
Abstract
Our understanding of, and approach to, pulmonary arterial hypertension has undergone a paradigm shift in the past decade. Once a condition thought to be dominated by increased vasoconstrictor tone and thrombosis, pulmonary arterial hypertension is now seen as a vasculopathy in which structural changes driven by excessive vascular cell growth and inflammation, with recruitment and infiltration of circulating cells, play a major role. Perturbations of a number of molecular mechanisms have been described, including pathways involving growth factors, cytokines, metabolic signaling, elastases, and proteases, that may underlie the pathogenesis of the disease. Elucidating their contribution to the pathophysiology of pulmonary arterial hypertension could offer new drug targets. The role of progenitor cells in vascular repair is also under active investigation. The right ventricular response to increased pressure load is recognized as critical to survival and the molecular mechanisms involved are attracting increasing interest. The challenge now is to integrate this new knowledge and explore how it can be used to categorize patients by molecular phenotype and tailor treatment more effectively.
Key Points
-
Pulmonary hypertension is a progressive disease of various origins, which has a poor prognosis and affects, in its different forms, more than 100 million people worldwide
-
Pulmonary arterial hypertension (PAH) is now considered to be a vasculopathy in which structural changes driven by excessive vascular cell growth and inflammation have a major role
-
A number of proproliferative signaling pathways involving growth factors, cytokines, metabolic signaling, and elastases and proteases have been identified in the pathophysiology of PAH
-
Clinical studies with tyrosine kinase inhibitors, serotonin antagonists, and soluble guanylate cyclase stimulators are underway in patients with PAH
-
The benefits of progenitor cells for vascular repair in PAH are under active investigation
-
The right ventricular response to increased pressure load is recognized as critical to survival in patients with PAH, and strategies for preserving myocardial function are increasingly attracting interest
Similar content being viewed by others

Introduction
Pulmonary hypertension is a progressive disease of various origins that is associated with a poor prognosis and results in right heart dysfunction. In all its variant presentations, this disease is estimated to affect up to 100 million people worldwide.1 According to the current classification of pulmonary hypertension, which was agreed upon at the 4th World Symposium on Pulmonary Hypertension in 2008, five categories of chronic pulmonary hypertension exist (Box 1).2
In this Review, we focus on molecular mechanisms involved in the pathogenesis of group 1, pulmonary arterial hypertension (PAH), which includes a variety of diseases that share several pathophysiological, histological, and prognostic features. Although rare, idiopathic PAH (IPAH; group 1.1) defines the group and is a disease in which there is neither a family history of, nor an identified risk factor for, PAH. In heritable PAH (group 1.2; formerly known as familial PAH), loss-of-function mutations in the transforming growth factor β/bone morphogenetic protein (TGF-β/BMP) receptor superfamily and, more rarely, in activin receptor-like kinase type 1 (ALK-1; also called ACVRL1) have been identified as underlying mechanisms. Drug-induced and toxin-induced forms of PAH are included in group 1.3, and group 1.4 comprises PAH associated with identified diseases (for example, HIV and schistosomiasis infections). Persistent pulmonary hypertension of the newborn is included in group 1.5, and patients with pulmonary veno-occlusive disease and/or pulmonary capillary hemangiomatosis are classified as being in group 1′.1,3,4,5
In this article, we highlight findings from patients with PAH. However, we also discuss preclinical data from a number of animal models of pulmonary hypertension. Notably, although the existing animal models inform on certain aspects of PAH, no animal model of pulmonary hypertension recapitulates the human condition. For example, mice exposed to hypoxia develop pulmonary hypertension and are widely used as a disease model, but although they display certain features in common with PAH, they are more closely related to group 3 pulmonary hypertension. Rats treated with the toxin monocrotaline, another frequently used model of pulmonary hypertension, may exhibit direct damage to the liver, kidneys, and heart, in addition to the pulmonary vasculature. For an in-depth discussion of the merits and limitations of the different animal models of pulmonary hypertension and their relevance to human PAH, the reader is referred to an article by Stenmark et al.6
Pathways of disease
The subcategories of PAH differ in their underlying causes. However, all are characterized by excessive pulmonary vasoconstriction and abnormal vascular remodeling processes that usually affect all vessel layers (intima, media, and adventitia) and result in severe loss of cross-sectional area and, therefore, increased right ventricular afterload (Figure 1).7 Large pulmonary artery compliance is also decreased, contributing to the strain on the right ventricle.8 Intimal changes include endothelial injury, endothelial cell proliferation, invasion of the intima by (myo)fibroblast-like cells, enhanced matrix deposition with intimal fibrosis and, sometimes, obstruction of the vascular lumen by unique plexiform lesions.9 Vascular smooth muscle cell (SMC) proliferation is a prominent feature of PAH. These structural changes suggest a switch from a quiescent state to a proliferative, apoptosis-resistant cellular phenotype. Lung vascular remodeling can be associated with chronic inflammatory events and recruitment of progenitor cell types, which might fuel proliferative changes (for example, in circulating fibrocytes and mesenchymal progenitor cells)10 or which might have an antiproliferative effect and thus offer a prospective therapeutic intervention (for example, endothelial progenitor cells).11

Putative therapeutic targets are indicated. Abbreviations: 5-HT, 5-hydroxytryptamin; K- and Ca-channels, potassium and calcium channels; AEC, alveolar epithelial cells; BMP, bone morphogenetic protein; cGMP, cyclic guanosine monophosphate; ECM, extracellular matrix; EGF, epidermal growth factor; EPC, endothelial progenitor cells; HIF, hypoxia inducible factor; MMPs, matrix metalloproteinases; NADPH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; PDE, phosphodiesterase; PDGF, platelet-derived growth factor; PGI2, prostaglandin I2; Rho-Ki, Rho kinases; ROS, reactive oxygen species; sGC, soluble guanylate cyclase; TGF, transforming growth factor-β; TK, tyrosine kinase; TKi, tyrosine kinase inhibitor; TRPC, transient receptor potential cation channels; VEGF, vascular endothelial growth factor.
Imbalance in vasoactive mediators
In patients with PAH, the generation of vasodilatory mediators, in particular prostaglandin I2, is reduced.12,13 Reduced levels of the vasodilatory and antiproliferative nitric oxide (NO; generated via NO synthases), and cyclic guanosine monophosphate (the second messenger downstream of NO) are also well described in pulmonary vascular disease, including PAH.14,15 Increased levels of asymmetric dimethylarginine (an endogenous inhibitor of NO synthase) might also have a functional role in PAH (Figure 2).16,17

Expression of certain growth factors is increased in pulmonary arterial hypertension, promoting cell proliferation, survival, and migration via a number of signaling pathways, and thus contributing to pulmonary vascular remodeling. Abbreviations: Akt, v-akt murine thymoma viral oncogene homolog; AP-1, activator protein-1; CaM, calmodulin; CamKII, calcium/calmodulin-dependent kinase II; CRE, cyclic adenosine monophosphate response element; DAG, diacylglycerol; Erk 1/2, extracellular signal-regulated kinase 1/2; FGF, fibroblast growth factor; GF, growth factor; Grb2, growth factor receptor-bound protein 2; IP3, inositol triphosphate; JAK-2, Janus-activated kinase 2; KSR, kinase suppressor of Ras; Mek 1/2, mitogen-activated protein kinase kinase 1/2; MLCK, myosin light chain kinase; PDGF, platelet-derived growth factor; PI3K, phosphoinositide-3-kinase; PKB, protein kinase B; PKC, protein kinase C; PLC, phospholipase C; Raf, rapidly growing fibrosarcoma; Ras, Ras protein; SOCS, suppressor of cytokine signaling; SOS, son of sevenless; STAT, signal transducer and activator of transcription.
In addition to reduced generation of vasodilatory mediators in patients with PAH, levels of the potent vasoconstrictors thromboxane and endothelin 1 are increased.12,13 Moreover, 5-hydroxytryptamine (5-HT) causes vasoconstriction, and abnormalities in endothelial cell–SMC cross-talk have been observed in humans with IPAH and a mouse model of hypoxia-induced pulmonary hypertension, with enhanced endothelial 5-HT production, transport, and paracrine activity in adjacent pulmonary vascular SMCs.18,19 Certain forms of drug-induced or toxin-induced PAH (group 1.3) also involve the 5-HT pathway. Fenfluramine and aminorex, anorexigenic drugs associated with an increased risk of PAH, are both 5-HT transporter substrates and increase extracellular concentrations of 5-HT.20
Abnormalities in both K+ channels and Ca2+ channels have been linked with pathological pulmonary vascular tone, dysregulation of cellular homeostasis, and induction of fibroproliferative sequelae, particularly in SMCs. A selective downregulation of pulmonary artery smooth muscle voltage-gated K+ channels has been described in tissue from humans with IPAH and from animal models of pulmonary hypertension, with subsequent membrane depolarization, opening of voltage-gated Ca2+ channels, and induction of muscle contraction via Ca2+–calmodulin and myosin light chain kinase.21,22,23 Moreover, Ca2+ signaling via transient receptor potential (TRP) ion channels has an important role in IPAH. TRPC3 and TRPC6 expression were found to be upregulated in pulmonary arterial SMCs from patients with IPAH, and inhibition of TRPC6 expression markedly reduced SMC proliferation.24 The acute hypoxic vasoconstrictor response was also reported to be exclusively dependent on TRPC6 in mice with hypoxia-induced pulmonary hypertension.25
Cell proliferation and vascular remodeling
Some vasoconstrictors are also able to exert proproliferative actions. Endothelin stimulates SMC proliferation via downstream signaling cascades involving endothelin type A and type B receptors.26 In addition to the vasoconstriction described above, which mainly occurs via the SMC 5-HT receptors, 5-HT also causes SMC proliferation, mostly via 5-HT-transporter-mediated 5-HT uptake.18,19,20 Sustained Ca2+ flux, whether from the extracellular space or from intracellular stores, may also exert mitogenic effects in SMCs, with Ca2+–calmodulin-dependent stimulation of the cell cycle, activation of mitogen-activated protein kinase pathways, and stimulation of the activating protein 1 family of transcription factors (upstream of several growth factors).27
Remodeling is seen in the large pulmonary arteries as well in the smaller distal pulmonary arteries. In mice with hypoxic pulmonary hypertension, compliance of the extralobar pulmonary arteries was found to be reduced as a result of narrowing and wall thickening, rather than vasoconstriction.28 Excessive vascular collagen might contribute to this phenotype; large pulmonary arteries from rats with hypoxic pulmonary hypertension showed increased collagen alongside decreased compliance,29 and expression of a degradation-resistant form of collagen in hypoxic mice prevented the mechanical properties of the extralobar pulmonary arteries returning to normal after removal from hypoxic conditions.30
Growth factors
Several growth factors are implicated in PAH and vascular remodeling (Figure 2). By binding and activating cell surface tyrosine kinase receptors, growth factors act as potent mitogens and chemoattractants for vascular cells, such as SMCs, fibroblasts, and endothelial cells. Activation of the tyrosine kinase receptor initiates major intracellular signaling cascades, resulting in cellular proliferation, migration, and resistance to apoptosis. Only the growth factors most clearly implicated in PAH will be discussed in this Review.
Vascular endothelial growth factor (VEGF) and VEGF receptor 2 (VEGFR-2) are expressed in the plexiform lesions of patients with PAH,31,32 indicating a role for VEGF in the pathogenesis of PAH. However, preclinical data have been inconsistent. VEGF is upregulated in rats with chronic hypoxic pulmonary hypertension, but downregulated in rats with monocrotaline-induced pulmonary hypertension.33,34 The inhibition of VEGFR in rats in vivo results in septal cell apoptosis and emphysema formation.35,36 However, when VEGFR inhibition is combined with chronic hypoxia in rats, severe pulmonary hypertension with angioproliferative lesions develops,37,38 and when rats with established pulmonary hypertension are treated with VEGF, the disease is aggravated.38 The controversies regarding the role of VEGF in the pathogenesis of pulmonary hypertension continue, as is evident from findings that gene-transfer-mediated VEGF overexpression in rats attenuates the development of experimental pulmonary hypertension (in monocrotaline-induced, pulmonary fibrosis, and hypoxic models),39,40,41 whereas inhibition of VEGF signaling by the small molecule multikinase inhibitor sorafenib in rats also attenuates experimental pulmonary hypertension (generated by monocrotaline or VEGFR inhibition combined with hypoxia).42,43 Overall, results indicate a unique and complex role for VEGF in pulmonary pathophysiology,44 but therapeutic applications are unlikely until the precise role of VEGF is elucidated.
Basic fibroblast growth factor (bFGF) is a ubiquitously expressed member of the FGF family. Substantial alterations in plasma and urine bFGF levels have been observed in patients with PAH.45 Upregulation of bFGF in response to hypoxia and shear stress is observed in rat and in ovine pulmonary arterial SMCs,46,47 and upregulation of FGF-2 occurs in lambs with pulmonary hypertension,48 but not hypoxic mice.49 Repeated intravenous FGF-2 small interfering RNA administration prevents (and nearly reverses) monocrotaline-induced pulmonary hypertension in rats, and pharmacological inhibition of FGF receptor 1 reverses established pulmonary hypertension.50 Thus, FGF-2 is a possible therapeutic target.
Pulmonary overexpression of TGF-α, a member of the epidermal growth factor (EGF) family, results in the development of pulmonary hypertension and vascular remodeling in mice,51 and might be associated with EGF-induced pulmonary vascular SMC proliferation.52,53 Inhibition of the EGF receptor attenuates monocrotaline-induced pulmonary hypertension in rats,54,55 but it does not improve chronic hypoxia-induced pulmonary hypertension in mice,54 undermining EGF as a therapeutic target in PAH.
Platelet-derived growth factor (PDGF) acts as a potent mitogen and chemoattractant for SMCs.56,57 Pulmonary expression of the PDGF ligand and receptor is increased in patients with severe PAH,58,59,60 and PDGF signaling is upregulated in the pulmonary arteries of patients with IPAH.60 Moreover, the activation of Wnt signaling (which regulates lung SMC development) in mice increases expression of PDGF receptors (PDGFRs) through the transcriptional activity of tenascin C.61 Upregulation of PDGFR has also been shown in lambs with pulmonary hypertension induced by partial ligation of the ductus arteriosus,62 and inhibition of PDGFR improves survival in rats with monocrotaline-induced pulmonary hypertension.42,59
Hepatocyte growth factor (HGF) signals through c-Met (a tyrosine kinase receptor). HGF expression decreases under hypoxic conditions in cell and organ culture systems,63 and HGF production is downregulated in monocrotaline-injected rats.64 Furthermore, HGF gene transfer attenuates the development of monocrotaline-induced pulmonary hypertension in rats.64,65 Thus, HGF might have a protective role in pulmonary vascular remodeling, but preclinical and clinical studies in this area are needed.
The TGF-β superfamily comprises a large series of cytokine growth factors that control many cellular functions, including proliferation, migration, differentiation, and extracellular matrix secretion and deposition. BMP receptor type 2 (BMPR2), ALK-1, and endoglin have been implicated as causal factors in hereditary and associated forms of PAH.66 Stem cell factor acts through its receptor c-kit, which is expressed on stem and progenitor cells as well as mast cells. Cells expressing c-kit are found in pulmonary artery walls, both in clinical PAH and in animals with hypoxic pulmonary hypertension.67,68,69,70 Further studies are needed to define the role of stem cell factor and c-kit.
Proteases and elastases
Matrix metalloproteinases (MMPs) have key roles in modulating structural extracellular matrix (ECM) proteins. They also enable release of growth factors from ECM-associated stores by revealing previously cryptic ECM binding sites, and by targeting non-ECM substrates. Upregulation of MMPs and endogenous vascular elastase activity occurs in remodeled lung vasculature in experimental and clinical settings of pulmonary hypertension,71,72,73 and inhibitors of MMPs and elastase exert beneficial effects in rats with monocrotaline-induced pulmonary hypertension.74,75 The broad spectrum of MMP activities and their roles in developmental biology and tissue homeostasis mandates a more detailed understanding of their physiological and pathophysiological functions in lung parenchyma and vasculature.
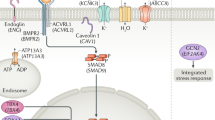
Bone morphogenetic protein receptor type 2
Mutations in the BMPR2 gene have been found in the majority of patients with heritable PAH and some forms of IPAH, providing a strong stimulus for further research in this field.76,77,78 To date, more than 140 distinct mutations have been identified in BMPR2 in patients with IPAH. Moreover, mutations in the gene for type I TGF-β superfamily receptor ALK-1 have been linked with severe PAH development in families with hereditary hemorrhagic telangiectasia.78 The mechanisms by which BMPR2 and ACVRL1 mutations disrupt physiological intracellular signaling are not well defined, and might vary between cell types. Some potential mechanisms suggested by in vitro studies include upregulation of proproliferative pathways involving p38 mitogen-activated protein kinase,79 reduced activation of the transcription factor Smad 1,80 and disruption of BMPR2 binding to Tctex-1 (a light chain of the motor complex dynein)81 and LIM kinase 1, which regulates actin dynamics.82 Further investigation is needed into the molecular mechanisms linking these mutations with the pulmonary vasculopathy, the growth of pulmonary arterial SMCs, and abnormal endothelial cell proliferation in PAH.83
Notch
Notch signaling is a conserved signaling mechanism, essential for cell-fate determination during embryonic development. Four mammalian Notch receptors (Notch 1–4) and five ligands (Jagged 1 and 2, and delta-like protein 1, 3, and 4) have been described. These receptors and ligands are membrane bound; therefore, signaling occurs by cell-to-cell contact, leading to proteolytic cleavages in the Notch receptor, including a final cleavage by γ secretase to release the receptor's intracellular domain. The intracellular domain of the Notch receptor then translocates to the nucleus and forms an active transcription activator complex with the DNA binding protein CBF-1 and activates transcription of the HES and HEY genes, its downstream targets (Figure 3).84

In the absence of activated Notch signaling, the DNA binding protein CBF-1/RBP-Jκ forms a complex with co-repressors to prevent transcription of target genes. Interaction of ligand (via its DSL motif) with the EGF-like repeats on the receptor triggers two successive proteolytic cleavages by TACE, followed by gamma secretase. These cleavages result in release of NICD, which translocates to the nucleus and interacts with CBF-1/RBP-Jκ, removing the co-repressors. Coactivators, including MAML, CBP, and other transcription factors, are subsequently recruited, leading to transcription of Notch target genes (the HES family of transcription repressors). Notch 3 is the predominantly expressed Notch receptor in vascular SMCs, including pulmonary artery SMCs. Arrows indicate changes observed in pulmonary arterial hypertension. Abbreviations: APH 1, anterior pharynx-defective 1; CBF-1, Cp-binding factor 1; CBP, CREB binding protein; CREB, cyclic adenosine monophosphate response element-binding; DSL, Delta Serrate Ligand; EGF, epidermal growth factor; MAML, mastermind-like protein; NICD, Notch Intracellular Domain; PEN 2, presenilin 2; RBP-Jκ, recombination signal sequence-binding protein-Jκ; SMC, smooth muscle cell; TACE, tumor necrosis factor α-converting enzyme; TBP, TATA box-binding protein; TF, transcription factor.
Notch signaling is involved in multiple aspects of vascular development, including vasculogenesis, angiogenesis, and differentiation of vascular SMCs.85 Notch 3 is expressed exclusively in adult human and rodent SMCs,86 and might regulate arterial SMC identity and maturation during development.87 Notch 3 acts to repress expression of contractile protein genes88 and to promote proliferation of vascular SMCs.89 During hypoxia, hypoxia-inducible factor 1α activates Notch-responsive genes, maintaining cells in an undifferentiated state.90 Notch 3 also regulates expression of PDGF receptor β in vascular SMCs.91
Notch 3 and its target gene, HES5, are expressed in lung biopsies from patients with nonfamilial PAH, and in the lungs of two rodent models of pulmonary hypertension (hypoxic mice and monocrotaline-treated rats).92 The extent of overexpression at the protein level correlates with disease severity in these rodent models, and lung immunofluorescent stains show confinement of Notch 3 and transcription factor HES-5 expression to the media of small pulmonary arteries and arterioles. Cultures of pulmonary arteriole SMCs from patients with PAH display higher levels of mRNA and protein for Notch 3 and transcription factor HES-5 than SMCs from healthy lungs. Overexpression of the Notch 3 intracellular domain leads to an increased growth rate of these SMCs, whereas knock down of HES5 obliterates this proliferative effect.92 Moreover, treatment with a γ-secretase inhibitor that blocks cleavage of the Notch receptor attenuates hypoxia-induced pulmonary hypertension in mice.92 In summary, these data indicate that upregulation of the Notch 3–transcription factor HES-5 signaling pathway is associated with development of PAH in humans and is needed for development of the disease in the rodent models of pulmonary hypertension.
Peroxisome proliferator-activated receptor γ
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors involved in physiological processes ranging from lipogenesis to inflammation.93 PPARγ is expressed in many tissues, including vascular endothelial cells, vascular SMCs, and macrophages,94,95 and has crucial roles in cell growth, inflammation, and angiogenesis.96,97,98 PPARγ is activated by several natural ligands,99 and is also stimulated by synthetic ligands termed thiazolidinediones (for example, troglitazone and rosiglitazone) that have been used as therapeutic agents in type 2 diabetes mellitus.100
PPARγ has a potential role in the pathogenesis of PAH. This transcription factor is expressed abundantly in pulmonary vascular endothelial cells in normal human lung tissue, but not in the angiogenic plexiform lesions of lungs from patients with PAH or in the vascular lesions of rats with severe pulmonary hypertension induced by inhibition of VEGFR combined with chronic hypoxia.101 Global deletion of PPARγ results in embryonic lethality,102 but the critical role of PPARγ in maintaining pulmonary vascular homeostasis was demonstrated in mice that spontaneously developed pulmonary hypertension as a result of conditional knockout of PPARγ in SMCs or in endothelial cells.103,104
If, as these reports suggest, reduced PPARγ in cells of the pulmonary vascular wall causes PAH, then the stimulation of PPARγ might alleviate PAH. Apolipoprotein E deficiency, in association with a high-fat diet, results in insulin resistance and PAH in mice, and PPARγ activation reversed PAH in this animal model.105 Endogenous expression of PPARγ in SMCs protects against the spontaneous development of PAH; studies using a PPARγ antagonist and PPARγ-deficient pulmonary artery SMCs showed that PPARγ is required for BMP-2-mediated inhibition of SMC proliferation.103 PPARγ activation also reduces levels of two factors critical in PAH pathogenesis: endothelin 1106 and asymmetric dimethylarginine (an endogenous NO synthase inhibitor).107,108 Activation of PPARγ by rosiglitazone induces significant expression of heme oxygenase 1 in primary cultured pulmonary arterial SMCs, which upregulates p21 (WAF1) expression, leading to inhibition of 5-HT-stimulated SMC proliferation.109 Moreover, PPARγ agonists partially reverse pulmonary hypertension induced by monocrotaline and chronic hypoxia in rats,110,111 attenuate the development of hypoxic pulmonary hypertension in mice, and reduce hypoxic Nox4 induction and generation of reactive oxygen species in the lung of mice.112 Concerns about the risks of cardiovascular events from the use of PPARγ agonists in older patients with diabetes temper enthusiasm for this therapeutic drug class in patients.113 Identifying downstream targets regulated by PPARs in pulmonary vasculature might enable development of PPAR-related therapeutic strategies for PAH and perhaps circumnavigate concerns over the adverse effects of PPARγ agonists reported in patients with diabetes.
Endothelial repair and angiogenesis
Bone-marrow-derived endothelial progenitor cells (EPCs) circulate in adult peripheral blood,114 and have a role in the maintenance of endothelial integrity and function and repair of endothelial injury. These cells also participate directly in postnatal vasculogenesis and angiogenesis in systemic vascular beds.115 Over a decade ago, Tuder et al. reported that plexiform lesions might result from deregulated growth of endothelial cells, and that the presence of perivascular inflammatory cells might influence lesion development.116 After VEGF and VEGFR-2 expression were demonstrated in plexiform lesions from patients with severe PAH, the lesions were hypothesized to perhaps be attributable to a process of disordered angiogenesis.32 Thus, proangiogenic factors (particularly progenitor cells) became a focus of research.
Animal studies involving transplantation of bone-marrow-derived cells suggest that bone-marrow progenitor cells are recruited during pulmonary vascular remodeling.117,118,119 Selective depletion of monocytes and fibrocytes attenuated hypoxia-induced pulmonary vascular remodeling in rats, indicating a role for fibrocytes in the remodeling process.120 By contrast, defective mobilization and recruitment of EPCs resulted in potentiation of hypoxic pulmonary hypertension in mice that lacked the erythropoietin receptor in nonerythroid lineages.121 Furthermore, bone-marrow-derived cells did not contribute substantially to pathological pulmonary arterial remodeling induced by monocrotaline with unilateral subpneumonectomy in rats.122 However, EPC dysfunction was sustained after chronic hypoxia-induced pulmonary hypertension in mice.123 It seems that compensatory EPC mobilization and recruitment might occur in PAH, but might not be sufficient to prevent disease development as the EPCs are dysfunctional.
Patients with Eisenmenger syndrome and IPAH have reduced numbers of EPCs.124,125 However, some investigators have found increased EPC numbers in patients with PAH,126,127 which may reflect an unsuccessful effort to repair damage to the pulmonary endothelium, as EPC function can be impaired in patients with PAH.126 Specifically, lung tissue from patients with IPAH showed increased expression of progenitor cell markers compared with controls, particularly within plexiform lesions.126 A higher level of circulating proangiogenic progenitor cells was also observed in patients with IPAH, compared with healthy individuals.126,127 Dysfunctional BMP2-mediated Wnt signaling128 might partly explain the altered endothelial cell behavior and angiogenesis; however, the precise mechanism of the (dys)regulation of angiogenesis in the pathogenesis of PAH remains unresolved. At the experimental level, the beneficial effects of proangiogenic therapies in animal models of pulmonary hypertension (associated with pulmonary fibrosis or induced by hypoxia or monocrotaline treatment) suggest that dysregulation of angiogenesis might be associated with pathogenesis.40,129,130 Impaired angiogenesis might be attributable to increased oxidative stress,131 among many other factors, but precise elucidation of the link between angiogenesis and PAH pathogenesis needs further study, and the role of stem and progenitor cells in the pathogenesis of PAH remains far from clear.
Inflammation as a trigger
Infectious organisms can affect the lung circulation directly by obliterating lung vessels or indirectly via inflammation. Inflammation has a key role in human PAH as well as in experimental models of pulmonary hypertension (Figure 4).132,133 Monocytes, macrophages, T lymphocytes, and dendritic cells are found in plexiform lesions, and other vascular lesions of PAH-affected human lungs.134 Furthermore, inflammation contributes to the growth of pulmonary plexiform lesions.135

In response to infection and inflammatory events, lung vascular cells produce inflammatory mediators (chemokines and cytokines), thereby recruiting inflammatory cells (macrophages, dendritic cells, mast cells, B cells, T cells, and T regulatory cells). With the coordination of inflammatory mediators, inflammatory cells might perpetuate the release of cytokines, chemokines, and growth factors. Finally, these processes lead to vascular remodeling in PAH by matrix remodeling, collagen deposition, vascular cell proliferation, migration, and in situ thrombosis. Abbreviations: CCL2, chemokine (C-C motif) ligand 2; CCL5, chemokine (C-C motif) ligand 5, also known as Regulated upon Activation, Normally T cell Expressed and Secreted (RANTES); CCR1, chemokine (C-C motif) receptor 1; FKN, fractalkine (also known as C-X3-C motif chemokine 1); IL, interleukin; MCP-1, monocyte chemotactic protein-1; PAH, pulmonary arterial hypertension.
PAH is associated with latent viral infections (for example, HIV, human herpes virus [HHV], Epstein–Barr virus, and cytomegalovirus).136 The causative role of viruses in PAH induction is not well characterized, but HHV-8—a vasculotropic herpes virus associated with angioproliferative disorders—has provided some insight. HHV-8 has a particular tropism for endothelial cells and B lymphocytes, and might regulate angiogenesis. In one study, HHV-8 latency-associated nuclear antigen 1 (LANA-1) was identified in the lung tissue of 10 of 16 (62.5%) patients with IPAH;137 however, other researchers have not found HHV-8 LANA-1.138 In vitro studies found that a HHV-8-encoded viral G-protein-coupled receptor induces MMP2 activation and promotes angiogenesis in human pulmonary artery endothelial cells, implicating this receptor as a causative agent in IPAH.139 Moreover, HHV-8 infection results in an apoptosis-resistant phenotype, which may be important in the development of plexiform lesions.140
PAH is also associated with schistosomiasis, a parasitic infection that causes substantial inflammation and pulmonary vascular disease. Over 200 million people are currently infected with schistosomiasis and up to 20% of those with long-term infection develop PAH.1,3,5,141 Lung tissue parasitic egg burden induces inflammatory and immune processes, and may underlie PAH pathogenesis in infected patients. Schistosoma eggs modulate regulatory T cell activity and express a novel TGF-β superfamily member: Schistosoma mansoni inhibin/activin (SmInAct).142 Infiltration of T and B cells, plus increased serum levels of interleukin (IL)-1, IL-6, connexin 36, fractalkine (FKN) and chemokine (C-C motif) ligand 5 (also known as Regulated upon Activation, Normally T cell Expressed and Secreted; RANTES), have been reported in the pulmonary vascular lesions of patients with Schistosoma-related PAH.1 Interestingly, the chronically infected schistosomiasis mouse model showed extensive pulmonary vascular remodeling in the absence of pulmonary hypertension and a correlation between cytokine levels (particularly IL-13) and the extent of remodeling.143
Cytokines
Substantial evidence indicates a role for inflammatory cytokines in IPAH. Increased circulating levels of monocyte chemoattractant protein 1, tumor necrosis factor, IL-1β, and IL-6 were found in patients with IPAH.144,145 T and B lymphocytes and macrophages are described in plexiform lesions characteristic of PAH.146
Animal models also support the role of inflammatory cytokines in the initiation and progression of PAH. Monocrotaline-treated rats showed increased IL-6 expression in the lungs, and rats injected with IL-6 developed pulmonary hypertension.147,148 Furthermore, transgenic overexpression of IL-6 led to severe pulmonary hypertension in mice,149 and IL-6-deficient mice were protected from hypoxia-driven experimental pulmonary hypertension.150 Both in-vitro and in-vivo models also suggest an interaction between BMPR2 and IL-6: transgenic mice overexpressing a dominant-negative BMPR2 had increased lung IL-6 expression and enhanced susceptibility to pulmonary hypertension,151 and increased IL-6 levels were found to suppress the expression of BMPR2 via induction of a microRNA cluster pathway.152
Chemokines
Chemokines have a major role in the various steps of leukocyte recruitment, including initial reversible adherence to the endothelium (rolling), activation, firm adherence, and extravasation into the inflamed tissue.153 Chemokine-dependent mechanisms leading to inflammatory cell recruitment in the lungs of patients with PAH have been analyzed.154,155
FKN (also known as C-X3-C motif chemokine 1) is a soluble chemotactic protein that is also found as a membrane-anchored cell-adhesion molecule on endothelial cells.156,157 The actions of FKN are mediated by CX3C chemokine receptor 1 (CX3CR1), a transmembrane receptor expressed by monocytes, microglial cells, neurons, natural killer cells, mast cells, and subpopulations of T lymphocytes. Unlike other chemokines, FKN can mediate the rapid-capture, integrin-independent adhesion, and activation of circulating CX3CR1+ leukocytes under high blood flow.158 FKN expression is increased in circulating CD4+ and CD8+ T lymphocytes from patients with PAH, in inflammatory cells surrounding plexiform lesions, and in pulmonary arterial SMCs of remodeled arteries.155,159 A polymorphism in CX3CR1 is associated with a reduced risk of acute coronary artery disease in humans, indicating that FKN has a critical role in monocyte and T cell recruitment to the vessel wall.160 Other cytokines overproduced in patients with PAH are RANTES and chemokine ligand 2 (CCL-2).154,161 CCL-2 overproduction is a feature of the abnormal pulmonary endothelial cell phenotype in IPAH and, compared with healthy controls, pulmonary artery SMCs from patients with IPAH exhibited stronger migratory and proliferative responses to CCL-2.161
Thrombosis
Thrombosis is a common feature of PAH, and research using calibrated automated thrombography demonstrated that patients with IPAH have a hypercoagulable phenotype.162 Tissue factor (which initiates coagulation) is upregulated in the pulmonary vasculature of rats with pulmonary hypertension induced by monocrotaline and pneumonectomy, and is also strongly expressed in the pulmonary vasculature of patients with IPAH.163 Increased plasma levels of fibrinopeptide A, a marker of fibrin generation, have been reported in patients with IPAH.164 Fibrinolysis might also be impaired, since patients with IPAH were shown to have increased levels of plasminogen activator inhibitor 1, compared with controls.164 Increased activity of von Willebrand factor, which is essential for the interaction between platelets and endothelial cells, has been noted in IPAH.164 Platelet aggregation might also be increased as a result of imbalance in vasoactive mediators; thromboxane A2 (levels of which are increased in patients with PAH) is proaggregatory, whereas NO and prostacyclin (levels of which are decreased in patients with PAH) inhibit aggregation.12,165
Warburg hypothesis: role of mitochondria
In 1924, Otto Warburg proposed that a shift in glucose metabolism from oxidative phosphorylation to glycolysis (despite adequate oxygen) was central to the growth of cancers, and this phenomenon became known as the 'Warburg effect' (Figure 5).166 Glycolysis is less efficient at the generation of ATP than at oxidative phosphorylation, which results in increased glucose uptake, a phenomenon that can be tracked using 18F-fluorodeoxyglucose (18F-FDG). Cancers typically show increased 18F-FDG uptake and data suggest that proliferating pulmonary vascular endothelial cells from patients with PAH have a similar phenotype.167 Reduced oxidative phosphorylation inhibits the electron transport chain and reduces the generation of superoxide, which might confer resistance to apoptosis and so perpetuate the phenotype. If so, then restoration of oxidative phosphorylation would be expected to render cells more susceptible to apoptosis and offer a novel strategy for treating cancer and vascular remodeling in PAH.

The PDH complex converts pyruvate, derived from glycolysis, to acetyl-coenzyme A in the mitochondrion, thus allowing it to enter the TCA Cycle and generate up to 36 moles of ATP per molecule of glucose in the presence of oxygen. Electron donors (mitochondrial NADH and FADH) produced by the TCA cycle pass electrons down a redox-potential gradient in the electron transport chain to molecular O2. This electron flux powers H+ ion extrusion, powering ATP synthase. SOD2 converts superoxide anion (produced at complexes I and III) to H2O2, which serves as a redox messenger signaling 'normoxia'. In hypoxia, there is activation of HIF-1 and PDK which inhibits PDH. If pyruvate remains in the cytoplasm, it may complete glycolysis, producing lactic acid and generating 2 moles of ATP per glucose molecule. Dicholoroacetate inhibits PDK and increases the ratio of glucose oxidation to glycolysis. Abbreviations: ATP, adenosine triphosphate; FADH, flavine adenine dinucleotide; HIF-1, hypoxia-inducible factor 1; NADH, nicotinamide adenine dinucleotide; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; SOD2, superoxide dismutase 2; TCA, tricarboxylic acid.
Dichloroacetate, a pyruvate dehydrogenase kinase inhibitor, has been used to test this hypothesis: dichloroacetate decreases tumor growth in vitro and in vivo, without affecting noncancer mitochondria and tissues.168 Studies of this drug in several experimental models of pulmonary hypertension have also been encouraging. Chronic dosing of dichloroacetate markedly attenuated pulmonary hypertension in rats exposed to hypoxia or monocrotaline, the fawn-hooded rat, which has deficient uptake of serotonin into platelets and develops pulmonary hypertension spontaneously, and mice overexpressing the serotonin transporter.169,170,171
Clinical studies in glioblastoma multiforme have begun,172 and a clinical trial of dichloroacetate in PAH is also underway.173 The right ventricle in patients with PAH also exhibits a metabolic shift to glycolysis, which might impair function.174 Inhibition of pyruvate dehydrogenase kinase might, therefore, have beneficial effects on myocardial function in addition to effects on pulmonary vascular resistance.175,176
Right ventricular hypertrophy
Right ventricular hypertrophy (RVH) develops when the right ventricle works against increased resistance in the pulmonary circulation. Cardiac hypertrophy—defined as an increase in myocyte size without cell replication—develops in PAH, at first as a compensatory mechanism. The right ventricle accumulates muscle mass (via elevated protein synthesis and an increase in myocyte size), and the right ventricle wall thickens.177 The right ventricle also assumes a more-rounded shape, compressing the left ventricle.178 Cardiac output is initially maintained, but with persistent elevated resistance, progressive contractile dysfunction occurs, leading to decompensation, dilatation, and right heart failure.177,179
Elevated pressure within the right ventricle increases right ventricular wall stress and myocardial oxygen demand.177 The increase in wall stress also impedes myocardial perfusion; impaired right coronary artery systolic flow and right ventricular ischemia have both been demonstrated in patients with PAH.180,181 Mechanical stretch is translated into intracellular signaling pathways via integrins,182 and integrin signaling to focal adhesion kinase has been implicated in right ventricular myocardial hypertrophy in rats with monocrotaline-induced pulmonary hypertension.183 Metabolic changes have been noted, with upregulation of glucose transporter protein 4 (GLUT4) in the right ventricle of rats with monocrotaline-induced pulmonary hypertension,184 and increased right ventricular uptake of 18F-FDG in patients with PAH.174
Little is currently known about the processes governing the transition from compensated RVH to decompensated right heart failure in PAH. The development of right heart failure might be accelerated by maladaptive neurohormonal signaling, formation of reactive oxygen and nitrogen species, and exaggerated inflammatory responses.177 Although elevated right ventricular afterload leads to RVH, experimental data suggest it might not be sufficient to cause progression to right heart failure. Rats with afterload increased by pulmonary arterial banding developed RVH but not right heart failure, whereas rats with pulmonary hypertension induced by VEGFR inhibition combined with hypoxia did develop right heart failure, indicating that the remodeled vasculature in PAH might release mediators that interfere with adaptive RVH.185 The monocrotaline rat model might also help to shed light on the transition from RVH to right heart failure. Rats treated with 30 mg/kg monocrotaline develop compensated RVH, whereas rats treated with a higher dose of 80 mg/kg monocrotaline develop right heart failure. This model has been used to demonstrate that right heart failure is associated with myocardial expression of tenascin C and downregulation of integrin alpha 6, and the investigators suggested that the deadhesive properties of tenascin C might cause myocardial slippage and thus lead to ventricular dilatation.186
Harnessing molecular technologies
The power of genetics to aid our understanding of disease mechanisms is illustrated by the revelation of a link between mutations in BMPR2 and PAH. Evolving technology and expertise in genomics, transcriptomics, and proteomics offer promise of further insights into the pathogenesis of this disease. These tools are being employed with enthusiasm for discovery research on serum and plasma samples, circulating cells, and human tissue;187,188 animal models provide an alternative source of tissue for understanding molecular mechanisms.187 For example, a screen to detect altered protein expression in mice with hypoxic pulmonary hypertension led to the identification of a novel protein with a role in IPAH. Four and a half LIM domains protein 1 (FHL-1), a protein involved in muscle development, was upregulated in the lungs after 24 h of hypoxia (that is, at the onset of pulmonary hypertension).187 Strong upregulation of FHL-1 was also observed in pulmonary arteries from patients with IPAH, and in vitro studies in human pulmonary arterial SMCs showed that FHL-1 promotes cellular migration and proliferation.187 Proteomic screening of plasma samples and lung tissue from patients with PAH and controls has also identified a range of differentially expressed proteins with roles in growth, proliferation, and metabolism, providing new avenues for future research.188,189 One protein found to be upregulated in lungs from patients with IPAH, chloride intracellular channel protein 4, is important for vascular endothelial cell integrity and merits further study.189
Advances in imaging also permit studies of disease processes in vivo.190 Molecular imaging using PET is an emerging illustration of a novel approach to investigating the pathology of PAH in vivo. PET imaging is widely used in oncology, and a variety of PET tracers in development permit measurement of cell metabolism, proliferation, apoptosis, tissue hypoxia, and angiogenesis in vivo. 18F-FDG is an example of a PET tracer that is used frequently. This glucose analog is transported into metabolically active cells by the same transporter as glucose and converted to 18F-FDG-6-phosphate. In contrast to glucose, however, 18F-FDG cannot be further metabolized and 18F-FDG-6-phosphate becomes trapped in cells at a rate proportional to glucose use. FDG-PET in patients with cancer exploits the observation that many cancers use glycolysis in the cytoplasm instead of mitochondrial glucose oxidation as a major energy source. Studies in patients with PAH show increased uptake in the lung,167 but at this time it is unclear whether this uptake represents vascular cell proliferation or inflammation. In an ongoing study in patients with IPAH,173 dichloroacetate is being used, and 18F-FDG uptake in lung and heart measured, to probe the relationship between aberrant glucose metabolism and cardiopulmonary hemodynamics. Studies using new tracers (for example, 18F-labeled fluorothymidine and 18F-labeled fluciclatide) might inform us further on the relationship between active proliferation and angiogenesis and the natural history of PAH. Interestingly, FDG uptake is increased in the right ventricle of patients with PAH, and uptake is reduced by targeted PAH therapy (such as prostacyclin).174 FDG uptake, alone or combined with cardiac MRI, might prove a useful biomarker of cardiac function in PAH.
Conclusions
PAH is a treatable disease. The rapid pace of advance in our understanding of the condition in the past few years reflects, at least in part, the modern strategy of pursuing molecular mechanisms across 'disease boundaries'. As this pursuit continues, new therapies and management strategies will emerge for robust examination in the clinical setting.
A key challenge going forward will be to integrate this new knowledge in a systems biology approach, to prioritize key molecular pathways and link to personalized therapeutic interventions. The current assessment of patients with PAH is based on clinical phenotyping and fails to take into consideration the fact that PAH is a heterogeneous disorder. In neoplastic disorders, molecular signature analysis has proven useful in assigning prognosis and likely response to treatment. This assignment should be the goal in PAH management; patients with PAH are widely recognized to vary considerably in their response to treatment. For example, a small, but important, minority of patients with IPAH exhibit a pronounced acute hemodynamic response to NO inhalation, which predicts an approximately 50% probability of a favorable long-term response to calcium-channel blockers.191 Ongoing work to understand the molecular basis of this response, by comparing genomic sequences from responders and nonresponders, might provide a noninvasive test to guide the prescription of calcium antagonists for patients with IPAH. Circulating protein biomarkers, such as B-type natriuretic peptide (BNP) or N-terminal-proBNP, and growth differentiation factor 15, can be used to stratify patients according to prognosis but have limited clinical utility for diagnosis or predicting optimal drug therapy. Genomic markers carry more information and offer greater promise, whether based on haplotypes, circulating cell transcriptomes, or microRNA profiles. The individualization of therapy in PAH is an exciting prospect, but we are still at the very beginning of this story.
Review criteria
Articles were selected from literature published in English and listed by PubMed Central. Search terms included, but were not limited to, “pulmonary arterial hypertension”, “hypoxia”, “monocrotaline”, “rat”, “mouse”, “human”, “proliferation”, “apoptosis”, “genetics”, “genomics”, “mRNA”, “transcriptomics”, “proteomics”, and “imaging”, in various combinations. No date limitations were used. All cited articles reflect established or emerging opinions discussed in various fora (for example, international meetings) by recognized experts in the field of pulmonary vascular disease.
References
Simonneau, G. et al. Updated clinical classification of pulmonary hypertension. J. Am. Coll. Cardiol. 54, S43–S54 (2009).
dos Santos Fernandes, C. J. et al. Survival in schistosomiasis-associated pulmonary arterial hypertension. J. Am. Coll. Cardiol. 56, 715–720 (2010).
Graham, B. B., Bandeira, A. P., Morrell, N. W., Butrous, G. & Tuder, R. M. Schistosomiasis-associated pulmonary hypertension: pulmonary vascular disease: the global perspective. Chest 137, 20S–29S (2010).
Lapa, M. et al. Cardiopulmonary manifestations of hepatosplenic schistosomiasis. Circulation 119, 1518–1523 (2009).
Butrous, G., Ghofrani, H. A. & Grimminger, F. Pulmonary vascular disease in the developing world. Circulation 118, 1758–1766 (2008).
Stenmark, K. R., Meyrick, B., Galie, N., Mooi, W. J. & McMurtry, I. F. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L1013–L1032 (2009).
Tuder, R. M. et al. Development and pathology of pulmonary hypertension. J. Am. Coll. Cardiol. 54, S3–S9 (2009).
Fourie, P. R., Coetzee, A. R. & Bolliger, C. T. Pulmonary artery compliance: its role in right ventricular-arterial coupling. Cardiovasc. Res. 26, 839–844 (1992).
Rabinovitch, M. Pathobiology of pulmonary hypertension. Annu. Rev. Pathol. 2, 369–399 (2007).
Frid, M. G. et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 168, 659–669 (2006).
Zhao, Y. D. et al. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells: efficacy of combined cell and eNOS gene therapy in established disease. Circ. Res. 96, 442–450 (2005).
Christman, B. W. et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N. Engl. J. Med. 327, 70–75 (1992).
Kreymborg, K. et al. Identification of right heart-enriched genes in a murine model of chronic outflow tract obstruction. J. Mol. Cell. Cardiol. 49, 598–605 (2010).
Ghofrani, H. A., Osterloh, I. H. & Grimminger, F. Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat. Rev. Drug Discov. 5, 689–702 (2006).
Wharton, J. et al. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am. J. Respir. Crit. Care Med. 172, 105–113 (2005).
Leiper, J. et al. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat. Med. 13, 198–203 (2007).
Pullamsetti, S. et al. Increased levels and reduced catabolism of asymmetric and symmetric dimethylarginines in pulmonary hypertension. FASEB J. 19, 1175–1177 (2005).
Launay, J. M. et al. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat. Med. 8, 1129–1135 (2002).
Eddahibi, S. et al. Cross talk between endothelial and smooth muscle cells in pulmonary hypertension: critical role for serotonin-induced smooth muscle hyperplasia. Circulation 113, 1857–1864 (2006).
Uchida, S. et al. An integrated approach for the systematic identification and characterization of heart-enriched genes with unknown functions. BMC Genomics 10, 100 (2009).
Yuan, X. J., Wang, J., Juhaszova, M., Gaine, S. P. & Rubin, L. J. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet 351, 726–727 (1998).
Mandegar, M. & Yuan, J. X. Role of K+ channels in pulmonary hypertension. Vascul. Pharmacol. 38, 25–33 (2002).
Moudgil, R., Michelakis, E. D. & Archer, S. L. The role of K+ channels in determining pulmonary vascular tone, oxygen sensing, cell proliferation, and apoptosis: Implications in hypoxic pulmonary vasoconstriction and pulmonary arterial hypertension. Microcirculation 13, 615–632 (2006).
Yu, Y. et al. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc. Natl Acad. Sci. U.S.A. 101, 13861–13866 (2004).
Weissmann, N. et al. Classical transient receptor potential channel 6 (TRPC6) is essential for hypoxic pulmonary vasoconstriction and alveolar gas exchange. Proc. Natl Acad. Sci. U.S.A. 103, 19093–19098 (2006).
Davie, N. et al. ETA and ETB receptors modulate the proliferation of human pulmonary artery smooth muscle cells. Am. J. Respir. Crit. Care Med. 165, 398–405 (2002).
Burg, E. D., Remillard, C. V. & Yuan, J. X. Potassium channels in the regulation of pulmonary artery smooth muscle cell proliferation and apoptosis: pharmacotherapeutic implications. Br. J. Pharmacol. 153, S99–S111 (2008).
Tabima, D. M. & Chesler, N. C. The effects of vasoactivity and hypoxic pulmonary hypertension on extralobar pulmonary artery biomechanics. J. Biomech. 43, 1864–1869 (2010).
Tozzi, C. A., Christiansen, D. L., Poiani, G. J. & Riley, D. J. Excess collagen in hypertensive pulmonary arteries decreases vascular distensibility. Am. J. Respir. Crit. Care Med. 149, 1317–1326 (1994).
Ooi, C. Y., Wang, Z., Tabima, D. M., Eickhoff, J. C. & Chesler, N. C. The role of collagen in extralobar pulmonary artery stiffening in response to hypoxia-induced pulmonary hypertension. Am. J. Physiol. Heart Circ. Physiol. 299, H1823–H1831 (2010).
Geiger, R. et al. Enhanced expression of vascular endothelial growth factor in pulmonary plexogenic arteriopathy due to congenital heart disease. J. Pathol. 191, 202–207 (2000).
Tuder, R. M. et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J. Pathol. 195, 367–374 (2001).
Partovian, C. et al. Heart and lung VEGF mRNA expression in rats with monocrotaline- or hypoxia-induced pulmonary hypertension. Am. J. Physiol. 275, H1948–H1956 (1998).
Partovian, C. et al. Cardiac and lung VEGF mRNA expression in chronically hypoxic and monocrotaline-treated rats. Chest 114, 45S–46S (1998).
Kasahara, Y. et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Invest. 106, 1311–1319 (2000).
Tuder, R. M. et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am. J. Respir. Cell Mol. Biol. 29, 88–97 (2003).
Taraseviciene-Stewart, L. et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 15, 427–438 (2001).
Taraseviciene-Stewart, L. et al. Simvastatin causes endothelial cell apoptosis and attenuates severe pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L668–L676 (2006).
Campbell, A. I., Zhao, Y., Sandhu, R. & Stewart, D. J. Cell-based gene transfer of vascular endothelial growth factor attenuates monocrotaline-induced pulmonary hypertension. Circulation 104, 2242–2248 (2001).
Farkas, L. et al. VEGF ameliorates pulmonary hypertension through inhibition of endothelial apoptosis in experimental lung fibrosis in rats. J. Clin. Invest. 119, 1298–1311 (2009).
Partovian, C. et al. Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am. J. Respir. Cell Mol. Biol. 23, 762–771 (2000).
Klein, M. et al. Combined tyrosine and serine/threonine kinase inhibition by sorafenib prevents progression of experimental pulmonary hypertension and myocardial remodeling. Circulation 118, 2081–2090 (2008).
Moreno-Vinasco, L. et al. Genomic assessment of a multikinase inhibitor, sorafenib, in a rodent model of pulmonary hypertension. Physiol. Genomics 33, 278–291 (2008).
Tuder, R. M. & Yun, J. H. Vascular endothelial growth factor of the lung: friend or foe. Curr. Opin. Pharmacol. 8, 255–260 (2008).
Benisty, J. I. et al. Elevated basic fibroblast growth factor levels in patients with pulmonary arterial hypertension. Chest 126, 1255–1261 (2004).
Li, P., Oparil, S., Sun, J. Z., Thompson, J. A. & Chen, Y. F. Fibroblast growth factor mediates hypoxia-induced endothelin-- a receptor expression in lung artery smooth muscle cells. J. Appl. Physiol. 95, 643–651 (2003).
Quinn, T. P., Schlueter, M., Soifer, S. J. & Gutierrez, J. A. Cyclic mechanical stretch induces VEGF and FGF-2 expression in pulmonary vascular smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 282, L897–L903 (2002).
Wedgwood, S. et al. Fibroblast growth factor 2 expression is altered in lambs with increased pulmonary blood flow and pulmonary hypertension. Pediatr. Res. 61, 32–36 (2007).
Kwapiszewska, G. et al. Expression profiling of laser-microdissected intrapulmonary arteries in hypoxia-induced pulmonary hypertension. Respir. Res. 6, 109 (2005).
Izikki, M. et al. Endothelial-derived FGF2 contributes to the progression of pulmonary hypertension in humans and rodents. J. Clin. Invest 119, 512–523 (2009).
Le Cras, T. D., Hardie, W. D., Fagan, K., Whitsett, J. A. & Korfhagen, T. R. Disrupted pulmonary vascular development and pulmonary hypertension in transgenic mice overexpressing transforming growth factor-alpha. Am. J. Physiol. Lung Cell. Mol. Physiol. 285, L1046–L1054 (2003).
Jones, P. L. & Rabinovitch, M. Tenascin-C is induced with progressive pulmonary vascular disease in rats and is functionally related to increased smooth muscle cell proliferation. Circ. Res. 79, 1131–1142 (1996).
Schultz, K., Fanburg, B. L. & Beasley, D. Hypoxia and hypoxia-inducible factor-1alpha promote growth factor-induced proliferation of human vascular smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 290, H2528–H2534 (2006).
Dahal, B. K. et al. Role of epidermal growth factor inhibition in experimental pulmonary hypertension. Am. J. Respir. Crit. Care Med. 181, 158–167 (2010).
Merklinger, S. L., Jones, P. L., Martinez, E. C. & Rabinovitch, M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation 112, 423–431 (2005).
Grimminger, F. & Schermuly, R. T. PDGF receptor and its antagonists: role in treatment of PAH. Adv. Exp. Med. Biol. 661, 435–446 (2010).
Yu, Y. et al. PDGF stimulates pulmonary vascular smooth muscle cell proliferation by upregulating TRPC6 expression. Am. J. Physiol. Cell Physiol. 284, C316–C330 (2003).
Humbert, M. et al. Platelet-derived growth factor expression in primary pulmonary hypertension: comparison of HIV seropositive and HIV seronegative patients. Eur. Respir. J. 11, 554–559 (1998).
Schermuly, R. T. et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J. Clin. Invest. 115, 2811–2821 (2005).
Perros, F. et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 178, 81–88 (2008).
Cohen, E. D. et al. Wnt signaling regulates smooth muscle precursor development in the mouse lung via a tenascin C/PDGFR pathway. J. Clin. Invest 119, 2538–2549 (2009).
Balasubramaniam, V. et al. Role of platelet-derived growth factor in vascular remodeling during pulmonary hypertension in the ovine fetus. Am. J. Physiol. Lung Cell Mol. Physiol. 284, L826–L833 (2003).
Hayashi, S. et al. Potential role of hepatocyte growth factor, a novel angiogenic growth factor, in peripheral arterial disease: downregulation of HGF in response to hypoxia in vascular cells. Circulation 100, II301–II308 (1999).
Ono, M. et al. Hepatocyte growth factor suppresses vascular medial hyperplasia and matrix accumulation in advanced pulmonary hypertension of rats. Circulation 110, 2896–2902 (2004).
Ono, M. et al. Gene transfer of hepatocyte growth factor with prostacyclin synthase in severe pulmonary hypertension of rats. Eur. J. Cardiothorac. Surg. 26, 1092–1097 (2004).
Machado, R. D. et al. Genetics and genomics of pulmonary arterial hypertension. J. Am. Coll. Cardiol. 54, S32–S42 (2009).
Heath, D. & Yacoub, M. Lung mast cells in plexogenic pulmonary arteriopathy. J. Clin. Pathol. 44, 1003–1006 (1991).
Mitani, Y. et al. Mast cell chymase in pulmonary hypertension. Thorax 54, 88–90 (1999).
Nicolls, M. R., Taraseviciene-Stewart, L., Rai, P. R., Badesch, D. B. & Voelkel, N. F. Autoimmunity and pulmonary hypertension: a perspective. Eur. Respir. J. 26, 1110–1118 (2005).
Tucker, A., McMurtry, I. F., Alexander, A. F., Reeves, J. T. & Grover, R. F. Lung mast cell density and distribution in chronically hypoxic animals. J. Appl. Physiol. 42, 174–178 (1977).
Rabinovitch, M. Elastase and the pathobiology of unexplained pulmonary hypertension. Chest 114, 213S–224S (1998).
Lepetit, H. et al. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur. Respir. J. 25, 834–842 (2005).
Benisty, J. I. et al. Matrix metalloproteinases in the urine of patients with pulmonary arterial hypertension. Chest 128 (Suppl. 6), 572S (2005).
Cowan, K. N. et al. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. J. Am. Coll. Cardiol. 35, 545A–545A (2000).
Cowan, K. N., Jones, P. L. & Rabinovitch, M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J. Clin. Invest. 105, 21–34 (2000).
Lane, K. B. et al. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. Nat. Genet. 26, 81–84 (2000).
Deng, Z. et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am. J. Hum. Genet. 67, 737–744 (2000).
Trembath, R. C. et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 345, 325–334 (2001).
Rudarakanchana, N. et al. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum. Mol. Genet. 11, 1517–1525 (2002).
Yang, X. et al. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ. Res. 96, 1053–1063 (2005).
Machado, R. D. et al. Functional interaction between BMPR-II and Tctex-1, a light chain of Dynein, is isoform-specific and disrupted by mutations underlying primary pulmonary hypertension. Hum. Mol. Genet. 12, 3277–3286 (2003).
Foletta, V. C. et al. Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J. Cell Biol. 162, 1089–1098 (2003).
Eickelberg, O. & Morty, R. E. Transforming growth factor beta/bone morphogenic protein signaling in pulmonary arterial hypertension: Remodeling revisited. Trends Cardiovasc. Med. 17, 263–269 (2007).
Artavanis-Tsakonas, S., Rand, M. D. & Lake, R. J. Notch signaling: cell fate control and signal integration in development. Science 284, 770–776 (1999).
Alva, J. A. & Iruela-Arispe, M. L. Notch signaling in vascular morphogenesis. Curr. Opin. Hematol. 11, 278–283 (2004).
Villa, N. et al. Vascular expression of Notch pathway receptors and ligands is restricted to arterial vessels. Mech. Dev. 108, 161–164 (2001).
Domenga, V. et al. Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev. 18, 2730–2735 (2004).
Proweller, A., Pear, W. S. & Parmacek, M. S. Notch signaling represses myocardin-induced smooth muscle cell differentiation. J. Biol. Chem. 280, 8994–9004 (2005).
Campos, A. H., Wang, W., Pollman, M. J. & Gibbons, G. H. Determinants of Notch-3 receptor expression and signaling in vascular smooth muscle cells: implications in cell-cycle regulation. Circ. Res. 91, 999–1006 (2002).
Gustafsson, M. V. et al. Hypoxia requires notch signaling to maintain the undifferentiated cell state. Dev. Cell 9, 617–628 (2005).
Jin, S. et al. Notch signaling regulates platelet-derived growth factor receptor-beta expression in vascular smooth muscle cells. Circ. Res. 102, 1483–1491 (2008).
Li, X. D. et al. Notch3 signaling promotes the development of pulmonary arterial hypertension. Nat. Med. 15, 1289–1297 (2009).
Issemann, I. & Green, S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 347, 645–650 (1990).
Rios-Vazquez, R., Marzoa-Rivas, R., Gil-Ortega, I. & Kaski, J. C. Peroxisome proliferator-activated receptor-gamma agonists for management and prevention of vascular disease in patients with and without diabetes mellitus. Am. J. Cardiovasc. Drugs 6, 231–242 (2006).
Duan, S. Z., Usher, M. G. & Mortensen, R. M. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ. Res. 102, 283–294 (2008).
Chinetti, G. et al. Activation of proliferator-activated receptors alpha and gamma induces apoptosis of human monocyte-derived macrophages. J. Biol. Chem. 273, 25573–25580 (1998).
Jiang, C., Ting, A. T. & Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391, 82–86 (1998).
Panigrahy, D. et al. PPARgamma ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J. Clin. Invest. 110, 923–932 (2002).
Takeda, K., Ichiki, T., Tokunou, T., Iino, N. & Takeshita, A. 15-Deoxy-delta 12,14-prostaglandin J2 and thiazolidinediones activate the MEK/ERK pathway through phosphatidylinositol 3-kinase in vascular smooth muscle cells. J. Biol. Chem. 276, 48950–48955 (2001).
Spiegelman, B. M. & Flier, J. S. Adipogenesis and obesity: rounding out the big picture. Cell 87, 377–389 (1996).
Ameshima, S. et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ. Res. 92, 1162–1169 (2003).
Barak, Y. et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 4, 585–595 (1999).
Hansmann, G. et al. An antiproliferative BMP-2/PPARgamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Invest. 118, 1846–1857 (2008).
Guignabert, C. et al. Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes PDGF receptor-beta-dependent pulmonary arterial muscularization. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L1082–L1090 (2009).
Hansmann, G. et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 115, 1275–1284 (2007).
Martin-Nizard, F. et al. Peroxisome proliferator-activated receptor activators inhibit oxidized low-density lipoprotein-induced endothelin-1 secretion in endothelial cells. J. Cardiovasc. Pharmacol. 40, 822–831 (2002).
Wakino, S. et al. Pioglitazone lowers systemic asymmetric dimethylarginine by inducing dimethylarginine dimethylaminohydrolase in rats. Hypertens. Res. 28, 255–262 (2005).
Kielstein, J. T. et al. Asymmetrical dimethylarginine in idiopathic pulmonary arterial hypertension. Arterioscler. Thromb. Vasc. Biol. 25, 1414–1418 (2005).
Li, M. et al. Heme oxygenase-1/p21WAF1 mediates peroxisome proliferator-activated receptor-gamma signaling inhibition of proliferation of rat pulmonary artery smooth muscle cells. FEBS J. 277, 1543–1550 (2010).
Matsuda, Y. et al. Effects of peroxisome proliferator-activated receptor gamma ligands on monocrotaline-induced pulmonary hypertension in rats [Japanese]. Nihon Kokyuki Gakkai Zasshi 43, 283–238 (2005).
Crossno, J. T., Jr et al. Rosiglitazone attenuates hypoxia-induced pulmonary arterial remodeling. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L885–L897 (2007).
Nisbet, R. E. et al. Rosiglitazone attenuates chronic hypoxia-induced pulmonary hypertension in a mouse model. Am. J. Respir. Cell Mol. Biol. 42, 482–490 (2010).
Juurlink, D. N. et al. Adverse cardiovascular events during treatment with pioglitazone and rosiglitazone: population based cohort study. BMJ 339, b2942 (2009).
Khakoo, A. Y. & Finkel, T. Endothelial progenitor cells. Annu. Rev. Med. 56, 79–101 (2005).
Asahara, T. & Kawamoto, A. Endothelial progenitor cells for postnatal vasculogenesis. Am. J. Physiol. Cell Physiol. 287, C572–C579 (2004).
Tuder, R. M., Groves, B., Badesch, D. B. & Voelkel, N. F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 144, 275–285 (1994).
Spees, J. L. et al. Bone marrow progenitor cells contribute to repair and remodeling of the lung and heart in a rat model of progressive pulmonary hypertension. FASEB J. 22, 1226–1236 (2008).
Hayashida, K. et al. Bone marrow-derived cells contribute to pulmonary vascular remodeling in hypoxia-induced pulmonary hypertension. Chest 127, 1793–1798 (2005).
Angelini, D. J. et al. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELM alpha) recruits bone marrow-derived cells to the murine pulmonary vasculature. PLoS ONE 5, e11251 (2010).
Frid, M. G. et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am. J. Pathol. 168, 659–669 (2006).
Satoh, K. et al. Important role of endogenous erythropoietin system in recruitment of endothelial progenitor cells in hypoxia-induced pulmonary hypertension in mice. Circulation 113, 1442–1450 (2006).
Sahara, M. et al. Diverse contribution of bone marrow-derived cells to vascular remodeling associated with pulmonary arterial hypertension and arterial neointimal formation. Circulation 115, 509–517 (2007).
Marsboom, G. et al. Sustained endothelial progenitor cell dysfunction after chronic hypoxia-induced pulmonary hypertension. Stem Cells 26, 1017–1026 (2008).
Diller, G. P. et al. Circulating endothelial progenitor cells in patients with Eisenmenger syndrome and idiopathic pulmonary arterial hypertension. Circulation 117, 3020–3030 (2008).
Junhui, Z. et al. Reduced number and activity of circulating endothelial progenitor cells in patients with idiopathic pulmonary arterial hypertension. Respir. Med. 102, 1073–1079 (2008).
Toshner, M. et al. Evidence of dysfunction of endothelial progenitors in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 180, 780–787 (2009).
Asosingh, K. et al. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am. J. Pathol. 172, 615–627 (2008).
de Jesus Perez, V. A. et al. Bone morphogenetic protein 2 induces pulmonary angiogenesis via Wnt-beta-catenin and Wnt-RhoA-Rac1 pathways. J. Cell Biol. 184, 83–99 (2009).
Howell, K., Costello, C. M., Sands, M., Dooley, I. & McLoughlin, P. L-Arginine promotes angiogenesis in the chronically hypoxic lung: a novel mechanism ameliorating pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 296, L1042–L1050 (2009).
Zhao, Y. D. et al. Microvascular regeneration in established pulmonary hypertension by angiogenic gene transfer. Am. J. Respir. Cell Mol. Biol. 35, 182–189 (2006).
Teng, R. J., Eis, A., Bakhutashvili, I., Arul, N. & Konduri, G. G. Increased superoxide production contributes to the impaired angiogenesis of fetal pulmonary arteries with in utero pulmonary hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 297, L184–L195 (2009).
Hassoun, P. M. et al. Inflammation, growth factors, and pulmonary vascular remodeling. J. Am. Coll. Cardiol. 54, S10–S19 (2009).
Pullamsetti, S. S. et al. Inflammation, immunological reaction and role of infection in pulmonary hypertension. Clin. Microbiol. Infect. 17, 7–14 (2011).
Tuder, R. M., Groves, B., Badesch, D. B. & Voelkel, N. F. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am. J. Pathol. 144, 275–285 (1994).
Cool, C. D., Kennedy, D., Voelkel, N. F. & Tuder, R. M. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum. Pathol. 28, 434–442 (1997).
Hamamdzic, D., Kasman, L. M. & LeRoy, E. C. The role of infectious agents in the pathogenesis of systemic sclerosis. Curr. Opin. Rheumatol. 14, 694–698 (2002).
Cool, C. D. et al. Expression of human herpesvirus 8 in primary pulmonary hypertension. N. Engl. J. Med. 349, 1113–1122 (2003).
Henke-Gendo, C., Mengel, M., Hoeper, M. M., Alkharsah, K. & Schulz, T. F. Absence of Kaposi's sarcoma-associated herpesvirus in patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 172, 1581–1585 (2005).
Shan, B. et al. Activation of proMMP-2 and Src by HHV8 vGPCR in human pulmonary arterial endothelial cells. J. Mol. Cell. Cardiol. 42, 517–525 (2007).
Bull, T. M. et al. Human herpesvirus-8 infection of primary pulmonary microvascular endothelial cells. Am. J. Respir. Cell Mol. Biol. 39, 706–716 (2008).
Graham, B. B. et al. Schistosomiasis-induced experimental pulmonary hypertension: role of interleukin-13 signaling. Am. J. Pathol. 177, 1549–1561 (2010).
Freitas, T. C., Jung, E. & Pearce, E. J. TGF-beta signaling controls embryo development in the parasitic flatworm Schistosoma mansoni. PLoS Pathog. 3, e52 (2007).
Crosby, A. et al. Pulmonary vascular remodeling correlates with lung eggs and cytokines in murine schistosomiasis. Am. J. Respir. Crit. Care Med. 181, 279–288.
Humbert, M. et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am. J. Respir. Crit. Care Med. 151, 1628–1631 (1995).
Itoh, T. et al. Increased plasma monocyte chemoattractant protein-1 level in idiopathic pulmonary arterial hypertension. Respirology 11, 158–163 (2006).
Tuder, R. M. & Voelkel, N. F. Pulmonary hypertension and inflammation. J. Lab. Clin. Med. 132, 16–24 (1998).
Bhargava, A., Kumar, A., Yuan, N., Gewitz, M. H. & Mathew, R. Monocrotaline induces interleukin-6 mRNA expression in rat lungs. Heart Dis. 1, 126–132 (1999).
Miyata, M. et al. Pulmonary hypertension in rats. 2. Role of interleukin-6. Int. Arch. Allergy Immunol. 108, 287–291 (1995).
Steiner, M. K. et al. Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 104, 236–244 (2009).
Savale, L. et al. Impact of interleukin-6 on hypoxia-induced pulmonary hypertension and lung inflammation in mice. Respir. Res. 10, 6 (2009).
Hagen, M. et al. Interaction of interleukin-6 and the BMP pathway in pulmonary smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 292, L1473–L1479 (2007).
Brock, M. et al. Interleukin-6 modulates the expression of the bone morphogenic protein receptor type II through a novel STAT3-microRNA cluster 17/92 pathway. Circ. Res. 104, 1184–1191 (2009).
Zlotnik, A. & Yoshie, O. Chemokines: a new classification system and their role in immunity. Immunity 12, 121–127 (2000).
Dorfmuller, P. et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 165, 534–539 (2002).
Balabanian, K. et al. CX3C chemokine fractalkine in pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 165, 1419–1425 (2002).
Bazan, J. F. et al. A new class of membrane-bound chemokine with a CX3C motif. Nature 385, 640–644 (1997).
Papadopoulos, E. J. et al. Fractalkine, a CX3C chemokine, is expressed by dendritic cells and is up-regulated upon dendritic cell maturation. Eur. J. Immunol. 29, 2551–2559 (1999).
Fong, A. M. et al. Fractalkine and CX3CR1 mediate a novel mechanism of leukocyte capture, firm adhesion, and activation under physiologic flow. J. Exp. Med. 188, 1413–1419 (1998).
Perros, F. et al. Fractalkine-induced smooth muscle cell proliferation in pulmonary hypertension. Eur. Respir. J. 29, 937–943 (2007).
McDermott, D. H. et al. Association between polymorphism in the chemokine receptor CX3CR1 and coronary vascular endothelial dysfunction and atherosclerosis. Circ. Res. 89, 401–407 (2001).
Sanchez, O. et al. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med. 176, 1041–1047 (2007).
Tournier, A. et al. Calibrated automated thrombography demonstrates hypercoagulability in patients with idiopathic pulmonary arterial hypertension. Thromb. Res. 126, e418–e422 (2010).
White, R. J. et al. Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L583–L590 (2007).
Johnson, S. R., Granton, J. T. & Mehta, S. Thrombotic arteriopathy and anticoagulation in pulmonary hypertension. Chest 130, 545–552 (2006).
Giaid, A. & Saleh, D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 333, 214–221 (1995).
Warburg, O. On metabolism of tumors. (Constable, London, 1930).
Xu, W. et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc. Natl Acad. Sci. U.S.A. 104, 1342–1347 (2007).
Bonnet, S. et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37–51 (2007).
Michelakis, E. D. et al. Dichloroacetate, a metabolic modulator, prevents and reverses chronic hypoxic pulmonary hypertension in rats: role of increased expression and activity of voltage-gated potassium channels. Circulation 105, 244–250 (2002).
McMurtry, M. S. et al. Dichloroacetate prevents and reverses pulmonary hypertension by inducing pulmonary artery smooth muscle cell apoptosis. Circ. Res. 95, 830–840 (2004).
Guignabert, C. et al. Dichloroacetate treatment partially regresses established pulmonary hypertension in mice with SM22alpha-targeted overexpression of the serotonin transporter. FASEB J. 23, 4135–4147 (2009).
Michelakis, E. D. et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2, 31ra34 (2010).
US NIH. ClinicalTrials.gov. Dichloroacetate (DCA) for the treatment of pulmonary arterial hypertension [online], (2010).
Oikawa, M. et al. Increased [18F]fluorodeoxyglucose accumulation in right ventricular free wall in patients with pulmonary hypertension and the effect of epoprostenol. J. Am. Coll. Cardiol. 45, 1849–1855 (2005).
Piao, L. et al. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J. Mol. Med. 88, 47–60 (2010).
Piao, L., Marsboom, G. & Archer, S. L. Mitochondrial metabolic adaptation in right ventricular hypertrophy and failure. J. Mol. Med. 88, 1011–1020 (2010).
Bogaard, H. J., Abe, K., Vonk Noordegraaf, A. & Voelkel, N. F. The right ventricle under pressure: cellular and molecular mechanisms of right-heart failure in pulmonary hypertension. Chest 135, 794–804 (2009).
Haworth, S. The cell and molecular biology of right ventricular dysfunction in pulmonary hypertension. Eur. Heart J. 9 (Suppl. H), H10–H16 (2007).
Pokreisz, P., Marsboom, G. & Janssens, S. Pressure overload-induced right ventricular dysfunction and remodelling in experimental pulmonary hypertension: the right heart revisited. Eur. Heart J. 9 (Suppl. H), H75–H84 (2007).
Gomez, A. et al. Right ventricular ischemia in patients with primary pulmonary hypertension. J. Am. Coll. Cardiol. 38, 1137–1142 (2001).
van Wolferen, S. A. et al. Right coronary artery flow impairment in patients with pulmonary hypertension. Eur. Heart J. 29, 120–127 (2008).
Katsumi, A., Orr, A. W., Tzima, E. & Schwartz, M. A. Integrins in mechanotransduction. J. Biol. Chem. 279, 12001–12004 (2004).
Umar, S. et al. Activation of signaling molecules and matrix metalloproteinases in right ventricular myocardium of rats with pulmonary hypertension. Pathol. Res. Pract. 203, 863–872 (2007).
Broderick, T. L. & King, T. M. Upregulation of GLUT-4 in right ventricle of rats with monocrotaline-induced pulmonary hypertension. Med. Sci. Monit. 14, BR261–BR264 (2008).
Bogaard, H. J. et al. Chronic pulmonary artery pressure elevation is insufficient to explain right heart failure. Circulation 120, 1951–1960 (2009).
Hessel, M., Steendijk, P., den Adel, B., Schutte, C. & van der Laarse, A. Pressure overload-induced right ventricular failure is associated with re-expression of myocardial tenascin-C and elevated plasma tenascin-C levels. Cell Physiol. Biochem. 24, 201–210 (2009).
Kwapiszewska, G. et al. Fhl-1, a new key protein in pulmonary hypertension. Circulation 118, 1183–1194 (2008).
Abdul-Salam, V. B. et al. Identification of plasma protein biomarkers associated with idiopathic pulmonary arterial hypertension. Proteomics 6, 2286–2294 (2006).
Abdul-Salam, V. B. et al. Proteomic analysis of lung tissues from patients with pulmonary arterial hypertension. Circulation 122, 2058–2067 (2010).
Champion, H. C., Michelakis, E. D. & Hassoun, P. M. Comprehensive invasive and noninvasive approach to the right ventricle-pulmonary circulation unit: state of the art and clinical and research implications. Circulation 120, 992–1007 (2009).
Sitbon, O. et al. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension. Circulation 111, 3105–3111 (2005).
Author information
Authors and Affiliations
Contributions
All authors contributed to researching data for the article, made a substantial contribution to the discussion of content, wrote, and reviewed and edited the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
R. T. Schermuly declares he has been a consultant for Actelion, Bayer Healthcare, Ergonex, and Solvay Pharmaceuticals, and has received speakers bureau (honoraria) from Actelion, Bayer Healthcare, GlaxoSmithKlein, Lilly, Solvay Pharmaceuticals, and Pfizer. R. T. Schermuly also declares he has received grant/research support from Actelion, Bayer Healthcare, Ergonex, Excellence Cluster Cardiopulmonary System, German Research Foundation, Gilead, Novartis, Solvay Pharmaceuticals, Pfizer, and the University of Giessen and Marburg Lung Center.
H. A. Ghofrani declares he has been a consultant for Bayer Healthcare, Ergonex, GlaxoSmithKlein, Novartis, and Pfizer, and has received speakers bureau (honoraria) from Bayer Healthcare, GlaxoSmithKlein, Novartis, and Pfizer. H. A. Ghofrani also declares he has received grant/research support from Bayer Healthcare, Excellence Cluster Cardiopulmonary System, German Research Foundation, Novartis, Pfizer, and the University of Giessen and Marburg Lung Center.
M. R. Wilkins declares he has been a consultant for Biomarin and Pfizer, has received speakers bureau (honoraria) from Bayer Healthcare, GlaxoSmithKlein, and Pfizer, and has received grant/research support from Biomarin.
F. Grimminger declares he has been a consultant for Bayer Healthcare, has received speakers bureau (honoraria) from Actelion, Encysive, and Pfizer, and has received grant/research support from Actelion, Excellence Cluster Cardiopulmonary System, GlaxoSmithKlein, German Research Foundation, Novartis, Parexel International GmbH, Pfizer, and the University of Giessen and Marburg Lung Center.
Rights and permissions
About this article
Cite this article
Schermuly, R., Ghofrani, H., Wilkins, M. et al. Mechanisms of disease: pulmonary arterial hypertension. Nat Rev Cardiol 8, 443–455 (2011). https://doi.org/10.1038/nrcardio.2011.87
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrcardio.2011.87
This article is cited by
-
Periodontitis exacerbates pulmonary hypertension by promoting IFNγ+ T cell infiltration in mice
International Journal of Oral Science (2024)
-
A single-cell atlas of lung homeostasis reveals dynamic changes during development and aging
Communications Biology (2024)
-
Molecular Insights into the Relationship Between Platelet Activation and Endothelial Dysfunction: Molecular Approaches and Clinical Practice
Molecular Biotechnology (2024)
-
Extracellular vesicles in venous thromboembolism and pulmonary hypertension
Journal of Nanobiotechnology (2023)
-
Inhibition of Hsp110-STAT3 interaction in endothelial cells alleviates vascular remodeling in hypoxic pulmonary arterial Hypertension model
Respiratory Research (2023)
