2006).
Shi, Z. & Hu, Z. A review of studies on animal reservoirs of the SARS coronavirus. Virus Res. 133, 74–87 (2008).
Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D. & Fouchier, R. A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 367, 1814–1820 (2012). This is the first report of the discovery of MERS-CoV as the likely cause of MERS.
de Groot, R. J. et al. Middle East respiratory syndrome coronavirus (MERS-CoV): announcement of the Coronavirus Study Group. J. Virol. 87, 7790–7792 (2013).
Hijawi, B. et al. Novel coronavirus infections in Jordan, April 2012: epidemiological findings from a retrospective investigation. East. Mediterr. Health J. 19, S12–S18 (2013).
Hui, D. S., Perlman, S. & Zumla, A. Spread of MERS to South Korea and China. Lancet Respir. Med. 3, 509–510 (2015).
World Health Organization. Middle East Respiratory Syndrome coronavirus (MERS-CoV) — Jordan [online], (2015).
Woo, P. C. et al. Comparative analysis of twelve genomes of three novel group 2c and group 2d coronaviruses reveals unique group and subgroup features. J. Virol. 81, 1574–1585 (2007).
Reusken, C. B. et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect. Dis. 13, 859–866 (2013). This article is the first serological study to show that dromedary camels are a likely animal reservoir of MERS-CoV.
Haagmans, B. L. et al. Middle East respiratory syndrome coronavirus in dromedary camels: an outbreak investigation. Lancet Infect. Dis. 14, 140–145 (2014). This article is the first virological study to show that dromedary camels are a likely animal reservoir of MERS-CoV.
Azhar, E. I. et al. Evidence for camel-to-human transmission of MERS coronavirus. N. Engl. J. Med. 370, 2499–2505 (2014). This is the first study to provide serological and virological evidence to support camel-to-human transmission of MERS-CoV.
Lau, S. K. et al. Genetic characterization of Betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of Pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: implications for the origin of the novel Middle East respiratory syndrome coronavirus. J. Virol. 87, 8638–8650 (2013).
Corman, V. M. et al. Rooting the phylogenetic tree of middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. J. Virol. 88, 11297–11303 (2014).
Wang, Q. et al. Bat origins of MERS-CoV supported by bat coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe 16, 328–337 (2014).
Wernery, U. et al. Acute middle East respiratory syndrome coronavirus infection in livestock Dromedaries, Dubai, 2014. Emerg. Infect. Dis. 21, 1019–1022 (2015).
Penttinen, P. M. et al. Taking stock of the first 133 MERS coronavirus cases globally — is the epidemic changing? Euro Surveill. 18, 20596 (2013).
Muller, M. A. et al. Presence of Middle East respiratory syndrome coronavirus antibodies in Saudi Arabia: a nationwide, cross-sectional, serological study. Lancet Infect. Dis. 15, 559–564 (2015).
Assiri, A. et al. Hospital outbreak of Middle East respiratory syndrome coronavirus. N. Engl. J. Med. 369, 407–416 (2013).
Al-Abdallat, M. M. et al. Hospital-associated outbreak of Middle East respiratory syndrome coronavirus: a serologic, epidemiologic, and clinical description. Clin. Infect. Dis. 59, 1225–1233 (2014).
Memish, Z. A., Zumla, A. I., Al-Hakeem, R. F., Al-Rabeeah, A. A. & Stephens, G. M. Family cluster of Middle East respiratory syndrome coronavirus infections. N. Engl. J. Med. 368, 2487–2494 (2013).
Health Protection Agency. Evidence of person-to-person transmission within a family cluster of novel coronavirus infections, United Kingdom, February 2013. Euro Surveill. 18, 20427 (2013).
Oboho, I. K. et al. 2014 MERS-CoV outbreak in Jeddah — a link to health care facilities. N. Engl. J. Med. 372, 846–854 (2015).
Hui, D. S. & Peiris, M. Middle East respiratory syndrome. Am. J. Respir. Crit. Care Med. 192, 278–279 (2015).
Zumla, A., Hui, D. S. & Perlman, S. Middle East respiratory syndrome. Lancet 386, 995–1007 (2015).
Hui, D. S. & Zumla, A. Advancing priority research on the Middle East respiratory syndrome coronavirus. J. Infect. Dis. 209, 173–176 (2014).
Hui, D. S., Memish, Z. A. & Zumla, A. Severe acute respiratory syndrome vs. the Middle East respiratory syndrome. Curr. Opin. Pulm. Med. 20, 233–241 (2014). This is a comprehensive review and comparison of SARS and MERS.
Arabi, Y. M. et al. Clinical course and outcomes of critically ill patients with Middle East respiratory syndrome coronavirus infection. Ann. Intern. Med. 160, 389–397 (2014).
Assiri, A. et al. Epidemiological, demographic, and clinical characteristics of 47 cases of Middle East respiratory syndrome coronavirus disease from Saudi Arabia: a descriptive study. Lancet Infect. Dis. 13, 752–761 (2013).
Eckerle, I., Muller, M. A., Kallies, S., Gotthardt, D. N. & Drosten, C. In-vitro renal epithelial cell infection reveals a viral kidney tropism as a potential mechanism for acute renal failure during Middle East Respiratory Syndrome (MERS) Coronavirus infection. Virol. J. 10, 359 (2013).
Saad, M. et al. Clinical aspects and outcomes of 70 patients with Middle East respiratory syndrome coronavirus infection: a single-center experience in Saudi Arabia. Int. J. Infect. Dis. 29, 301–306 (2014).
Al-Tawfiq, J. A. et al. Middle East respiratory syndrome coronavirus: a case-control study of hospitalized patients. Clin. Infect. Dis. 59, 160–165 (2014).
Drosten, C. et al. Transmission of MERS-coronavirus in household contacts. N. Engl. J. Med. 371, 828–835 (2014).
Breban, R., Riou, J. & Fontanet, A. Interhuman transmissibility of Middle East respiratory syndrome coronavirus: estimation of pandemic risk. Lancet 382, 694–699 (2013).
Cauchemez, S. et al. Middle East respiratory syndrome coronavirus: quantification of the extent of the epidemic, surveillance biases, and transmissibility. Lancet Infect. Dis. 14, 50–56 (2014).
Poletto, C. et al. Assessment of the Middle East respiratory syndrome coronavirus (MERS-CoV) epidemic in the Middle East and risk of international spread using a novel maximum likelihood analysis approach. Euro Surveill. 19, 20824 (2014).
Anderson, R. M. et al. Epidemiology, transmission dynamics and control of SARS: the 2002–2003 epidemic. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359, 1091–1105 (2004).
Wallinga, J. & Teunis, P. Different epidemic curves for severe acute respiratory syndrome reveal similar impacts of control measures. Am. J. Epidemiol. 160, 509–516 (2004).
Cotten, M. et al. Transmission and evolution of the Middle East respiratory syndrome coronavirus in Saudi Arabia: a descriptive genomic study. Lancet 382, 1993–2002 (2013).
Cotten, M. et al. Spread, circulation, and evolution of the Middle East respiratory syndrome coronavirus. mBio 5, e01062-13 (2014).
Hui, D. S. & Sung, J. J. Severe acute respiratory syndrome. Chest 124, 12–15 (2003).
Cheng, V. C., Tang, B. S., Wu, A. K., Chu, C. M. & Yuen, K. Y. Medical treatment of viral pneumonia including SARS in immunocompetent adult. J. Infect. 49, 262–273 (2004).
Cheng, V. C., Chan, J. F., To, K. K. & Yuen, K. Y. Clinical management and infection control of SARS: lessons learned. Antiviral Res. 100, 407–419 (2013).
Wong, S. S. & Yuen, K. Y. The management of coronavirus infections with particular reference to SARS. J. Antimicrob. Chemother. 62, 437–441 (2008).
Chan, K. S. et al. Treatment of severe acute respiratory syndrome with lopinavir/ritonavir: a multicentre retrospective matched cohort study. Hong Kong Med. J. 9, 399–406 (2003).
Chu, C. M. et al. Role of lopinavir/ritonavir in the treatment of SARS: initial virological and clinical findings. Thorax 59, 252–256 (2004). This combined in vitro and in vivo study examines the use of lopinavir/ritonavir and ribavirin for the treatment of SARS.
Loutfy, M. R. et al. Interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. JAMA 290, 3222–3228 (2003).
Wang, H. et al. Fatal aspergillosis in a patient with SARS who was treated with corticosteroids. N. Engl. J. Med. 349, 507–508 (2003).
Griffith, J. F. et al. Osteonecrosis of hip and knee in patients with severe acute respiratory syndrome treated with steroids. Radiology 235, 168–175 (2005).
Tsang, O. T. et al. Coronavirus-positive nasopharyngeal aspirate as predictor for severe acute respiratory syndrome mortality. Emerg. Infect. Dis. 9, 1381–1387 (2003).
Lee, N. et al. Effects of early corticosteroid treatment on plasma SARS-associated Coronavirus RNA concentrations in adult patients. J. Clin. Virol. 31, 304–309 (2004).
Cheng, Y. et al. Use of convalescent plasma therapy in SARS patients in Hong Kong. Eur. J. Clin. Microbiol. Infect. Dis. 24, 44–46 (2005). This article reports the largest clinical study of convalescent-phase plasma for the treatment of SARS.
Soo, Y. O. et al. Retrospective comparison of convalescent plasma with continuing high-dose methylprednisolone treatment in SARS patients. Clin. Microbiol. Infect. 10, 676–678 (2004).
Mair-Jenkins, J. et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. J. Infect. Dis. 211, 80–90 (2015).
Omrani, A. S. et al. Ribavirin and interferonα-2a for severe Middle East respiratory syndrome coronavirus infection: a retrospective cohort study. Lancet Infect. Dis. 14, 1090–1095 (2014). This is currently the largest clinical study of the treatment effects of ribavirin and interferon alfa-2a for MERS.
Al-Tawfiq, J. A., Momattin, H., Dib, J. & Memish, Z. A. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: an observational study. Int. J. Infect. Dis. 20, 42–46 (2014).
Khalid, M. et al. Ribavirin and interferon-α-2b as primary and preventive treatment for Middle East respiratory syndrome coronavirus (MERS-CoV): a preliminary report of two cases. Antivir. Ther. 20, 87–91 (2015).
Shalhoub, S. et al. IFN-α2a or IFN-β1a in combination with ribavirin to treat Middle East respiratory syndrome coronavirus pneumonia: a retrospective study. J. Antimicrob. Chemother. 70, 2129–2132 (2015).
Spanakis, N. et al. Virological and serological analysis of a recent Middle East respiratory syndrome coronavirus infection case on a triple combination antiviral regimen. Int. J. Antimicrob. Agents 44, 528–532 (2014).
van Boheemen, S. et al. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3, e00473-12 (2012).
Lu, L. et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 5, 3067 (2014).
Gao, J. et al. Structure of the fusion core and inhibition of fusion by a heptad repeat peptide derived from the S protein of Middle East respiratory syndrome coronavirus. J. Virol. 87, 13134–13140 (2013).
Jiang, L. et al. Potent neutralization of MERS-CoV by human neutralizing monoclonal antibodies to the viral spike glycoprotein. Sci. Transl. Med. 6, 234ra59 (2014).
Ying, T. et al. Exceptionally potent neutralization of Middle East respiratory syndrome coronavirus by human monoclonal antibodies. J. Virol. 88, 7796–7805 (2014).
Tang, X. C. et al. Identification of human neutralizing antibodies against MERS-CoV and their role in virus adaptive evolution. Proc. Natl Acad. Sci. USA 111, E2018–E2026 (2014).
Du, L. et al. A conformation-dependent neutralizing monoclonal antibody specifically targeting receptor-binding domain in middle East respiratory syndrome coronavirus spike protein. J. Virol. 88, 7045–7053 (2014).
Yuan, K. et al. Suppression of SARS-CoV entry by peptides corresponding to heptad regions on spike glycoprotein. Biochem. Biophys. Res. Commun. 319, 746–752 (2004).
Channappanavar, R. et al. Protective effect of intranasal regimens containing peptidic Middle East Respiratory Syndrome coronavirus fusion inhibitor against MERS-CoV infection. J. Infect. Dis. 212, 1894–1903 (2003). This is the first in vivo treatment evaluation of an anti-MERS-CoV-S2 antiviral peptide in MERS-CoV-infected mice.
Pascal, K. E. et al. Pre- and postexposure efficacy of fully human antibodies against Spike protein in a novel humanized mouse model of MERS-CoV infection. Proc. Natl Acad. Sci. USA 112, 8738–8743 (2015).
Li, W. et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426, 450–454 (2003). This paper identifies ACE2 as the functional receptor for SARS-CoV.
Raj, V. S. et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495, 251–254 (2013). This paper identifies DPP4 as the functional receptor for MERS-CoV.
Huang, X. et al. Human coronavirus HKU1 spike protein uses O-acetylated sialic acid as an attachment receptor determinant and employs hemagglutinin-esterase protein as a receptor-destroying enzyme. J. Virol. 89, 7202–7213 (2015).
Yeager, C. L. et al. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature 357, 420–422 (1992).
Vlasak, R., Luytjes, W., Spaan, W. & Palese, P. Human and bovine coronaviruses recognize sialic acid-containing receptors similar to those of influenza C viruses. Proc. Natl Acad. Sci. USA 85, 4526–4529 (1988).
Hofmann, H. et al. Human coronavirus NL63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc. Natl Acad. Sci. USA 102, 7988–7993 (2005).
Gierer, S. et al. The spike protein of the emerging betacoronavirus EMC uses a novel coronavirus receptor for entry, can be activated by TMPRSS2, and is targeted by neutralizing antibodies. J. Virol. 87, 5502–5511 (2013).
Qian, Z., Dominguez, S. R. & Holmes, K. V. Role of the spike glycoprotein of human Middle East respiratory syndrome coronavirus (MERS-CoV) in virus entry and syncytia formation. PLoS ONE 8, e76469 (2013).
Shirato, K., Kawase, M. & Matsuyama, S. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J. Virol. 87, 12552–12561 (2013).
Millet, J. K. & Whittaker, G. R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl Acad. Sci. USA 111, 15214–15219 (2014).
Zhou, Y. et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 116, 76–84 (2015).
Lundin, A. et al. Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the middle East respiratory syndrome virus. PLoS Pathog. 10, e1004166 (2014).
Knoops, K. et al. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6, e226 (2008).
Barnard, D. L. & Kumaki, Y. Recent developments in anti-severe acute respiratory syndrome coronavirus chemotherapy. Future Virol. 6, 615–631 (2011).
Kilianski, A. & Baker, S. C. Cell-based antiviral screening against coronaviruses: developing virus-specific and broad-spectrum inhibitors. Antiviral Res. 101, 105–112 (2014). This is a comprehensive review of the cell-based antiviral screening platforms used for anti-CoV drug development.
Cinatl, J. et al. Treatment of SARS with human interferons. Lancet 362, 293–294 (2003).
So, L. K. et al. Development of a standard treatment protocol for severe acute respiratory syndrome. Lancet 361, 1615–1617 (2003).
Pfefferle, S. et al. The SARS–coronavirus–host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 7, e1002331 (2011).
de Wilde, A. H. et al. MERS-coronavirus replication induces severe in vitro cytopathology and is strongly inhibited by cyclosporin A or interferon-α treatment. J. Gen. Virol. 94, 1749–1760 (2013).
Tanaka, Y., Sato, Y. & Sasaki, T. Suppression of coronavirus replication by cyclophilin inhibitors. Viruses 5, 1250–1260 (2013).
Falzarano, D. et al. Inhibition of novel β coronavirus replication by a combination of interferon-α2b and ribavirin. Sci. Rep. 3, 1686 (2013).
Chan, J. F. et al. Broad-spectrum antivirals for the emerging Middle East respiratory syndrome coronavirus. J. Infect. 67, 606–616 (2013). This article reports the first drug repurposing programme for MERS using high-throughput screening.
de Wilde, A. H. et al. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of Middle East respiratory syndrome coronavirus replication in cell culture. Antimicrob. Agents Chemother. 58, 4875–4884 (2014).
Dyall, J. et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother. 58, 4885–4893 (2014).
Kindrachuk, J. et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 59, 1088–1099 (2015).
Liu, Q. et al. Testing of Middle East respiratory syndrome coronavirus replication inhibitors for their ability to block viral entry. Antimicrob. Agents Chemother. http://dx.doi.org/10.1128/AAC.03977-14 (2014).
Elshabrawy, H. A. et al. Identification of a broad-spectrum antiviral small molecule against severe acute respiratory syndrome coronavirus and Ebola, Hendra, and Nipah viruses by using a novel high-throughput screening assay. J. Virol. 88, 4353–4365 (2014).
Chan, J. F. et al. Treatment with lopinavir/ritonavir or interferon-β1b improves outcome of MERS-CoV infection in a non-human primate model of common marmoset. J. Infect. Dis. 212, 1904–1913 (2015). This is the first in vivo treatment evaluation of repurposed drugs (lopinavir–ritonavir, interferon beta-1b and mycophenolate mofetil) in MERS-CoV-infected common marmosets.
Faure, E. et al. Distinct immune response in two MERS-CoV-infected patients: can we go from bench to bedside? PLoS ONE 9, e88716 (2014).
AlGhamdi, M., Mushtaq, F., Awn, N. & Shalhoub, S. MERS CoV infection in two renal transplant recipients: case report. Am. J. Transplant. 15, 1101–1104 (2015).
Fukushima, A. et al. Development of a chimeric DNA–RNA hammerhead ribozyme targeting SARS virus. Intervirology 52, 92–99 (2009).
Rider, T. H. et al. Broad-spectrum antiviral therapeutics. PLoS ONE 6, e22572 (2011).
Mielech, A. M., Kilianski, A., Baez-Santos, Y. M., Mesecar, A. D. & Baker, S. C. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology 450–451, 64–70 (2014).
Barretto, N. et al. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79, 15189–15198 (2005).
Mielech, A. M., Chen, Y., Mesecar, A. D. & Baker, S. C. Nidovirus papain-like proteases: multifunctional enzymes with protease, deubiquitinating and deISGylating activities. Virus Res. 194, 184–190 (2014).
Hilgenfeld, R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 281, 4085–4096 (2014).
Baez-Santos, Y. M., St John, S. E. & Mesecar, A. D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 115, 21–38 (2015).
Baez-Santos, Y. M. et al. X-ray structural and biological evaluation of a series of potent and highly selective inhibitors of human coronavirus papain-like proteases. J. Med. Chem. 57, 2393–2412 (2014).
Ratia, K. et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl Acad. Sci. USA 105, 16119–16124 (2008).
Lee, H. et al. Inhibitor recognition specificity of MERS-CoV papain-like protease may differ from that of SARS-CoV. ACS Chem. Biol. 10, 1456–1465 (2015).
Chaudhuri, R. et al. Comparison of SARS and NL63 papain-like protease binding sites and binding site dynamics: inhibitor design implications. J. Mol. Biol. 414, 272–288 (2011).
Adedeji, A. O. & Sarafianos, S. G. Antiviral drugs specific for coronaviruses in preclinical development. Curr. Opin. Virol. 8, 45–53 (2014).
Yang, H. et al. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 3, e324 (2005).
Ren, Z. et al. The newly emerged SARS-like coronavirus HCoV-EMC also has an 'Achilles' heel': current effective inhibitor targeting a 3C-like protease. Protein Cell 4, 248–250 (2013).
Chen, F. et al. In vitro susceptibility of 10 clinical isolates of SARS coronavirus to selected antiviral compounds. J. Clin. Virol. 31, 69–75 (2004).
Nukoolkarn, V., Lee, V. S., Malaisree, M., Aruksakulwong, O. & Hannongbua, S. Molecular dynamic simulations analysis of ritonavir and lopinavir as SARS-CoV 3CLpro inhibitors. J. Theor. Biol. 254, 861–867 (2008).
Olschlager, S., Neyts, J. & Gunther, S. Depletion of GTP pool is not the predominant mechanism by which ribavirin exerts its antiviral effect on Lassa virus. Antiviral Res. 91, 89–93 (2011).
Falzarano, D. et al. Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV-infected rhesus macaques. Nat. Med. 19, 1313–1317 (2013). This is the first in vivo treatment evaluation of ribavirin and interferon alfa-2b in MERS-CoV-infected rhesus macaques.
Warren, T. K. et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 508, 402–405 (2014).
Peters, H. L. et al. Design, synthesis and evaluation of a series of acyclic fleximer nucleoside analogues with anti-coronavirus activity. Bioorg. Med. Chem. Lett. 25, 2923–2926 (2015).
Lu, A. et al. Attenuation of SARS coronavirus by a short hairpin RNA expression plasmid targeting RNA-dependent RNA polymerase. Virology 324, 84–89 (2004).
Wang, Z. et al. Inhibition of severe acute respiratory syndrome virus replication by small interfering RNAs in mammalian cells. J. Virol. 78, 7523–7527 (2004).
Tanner, J. A. et al. The adamantane-derived bananins are potent inhibitors of the helicase activities and replication of SARS coronavirus. Chem. Biol. 12, 303–311 (2005).
Kim, M. K. et al. 2,6-bis-arylmethyloxy-5-hydroxychromones with antiviral activity against both hepatitis C virus (HCV) and SARS-associated coronavirus (SCV). Eur. J. Med. Chem. 46, 5698–5704 (2011).
Adedeji, A. O. et al. Severe acute respiratory syndrome coronavirus replication inhibitor that interferes with the nucleic acid unwinding of the viral helicase. Antimicrob. Agents Chemother. 56, 4718–4728 (2012).
Adedeji, A. O. et al. Evaluation of SSYA10-001 as a replication inhibitor of severe acute respiratory syndrome, mouse hepatitis, and Middle East respiratory syndrome coronaviruses. Antimicrob. Agents Chemother. 58, 4894–4898 (2014).
Zhao, J. et al. Rapid generation of a mouse model for Middle East respiratory syndrome. Proc. Natl Acad. Sci. USA 111, 4970–4975 (2014). This article reports the development of a rapidly generated mouse model using mice recently transduced by adenoviral vectors expressing human DPP4 for MERS.
Yang, Z. Y. et al. Evasion of antibody neutralization in emerging severe acute respiratory syndrome coronaviruses. Proc. Natl Acad. Sci. USA 102, 797–801 (2005).
Weingartl, H. et al. Immunization with modified vaccinia virus Ankara-based recombinant vaccine against severe acute respiratory syndrome is associated with enhanced hepatitis in ferrets. J. Virol. 78, 12672–12676 (2004).
Coughlin, M. M. & Prabhakar, B. S. Neutralizing human monoclonal antibodies to severe acute respiratory syndrome coronavirus: target, mechanism of action, and therapeutic potential. Rev. Med. Virol. 22, 2–17 (2012).
Li, Y. et al. A humanized neutralizing antibody against MERS-CoV targeting the receptor-binding domain of the spike protein. Cell Res. 25, 1237–1249 (2015).
Corti, D. et al. Prophylactic and postexposure efficacy of a potent human monoclonal antibody against MERS coronavirus. Proc. Natl Acad. Sci. USA 112, 10473–10478 (2015). This is one of the few studies demonstrating the effects of mAbs against MERS-CoV S in a mouse model.
Sainz, B. et al. Inhibition of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) infectivity by peptides analogous to the viral spike protein. Virus Res. 120, 146–155 (2006).
Zheng, B. J. et al. Synthetic peptides outside the spike protein heptad repeat regions as potent inhibitors of SARS-associated coronavirus. Antiviral Ther. 10, 393–403 (2005).
Liu, I. J. et al. Identification of a minimal peptide derived from heptad repeat (HR) 2 of spike protein of SARS-CoV and combination of HR1-derived peptides as fusion inhibitors. Antiviral Res. 81, 82–87 (2009).
Kilby, J. M. et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat. Med. 4, 1302–1307 (1998).
Greenberg, M. L. & Cammack, N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J. Antimicrob. Chemother. 54, 333–340 (2004).
Izumi, K. et al. Mechanism of resistance to S138A substituted enfuvirtide and its application to peptide design. Int. J. Biochem. Cell Biol. 45, 908–915 (2013).
Zhang, Y. et al. Silencing SARS-CoV Spike protein expression in cultured cells by RNA interference. FEBS Lett. 560, 141–146 (2004).
Wu, C. J., Huang, H. W., Liu, C. Y., Hong, C. F. & Chan, Y. L. Inhibition of SARS-CoV replication by siRNA. Antiviral Res. 65, 45–48 (2005).
Tang, Q., Li, B., Woodle, M. & Lu, P. Y. Application of siRNA against SARS in the rhesus macaque model. Methods Mol. Biol. 442, 139–158 (2008).
Li, B. J. et al. Using siRNA in prophylactic and therapeutic regimens against SARS coronavirus in Rhesus macaque. Nat. Med. 11, 944–951 (2005). This article described the in vivo treatment evaluation of siRNAs for SARS in rhesus macaques.
O'Keefe, B. R. et al. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family Coronaviridae. J. Virol. 84, 2511–2521 (2010).
Barton, C. et al. Activity of and effect of subcutaneous treatment with the broad-spectrum antiviral lectin griffithsin in two laboratory rodent models. Antimicrob. Agents Chemother. 58, 120–127 (2014).
Yang, Y. et al. The structural and accessory proteins M, ORF 4a, ORF 4b, and ORF 5 of Middle East respiratory syndrome coronavirus (MERS-CoV) are potent interferon antagonists. Protein Cell 4, 951–961 (2013).
Cui, L. et al. The nucleocapsid protein of coronaviruses acts as a viral suppressor of RNA silencing in mammalian cells. J. Virol. 89, 9029–9043 (2015).
Niemeyer, D. et al. Middle East respiratory syndrome coronavirus accessory protein 4a is a type I interferon antagonist. J. Virol. 87, 12489–12495 (2013).
Siu, K. L. et al. Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J. Virol. 88, 4866–4876 (2014).
He, M. L. et al. Development of interfering RNA agents to inhibit SARS-associated coronavirus infection and replication. Hong Kong Med. J. 3 (Suppl. 4), 28–31 (2009).
Akerstrom, S., Mirazimi, A. & Tan, Y. J. Inhibition of SARS-CoV replication cycle by small interference RNAs silencing specific SARS proteins, 7a/7b, 3a/3b and S. Antiviral Res. 73, 219–227 (2007).
Pervushin, K. et al. Structure and inhibition of the SARS coronavirus envelope protein ion channel. PLoS Pathog. 5, e1000511 (2009).
Wilson, L., Gage, P. & Ewart, G. Hexamethylene amiloride blocks E protein ion channels and inhibits coronavirus replication. Virology 353, 294–306 (2006).
Lin, S. Y. et al. Structural basis for the identification of the N-terminal domain of coronavirus nucleocapsid protein as an antiviral target. J. Med. Chem. 57, 2247–2257 (2014).
Wolf, M. C. et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl Acad. Sci. USA 107, 3157–3162 (2010).
Vigant, F. et al. A mechanistic paradigm for broad-spectrum antivirals that target virus-cell fusion. PLoS Pathog. 9, e1003297 (2013).
Hollmann, A., Castanho, M. A., Lee, B. & Santos, N. C. Singlet oxygen effects on lipid membranes: implications for the mechanism of action of broad-spectrum viral fusion inhibitors. Biochem. J. 459, 161–170 (2014).
Hollmann, A. et al. Effects of singlet oxygen generated by a broad-spectrum viral fusion inhibitor on membrane nanoarchitecture. Nanomedicine 11, 1163–1167 (2015).
Menachery, V. D. et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio 5, e01174–e01114 (2014).
Lau, S. K. et al. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: implications for pathogenesis and treatment. J. Gen. Virol. 94, 2679–2690 (2013).
Josset, L. et al. Cell host response to infection with novel human coronavirus EMC predicts potential antivirals and important differences with SARS coronavirus. mBio 4, e00165–e00113 (2013).
Haagmans, B. L. et al. Pegylated interferon-α protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat. Med. 10, 290–293 (2004).
Hart, B. J. et al. Interferon-β and mycophenolic acid are potent inhibitors of Middle East respiratory syndrome coronavirus in cell-based assays. J. Gen. Virol. 95, 571–577 (2014).
Rosenfeld, M. R. et al. A multi-institution Phase II study of poly-ICLC and radiotherapy with concurrent and adjuvant temozolomide in adults with newly diagnosed glioblastoma. Neuro Oncol. 12, 1071–1077 (2010).
Okada, H. et al. Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with α-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma. J. Clin. Oncol. 29, 330–336 (2011).
Rossignol, J. F. Nitazoxanide: a first-in-class broad-spectrum antiviral agent. Antiviral Res. 110, 94–103 (2014).
Haffizulla, J. et al. Effect of nitazoxanide in adults and adolescents with acute uncomplicated influenza: a double-blind, randomised, placebo-controlled, Phase 2b/3 trial. Lancet Infect. Dis. 14, 609–618 (2014).
Rossignol, J. F., Kabil, S. M., El-Gohary, Y., Elfert, A. & Keeffe, E. B. Clinical trial: randomized, double-blind, placebo-controlled study of nitazoxanide monotherapy for the treatment of patients with chronic hepatitis C genotype 4. Aliment. Pharmacol. Ther. 28, 574–580 (2008).
Wohlford-Lenane, C. L. et al. Rhesus theta-defensin prevents death in a mouse model of severe acute respiratory syndrome coronavirus pulmonary disease. J. Virol. 83, 11385–11390 (2009).
Wiley, J. A. et al. Inducible bronchus-associated lymphoid tissue elicited by a protein cage nanoparticle enhances protection in mice against diverse respiratory viruses. PLoS ONE 4, e7142 (2009).
Carbajo-Lozoya, J. et al. Human coronavirus NL63 replication is cyclophilin A-dependent and inhibited by non-immunosuppressive cyclosporine A-derivatives including Alisporivir. Virus Res. 184, 44–53 (2014).
Ohnuma, K. et al. Inhibition of Middle East respiratory syndrome coronavirus infection by anti-CD26 monoclonal antibody. J. Virol. 87, 13892–13899 (2013).
Huentelman, M. J. et al. Structure-based discovery of a novel angiotensin-converting enzyme 2 inhibitor. Hypertension 44, 903–906 (2004).
Han, D. P., Penn-Nicholson, A. & Cho, M. W. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology 350, 15–25 (2006).
Chen, Y. T. et al. In vitro and in vivo studies of the trypanocidal properties of WRR-483 against Trypanosoma cruzi. PLoS Negl. Trop. Dis. 4, e825 (2010).
Ndao, M. et al. A cysteine protease inhibitor rescues mice from a lethal Cryptosporidium parvum infection. Antimicrob. Agents Chemother. 57, 6063–6073 (2013).
Vermeire, J. J., Lantz, L. D. & Caffrey, C. R. Cure of hookworm infection with a cysteine protease inhibitor. PLoS Negl. Trop. Dis. 6, e1680 (2012).
Sai, J. K. et al. Efficacy of camostat mesilate against dyspepsia associated with non-alcoholic mild pancreatic disease. J. Gastroenterol. 45, 335–341 (2010).
Talukdar, R. & Tandon, R. K. Pancreatic stellate cells: new target in the treatment of chronic pancreatitis. J. Gastroenterol. Hepatol. 23, 34–41 (2008).
Burkard, C. et al. ATP1A1-mediated Src signaling inhibits coronavirus entry into host cells. J. Virol. 89, 4434–4448 (2015).
Savarino, A., Boelaert, J. R., Cassone, A., Majori, G. & Cauda, R. Effects of chloroquine on viral infections: an old drug against today's diseases? Lancet Infect. Dis. 3, 722–727 (2003).
Keyaerts, E., Vijgen, L., Maes, P., Neyts, J. & Van Ranst, M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem. Biophys. Res. Commun. 323, 264–268 (2004).
Vincent, M. J. et al. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2, 69 (2005).
Kono, M. et al. Inhibition of human coronavirus 229E infection in human epithelial lung cells (L132) by chloroquine: involvement of p38 MAPK and ERK. Antiviral Res. 77, 150–152 (2008).
Madrid, P. B. et al. A systematic screen of FDA-approved drugs for inhibitors of biological threat agents. PloS ONE 8, e60579 (2013).
Barnard, D. L. et al. Evaluation of immunomodulators, interferons and known in vitro SARS-coV inhibitors for inhibition of SARS-coV replication in BALB/c mice. Antiviral Chem. Chemother. 17, 275–284 (2006).
Zhang, N., Jiang, S. & Du, L. Current advancements and potential strategies in the development of MERS-CoV vaccines. Expert Rev. Vaccines 13, 761–774 (2014).
Papaneri, A. B. et al. Middle East respiratory syndrome: obstacles and prospects for vaccine development. Expert Rev. Vaccines 14, 949–962 (2015).
Almazan, F. et al. Engineering a replication-competent, propagation-defective Middle East respiratory syndrome coronavirus as a vaccine candidate. mBio 4, e00650–00613 (2013).
Wang, L. et al. Evaluation of candidate vaccine approaches for MERS-CoV. Nat. Commun. 6, 7712 (2015). This is the first in vivo evaluation of candidate MERS-CoV vaccines in rhesus macaques.
Song, F. et al. Middle East respiratory syndrome coronavirus spike protein delivered by modified vaccinia virus Ankara efficiently induces virus-neutralizing antibodies. J. Virol. 87, 11950–11954 (2013).
Volz, A. et al. Protective efficacy of recombinant modified vaccinia virus Ankara delivering Middle East respiratory syndrome coronavirus spike glycoprotein. J. Virol. 89, 8651–8656 (2015).
Kim, E. et al. Immunogenicity of an adenoviral-based Middle East respiratory syndrome coronavirus vaccine in BALB/c mice. Vaccine 32, 5975–5982 (2014).
Guo, X. et al. Systemic and mucosal immunity in mice elicited by a single immunization with human adenovirus type 5 or 41 vector-based vaccines carrying the spike protein of Middle East respiratory syndrome coronavirus. Immunology 145, 476–484 (2015).
Czub, M., Weingartl, H., Czub, S., He, R. & Cao, J. Evaluation of modified vaccinia virus Ankara based recombinant SARS vaccine in ferrets. Vaccine 23, 2273–2279 (2005).
Coleman, C. M. et al. Purified coronavirus spike protein nanoparticles induce coronavirus neutralizing antibodies in mice. Vaccine 32, 3169–3174 (2014).
Zhao, G. et al. A safe and convenient pseudovirus-based inhibition assay to detect neutralizing antibodies and screen for viral entry inhibitors against the novel human coronavirus MERS-CoV. Virol. J. 10, 266 (2013).
Du, L. et al. Identification of a receptor-binding domain in the S protein of the novel human coronavirus Middle East respiratory syndrome coronavirus as an essential target for vaccine development. J. Virol. 87, 9939–9942 (2013).
Mou, H. et al. The receptor binding domain of the new Middle East respiratory syndrome coronavirus maps to a 231-residue region in the spike protein that efficiently elicits neutralizing antibodies. J. Virol. 87, 9379–9383 (2013).
Du, L. et al. A truncated receptor-binding domain of MERS-CoV spike protein potently inhibits MERS-CoV infection and induces strong neutralizing antibody responses: implication for developing therapeutics and vaccines. PloS ONE 8, e81587 (2013).
Ma, C. et al. Intranasal vaccination with recombinant receptor-binding domain of MERS-CoV spike protein induces much stronger local mucosal immune responses than subcutaneous immunization: Implication for designing novel mucosal MERS vaccines. Vaccine 32, 2100–2108 (2014).
Ma, C. et al. Searching for an ideal vaccine candidate among different MERS coronavirus receptor-binding fragments-The importance of immunofocusing in subunit vaccine design. Vaccine 32, 6170–6176 (2014).
Lan, J. et al. Tailoring subunit vaccine immunity with adjuvant combinations and delivery routes using the Middle East respiratory coronavirus (MERS-CoV) receptor-binding domain as an antigen. PloS ONE 9, e112602 (2014).
Zhang, N. et al. Identification of an ideal adjuvant for receptor-binding domain-based subunit vaccines against Middle East respiratory syndrome coronavirus. Cell. Mol. Immunol. http://dx.doi.org/10.1038/cmi.2015.03 (2015).
Lassnig, C. et al. Development of a transgenic mouse model susceptible to human coronavirus 229E. Proc. Natl Acad. Sci. USA 102, 8275–8280 (2005).
Dijkman, R. et al. Seroconversion to HCoV-NL63 in rhesus macaques. Viruses 1, 647–656 (2009).
Jacomy, H., Fragoso, G., Almazan, G., Mushynski, W. E. & Talbot, P. J. Human coronavirus OC43 infection induces chronic encephalitis leading to disabilities in BALB/C mice. Virology 349, 335–346 (2006).
Sutton, T. C. & Subbarao, K. Development of animal models against emerging coronaviruses: from SARS to MERS coronavirus. Virology 479–480, 247–258 (2015).
Fouchier, R. A. et al. Aetiology: Koch's postulates fulfilled for SARS virus. Nature 423, 240 (2003).
McCray, P. B. et al. Lethal infection of K18-hACE2 mice infected with severe acute respiratory syndrome coronavirus. J. Virol. 81, 813–821 (2007). This article describes the establishment of the ACE2-transgenic mouse model for SARS.
Tseng, C. T. et al. Severe acute respiratory syndrome coronavirus infection of mice transgenic for the human Angiotensin-converting enzyme 2 virus receptor. J. Virol. 81, 1162–1173 (2007).
Roberts, A. et al. A mouse-adapted SARS-coronavirus causes disease and mortality in BALB/c mice. PLoS Pathog. 3, e5 (2007).
Munster, V. J., de Wit, E. & Feldmann, H. Pneumonia from human coronavirus in a macaque model. N. Engl. J. Med. 368, 1560–1562 (2013).
Yao, Y. et al. An animal model of MERS produced by infection of rhesus macaques with MERS coronavirus. J. Infect. Dis. 209, 236–242 (2014).
Falzarano, D. et al. Infection with MERS-CoV causes lethal pneumonia in the common marmoset. PLoS Pathog. 10, e1004250 (2014). This article establishes the common marmoset model for MERS, which resembles severe MERS in humans.
Raj, V. S. et al. Adenosine deaminase acts as a natural antagonist for dipeptidyl peptidase 4-mediated entry of the Middle East respiratory syndrome coronavirus. J. Virol. 88, 1834–1838 (2014).
de Wit, E. et al. The Middle East respiratory syndrome coronavirus (MERS-CoV) does not replicate in Syrian hamsters. PLoS One. 8, e69127 (2013).
Coleman, C. M., Matthews, K. L., Goicochea, L. & Frieman, M. B. Wild-type and innate immune-deficient mice are not susceptible to the Middle East respiratory syndrome coronavirus. J. General Virol. 95, 408–412 (2014).
Agrawal, A. S. et al. Generation of a transgenic mouse model of Middle East respiratory syndrome coronavirus infection and disease. J. Virol. 89, 3659–3670 (2015). This article establishes the DPP4-transgenic mouse model for MERS.
Hung, I. F. et al. Viral loads in clinical specimens and SARS manifestations. Emerg. Infect. Dis. 10, 1550–1557 (2004).
Abd El Wahed, A., Patel, P., Heidenreich, D., Hufert, F. T. & Weidmann, M. Reverse transcription recombinase polymerase amplification assay for the detection of middle East respiratory syndrome coronavirus. PLoS Curr. 5, currents.outbreaks. 62df1c7c75ffc96cd59034531e2e8364 (2013).
Shirato, K. et al. Detection of Middle East respiratory syndrome coronavirus using reverse transcription loop-mediated isothermal amplification (RT-LAMP). Virol. J. 11, 139 (2014).
Song, D. et al. Development and validation of a rapid immunochromatographic assay for detection of Middle East respiratory syndrome coronavirus antigen in dromedary camels. J. Clin. Microbiol. 53, 1178–1182 (2015).
Chen, Y. et al. A sensitive and specific antigen detection assay for Middle East respiratory syndrome coronavirus. Emerg. Microbes Infect. 4, e26 (2015).
Sridhar, S. et al. A systematic approach to novel virus discovery in emerging infectious disease outbreaks. J. Mol. Diagn. 17, 230–241 (2015).
Corman, V. M. et al. Assays for laboratory confirmation of novel human coronavirus (hCoV-EMC) infections. Euro Surveill. 17, 20334 (2012).
Corman, V. M. et al. Detection of a novel human coronavirus by real-time reverse-transcription polymerase chain reaction. Euro Surveill. 17, 20285 (2012).
Chan, J. F. et al. Development and evaluation of novel real-time RT-PCR assays with locked nucleic acid probes targeting the leader sequences of human pathogenic coronaviruses. J. Clin. Microbiol. 53, 2722–2726 (2015).
Chan, J. F. et al. Differential cell line susceptibility to the emerging novel human betacoronavirus 2c EMC/2012: implications for disease pathogenesis and clinical manifestation. J. Infect. Dis. 207, 1743–1752 (2013).
Pyrc, K. et al. Culturing the unculturable: human coronavirus HKU1 infects, replicates, and produces progeny virions in human ciliated airway epithelial cell cultures. J. Virol. 84, 11255–11263 (2010).
Dijkman, R. et al. Isolation and characterization of current human coronavirus strains in primary human epithelial cell cultures reveal differences in target cell tropism. J. Virol. 87, 6081–6090 (2013).
Dominguez, S. R., Travanty, E. A., Qian, Z. & Mason, R. J. Human coronavirus HKU1 infection of primary human type II alveolar epithelial cells: cytopathic effects and innate immune response. PLoS ONE 8, e70129 (2013).
Dominguez, S. R. et al. Isolation, propagation, genome analysis and epidemiology of HKU1 betacoronaviruses. J. General Virol. 95, 836–848 (2014).
Zhou, J. et al. Active replication of Middle East respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: implications for pathogenesis. J. Infect. Dis. 209, 1331–1342 (2014).
Chu, H. et al. Middle East respiratory syndrome coronavirus efficiently infects human primary T lymphocytes and activates the extrinsic and intrinsic apoptosis pathways. J. Infect. Dis. http://dx.doi.org/10.1093/infdis/jiv380 (2015).
Chu, H. et al. Productive replication of Middle East respiratory syndrome coronavirus in monocyte-derived dendritic cells modulates innate immune response. Virology 454–455, 197–205 (2014).
Chan, R. W. et al. Tropism of and innate immune responses to the novel human betacoronavirus lineage C virus in human ex vivo respiratory organ cultures. J. Virol. 87, 6604–6614 (2013).
Chan, R. W. et al. Tropism and replication of Middle East respiratory syndrome coronavirus from dromedary camels in the human respiratory tract: an in-vitro and ex-vivo study. Lancet Respir.Med. 2, 813–822 (2014).
Muller, M. A. et al. Human coronavirus EMC does not require the SARS-coronavirus receptor and maintains broad replicative capability in mammalian cell lines. mBio 3, e00515-12 (2012).
Eckerle, I. et al. Replicative capacity of MERS coronavirus in livestock cell lines. Emerg. Infect. Dis. 20, 276–279 (2014).
Chan, K. H. et al. Cross-reactive antibodies in convalescent SARS patients' sera against the emerging novel human coronavirus EMC (2012) by both immunofluorescent and neutralizing antibody tests. J. Infect. 67, 130–140 (2013).
Perera, R. A. et al. Seroepidemiology for MERS coronavirus using microneutralisation and pseudoparticle virus neutralisation assays reveal a high prevalence of antibody in dromedary camels in Egypt, June 2013. Euro Surveill. 18, 20574 (2013).
Hung, I. F. et al. Hyperimmune IV immunoglobulin treatment: a multicenter double-blind randomized controlled trial for patients with severe 2009 influenza A(H1N1) infection. Chest 144, 464–473 (2013).
Hung, I. F. et al. Convalescent plasma treatment reduced mortality in patients with severe pandemic influenza A (H1N1) 2009 virus infection. Clin. Infect. Dis. 52, 447–456 (2011).
WHO MERS-CoV Research Group. State of knowledge and data gaps of Middle East respiratory syndrome coronavirus (MERS-CoV) in humans. PLoS Curr. 5, ecurrents.outbreaks.0bf719e352e7478f8ad85fa30127ddb8 (2013).
Kilianski, A., Mielech, A. M., Deng, X. & Baker, S. C. Assessing activity and inhibition of Middle East respiratory syndrome coronavirus papain-like and 3C-like proteases using luciferase-based biosensors. J. Virol. 87, 11955–11962 (2013).
Agnihothram, S. et al. A mouse model for Betacoronavirus subgroup 2c using a bat coronavirus strain HKU5 variant. mBio 5, e00047–00014 (2014).
Reichard, O., Yun, Z. B., Sonnerborg, A. & Weiland, O. Hepatitis C viral RNA titers in serum prior to, during, and after oral treatment with ribavirin for chronic hepatitis C. J. Med. Virol. 41, 99–102 (1993).
Hall, C. B., Walsh, E. E., Hruska, J. F., Betts, R. F. & Hall, W. J. Ribavirin treatment of experimental respiratory syncytial viral infection. A controlled double-blind study in young adults. JAMA 249, 2666–2670 (1983).
Ascioglu, S., Leblebicioglu, H., Vahaboglu, H. & Chan, K. A. Ribavirin for patients with Crimean-Congo haemorrhagic fever: a systematic review and meta-analysis. J. Antimicrob. Chemother. 66, 1215–1222 (2011).
Bausch, D. G., Hadi, C. M., Khan, S. H. & Lertora, J. J. Review of the literature and proposed guidelines for the use of oral ribavirin as postexposure prophylaxis for Lassa fever. Clin. Infect. Dis. 51, 1435–1441 (2010).
Lee, C. et al. Aryl diketoacids (ADK) selectively inhibit duplex DNA-unwinding activity of SARS coronavirus NTPase/helicase. Bioorg. Med. Chem. Lett. 19, 1636–1638 (2009).
Liao, H. I. et al. mRNA display design of fibronectin-based intrabodies that detect and inhibit severe acute respiratory syndrome coronavirus nucleocapsid protein. J. Biol. Chem. 284, 17512–17520 (2009).
Simmons, G. et al. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl Acad. Sci. USA 102, 11876–11881 (2005).
Hatesuer, B. et al. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 9, e1003774 (2013).
Abe, M. et al. TMPRSS2 is an activating protease for respiratory parainfluenza viruses. J. Virol. 87, 11930–11935 (2013).
Bertram, S. et al. TMPRSS2 activates the human coronavirus 229E for cathepsin-independent host cell entry and is expressed in viral target cells in the respiratory epithelium. J. Virol. 87, 6150–6160 (2013).
Lokugamage, K. G. et al. Chimeric coronavirus-like particles carrying severe acute respiratory syndrome coronavirus (SCoV) S protein protect mice against challenge with SCoV. Vaccine 26, 797–808 (2008).
Lu, X. et al. Immune responses against severe acute respiratory syndrome coronavirus induced by virus-like particles in mice. Immunology 122, 496–502 (2007).
See, R. H. et al. Comparative evaluation of two severe acute respiratory syndrome (SARS) vaccine candidates in mice challenged with SARS coronavirus. J. General Virol. 87, 641–650 (2006).
Spruth, M. et al. A double-inactivated whole virus candidate SARS coronavirus vaccine stimulates neutralising and protective antibody responses. Vaccine 24, 652–661 (2006).
Qin, E. et al. Immunogenicity and protective efficacy in monkeys of purified inactivated Vero-cell SARS vaccine. Vaccine 24, 1028–1034 (2006).
Zhou, J. et al. Immunogenicity, safety, and protective efficacy of an inactivated SARS-associated coronavirus vaccine in rhesus monkeys. Vaccine 23, 3202–3209 (2005).
Lamirande, E. W. et al. A live attenuated severe acute respiratory syndrome coronavirus is immunogenic and efficacious in golden Syrian hamsters. J. Virol. 82, 7721–7724 (2008).
Netland, J. et al. Immunization with an attenuated severe acute respiratory syndrome coronavirus deleted in E protein protects against lethal respiratory disease. Virology 399, 120–128 (2010).
Fett, C., DeDiego, M. L., Regla-Nava, J. A., Enjuanes, L. & Perlman, S. Complete protection against severe acute respiratory syndrome coronavirus-mediated lethal respiratory disease in aged mice by immunization with a mouse-adapted virus lacking E protein. J. Virol. 87, 6551–6559 (2013).
Stadler, K. et al. SARS — beginning to understand a new virus. Nat. Rev. Microbiol. 1, 209–218 (2003).
Peiris, J. S., Guan, Y. & Yuen, K. Y. Severe acute respiratory syndrome. Nat. Med. 10, (Suppl. 12), 88–97 (2004).
Haagmans, B. L. et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 351, 77–81 (2016).
Acknowledgements
The research of J. F.-W. C. and K.-Y. Y. is partly supported by funding from Respiratory Viral Research Foundation Limited; the Consultancy Service for Enhancing Laboratory Surveillance of Emerging Infectious Disease of the Department of Health; the Health and Medical Research Fund (15140762) of the Food and Health Bureau; the National Natural Science Foundation of China/Research Grants Council Joint Research Scheme (N_HKU728/14); and the Theme-based Research Scheme (T11 707/15 R) of the Research Grants Council, Hong Kong Special Administrative Region of the People's Republic of China. A.Z. acknowledges support from the National Institutes of Health Research (NIHR), Biomedical Research Centre at UCL Hospital, London, UK. The funding sources had no role in study design, data collection, analysis, or interpretation or writing of the report.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
J. F.-W. C. has received travel grants from Pfizer Corporation Hong Kong and Astellas Pharma Hong Kong Corporation Limited. The funding sources had no role in study design, data collection, analysis or interpretation or writing of the report. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Supplementary information
Supplementary information S1 (figure)
Global Distribution of SARS and MERS (PDF 1587 kb)
Supplementary information S2 (table)
Comparisons of the clinical and laboratory features between SARS and MERS (PDF 139 kb)
Glossary
- Zoonotic virus
-
A virus that can transmit an infectious disease from animals (usually vertebrates) to humans.
- Spike glycoprotein (S)
-
A major immunogenic antigen of coronaviruses that is essential for interactions between a virus and host cell receptor, and is an important therapeutic target.
- Syncytium
-
A multinucleated cell resulting from the fusion of the host membranes of neighbouring cells infected by various viruses, including CoVs.
- Convalescent-phase plasma
-
Plasma that contains neutralizing antibodies against a microorganism and is obtained from patients recovering from the infection.
- Viroporin
-
A small integral membrane protein that is localized primarily within the endoplasmic reticulum and plasma membranes of host cells, and has the characteristic ability to form ion channels or pores.
- Protein cage nanoparticles
-
Nanoscale delivery platforms, made of biomaterials and/or proteins, that are used for various biomedical applications including the delivery of therapeutic cargo molecules.
- Koch's postulates
-
Criteria used to establish a causative relationship between a microorganism and a disease.
Rights and permissions
About this article

Cite this article
Zumla, A., Chan, J., Azhar, E. et al. Coronaviruses — drug discovery and therapeutic options. Nat Rev Drug Discov 15, 327–347 (2016). https://doi.org/10.1038/nrd.2015.37
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrd.2015.37
This article is cited by
-
Synergistic inhibition effects of andrographolide and baicalin on coronavirus mechanisms by downregulation of ACE2 protein level
Scientific Reports (2024)
-
Computational insight of repurpose drug for treatment of COVID-19: a CDFT approach
Theoretical Chemistry Accounts (2024)
-
Nanostructures for prevention, diagnosis, and treatment of viral respiratory infections: from influenza virus to SARS-CoV-2 variants
Journal of Nanobiotechnology (2023)
-
Systems pharmacology dissection of pharmacological mechanisms of Xiaochaihu decoction against human coronavirus
BMC Complementary Medicine and Therapies (2023)
-
Cytokine storm and translating IL-6 biology into effective treatments for COVID-19
Frontiers of Medicine (2023)
